

Abstract
The importance of protonation states and proton transfer in pyridoxal 5′-phosphate (PLP)-chemistry can hardly be overstated. Although experimental approaches to investigate pKa values can provide general guidance for assigning proton locations, only static pictures of the chemical species are available. To obtain the overall protein dynamics for the interpretation of detailed enzyme catalysis in this study, guided by information from solid-state NMR, we performed molecular dynamics (MD) simulations for the PLP-dependent enzyme tryptophan synthase (TRPS), whose catalytic mechanism features multiple quasi-stable intermediates. The primary objective of this work is to elucidate how the position of a single proton on the reacting substrate affects local and global protein dynamics during the catalytic cycle. In general, proteins create a chemical environment and an ensemble of conformational motions to recognize different substrates with different protonations. The study of these interactions in TRPS shows that functional groups on the reacting substrate, such as the phosphoryl group, pyridine nitrogen, phenolic oxygen and carboxyl group, of each PLP-bound intermediate play a crucial role in constructing an appropriate molecular interface with TRPS. In particular, the protonation states of the ionizable groups on the PLP cofactor may enhance or weaken the attractions between the enzyme and substrate. In addition, remodulation of the charge distribution for the intermediates may help generate a suitable environment for chemical reactions. The results of our study enhance knowledge of protonation states for several PLP intermediates and help to elucidate their effects on protein dynamics in the function of TRPS and other PLP-dependent enzymes.
Keywords: proton switch, internal aldimine, aminoacrylate, indoline quinonoid, conformational change, water dynamics, charge remodulation, channeling, allosteric regulation
Introduction
In many enzymes, the chemistry of catalysis is based on simple acid-base reactions involving a series of proton transfers.1–4 The protonation state of a specific atom can act as a promoter or mediator to initiate chemical reactions in enzyme active sites. Protonation/deprotonation of atoms at the catalytic site, coenzyme, and/or the bound substrate may affect structural stability, induce conformational changes, and promote or reduce enzymatic activity.5,6 Thus, determining protein structures, including correctly identifying proton positions, is critical for understanding enzyme-catalyzed pathways at a chemical level. High-resolution crystal structures usually define the spatial location and nature of the protein residues and cofactors that interact with reacting substrates. However, the resolution of most protein structures, typically 1.5–2.5 Å, is insufficient to reliably determine the protonation states of key ionizable groups in the active site.
The efficacy of 13C- and 15N-enriched substrates for determining protonation and hybridization states by solid-state NMR (ssNMR) has been exploited to map electrostatic and chemical environments at the catalytic sites of enzymes.7,8 The increasing relevance of ab initio calculations to the analysis of enzyme catalytic mechanisms provides a new level of quantification that is reshaping bioorganic mechanistic concepts.9 The combination of isotopic labeling in ssNMR experiments with computational studies grounded in quantum theory can be tailored to a scale where only protein residues in and around the active site and atoms of the substrate need be considered to generate atomic level models relevant to catalysis. With this detailed chemical information about the active site in hand, full structural models of the protein can be explored to determine how the position of a single proton can change the overall protein dynamics and further activate or inactivate enzyme catalysis.
Bacterial tryptophan synthase (TRPS), an α2β2 tetrameric enzyme that catalyzes the last two steps in the synthesis of L-tryptophan, serves as our model system [Fig. 1(top)]. Since the 1950s, TRPS has been exploited as a paradigm for understanding the catalytic and regulatory mechanisms of enzyme complexes,10–12 and it has recently been implicated as a target in the development of drugs for infectious diseases and herbicides.13,14 Catalysis in the α- and β-subunits is regulated via allosteric interactions that switch the protein from an open, inactive conformation to a closed, active conformation.12,15–19 Closing of the α-subunit is associated with large motions in α-loop L6 (αL6, residues α179–193) that switch the subunit from a disordered (open) to an ordered (closed) state, while the open-to-closed transition of the β-subunit involves motion of the communication (COMM) domain (residues β102–189). Measurement of two distances (αL6: βH6 of the COMM domain and αL6: αL2) determines whether or not the α-subunit is considered open, partially closed, or closed. The β-subunit is considered closed upon formation of a salt-bridge between βArg141 and βAsp305.20 Switching between the conformations is modulated by the binding of substrates to the α-site, binding of monovalent cations (MVCs), and covalent interconversions between intermediates formed at the β-site. Although both the α- and β-subunits of TRPS can have open or closed conformations, catalysis likely occurs only within the closed conformations.21 Substrates and products bind and dissociate via intermediates displaying open conformations (e.g., the internal aldimine (E(Ain)), while the α-aminoacrylate and quinonoid forms have completely closed conformations.
Figure 1.

Overall structure and chemical reactions of tryptophan synthase (TRPS). (top) TRPS is composed of an α-subunit (purple) and β-subunit (yellow). The two ligands binding to each subunit are shown in bead representation. The open, partially closed, and fully closed conformations of the α-subunit are controlled by αL2 (pink), αL6 (green), and βHelix-6 of the COMM domain (blue). The residues βArg141 and βAsp305, highlighted in red, are associated with the open and closed β-site conformations. The tunnel used to channel indole from the α-site to the β-site is marked as a cyan dashed line. (bottom) The α- and β-reactions of TRPS.
The detailed interactions and driving forces that induce conformational changes in TRPS have been investigated in early crystal studies and molecular dynamics (MD) simulations.22–25 Much focus has been placed on the hydrophobic tunnel of about 25 Å that connects the two active sites in crystal structures of TRPS [Fig. 1(top)]. Indole generated by the cleavage of indole-3-glycerol phosphate (IGP) at the α-site is transferred via this tunnel into the β-site where the synthesis of L-Trp is completed. Substrate channeling may have many advantages over the free diffusion of reaction intermediates via the bulk solvent. For example, channeling may reduce the transit time for transferring the reaction intermediate from one active site to the next.26–28 However, it is likely that the transfer of indole from the α-site to the β-site in TRPS evolved to prevent the escape of indole into solution, as the catalytic turnover rate for the enzyme is much too slow to benefit from the rapid diffusion of indole into the β-site via the tunnel.
Figure 1(bottom) depicts the chemical pathway for the biosynthesis of L-Trp in the pyridoxal 5′-phosphate (PLP; Vitamin-B6)–dependent TRPS bienzyme complex. The α-subunit catalyzes the α-reaction, consisting of cleavage of IGP to yield indole and glyceraldehyde-3-phosphate (G3P), Eq. (1).17 The β-subunit catalyzes the condensation of indole with L-Ser to produce L-Trp in a pathway involving at least nine different intermediates formed from PLP and the reacting substrates, Eq. (2).29,30
| (1) |
| (2) |
In stage I of the β-reaction, L-Ser reacts with the cofactor in its E(Ain) resting form, producing in sequence the species gem-diamine, E(GD1); L-Ser external aldimine, E(Aex1); quinonoid, E(Q1); and α-aminoacrylate Schiff base (SB), E(A-A), and a water molecule. In stage II, indole, channeled from the α-site, makes a nucleophilic attack on E(A-A), giving the intermediates E(Q2), E(Q3), E(Aex2), E(GD2), and, finally, L-Trp. TRPS complexes with ligands bound to the α-site readily crystallize and, when soaked with substrates and substrate analogues for the β-reaction, give quasi-stable, catalytically active species equivalent to intermediates formed along the β-reaction pathway. For example, indoline and 2-aminophenol (2AP) both have been shown to behave as indole analogues, reacting rapidly with E(A-A) to produce the long-lived indoline quinonoid, E(Q)indoline, and 2AP quinonoid, E(Q)2AP, species, respectively. The E(Q)indoline intermediate slowly converts to the novel amino acid dihydroiso-L-tryptophan (DIT), while the E(Q)2AP species is stable and does not appear to turn over to form product(s) (Supporting Information Scheme S1).31,32 The compounds α-D,L-glycerol phosphate (GP), and the two high-affinity α-site binders, N-(4′-trifluorome-thoxybenzoyl)-2-aminoethyl phosphate (F6) and N-(4′-trifluoromethoxybenzenesulfonyl)-2-aminoethyl phosphate (F9), have been extensively studied and used as analogues for IGP.33 X-ray crystal structures are available for F6 and F9 complexes of the TRPS E(Ain), E(A-A), and both the E(Q)indoline and E(Q)2AP derivatives. Although these structures range in resolution from ~1.75 to ~1.20 Å, localizing the protons on the ionizable groups in the β-site by X-ray crystallography remains elusive. Recent ssNMR studies provided chemical shifts for selected atoms of the E(Ain) intermediate and have established that the SB linkage of the Lys87 ε-imine nitrogen is protonated while the pyridine nitrogen (PN) is deprotonated for this species.34 However, for the E(A-A), E(Q)indoline, and E(Q)2AP, previously published32 and additional chemical shift data35 (unpublished data; manuscript in preparation) seem to support the idea that the proton shifts from the SB nitrogen to the phenolic oxygen (PO), at least as the majority species. However, the predominant protonated PO structures appear to participate in proton exchange both with the carboxylate oxygen (CO) and, to a lesser extent, the SB nitrogen in the E(Q)indoline and E(Q)2AP forms (Scheme 1; Supporting Information Fig. S1). As a result, these various states of protonation must be considered when crafting MD models.
Scheme 1.
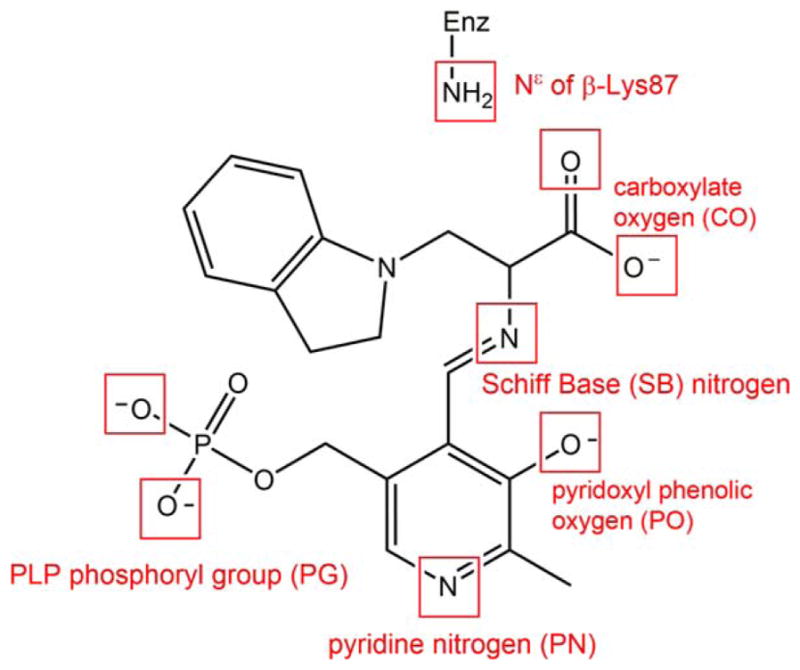
Example of the potential sites of protonation on an indoline quinonoid substrate. The ionizable groups, shown in a red box, include an atom that can be protonated or deprotonated.
The combination of X-ray crystallography and ssNMR provides an important platform for understanding structural frameworks at active sites by revealing chemically rich details about the interactions between enzyme residues and the reacting substrates. Yet, we still lack understanding of how the transfer of a proton among different loci changes TRPS dynamics and the relative stability of intermediates within the β-site. In this study, we investigate the dynamic behavior of TRPS for different intermediates – E(Ain), E(A-A), E(Q)indoline, and E(Q)2AP – formed during the β-reaction via MD simulations, including all-atom information and explicit solvent motions. Simulations probe the effects of different protonation states for the PLP ionizable groups and the covalently attached β-site substrates/analogues on the overall enzyme dynamics and stability. By examining dynamic motions of the TRPS complexes, we elucidate protein–substrate inter- and intra-molecular interactions and the changes in motions due to switching of proton positions in the reacting species. We also investigate key interactions among water molecules and TRPS substrates with open β-site conformations. We discuss the implications of these MD studies for understanding the roles played by protonation states and water interactions in the mechanism of TRPS.
Results and Discussion
Determining the protonation states of ionizable groups in β-reaction intermediates is essential for understanding the most important remaining questions concerning the mechanism of the acid-base chemical reactions in TRPS catalysis. In this respect, we aim to explore the detailed dynamics of model structures that differ only in placement of one proton in the β-active site. Seven different locations were alternately protonated/deprotonated for simulations: the phosphoryl group (PG), pyridoxyl nitrogen (PN), pyridoxyl oxygen (PO), SB nitrogen, both carboxyl oxygens (CO) of the L-Ser substrate, and the neighboring ε-nitrogen of βLys87 (Scheme 1). Guided by chemical intuition, not all of these sites can be simultaneously protonated. Therefore, we focus on structures relevant to one proton exchanging positions between the PO, SB, and CO sites. Doubly-protonated species that contain an additional proton on the PN or PG are also considered to study the specific interactions between the PLP ligand and enzyme residues. Thus, 17 total possible ligand-substrate protonation schemes are considered. To simplify the notation, we designate structural models to indicate specific protonation states using a system of abbreviations; for example, a proton at the PO position in E(Ain) is denoted as E(Ain):PO, while a proton at the SB position in E(A-A) is designated E(A-A):SB. The different protonation states considered here for intermediates in the TRPS β-reactions are listed in Scheme 2.
Scheme 2.

Substrates investigated in this molecular dynamics (MD) simulation study. The intermediates include the changes in proton on each ionizable group. The predominant protonation states from solid-state NMR reports are shown in boxes.
In this study, we assume that crystal structures provide equilibrium coordinates for all heavy atoms inclusive of the protein and α- and β-substrates; hence, the structures obtained from the PDB show optimal geometries in global energy minima. Using X-ray structures as templates, we manually assign protonation states for the β-reaction intermediates based on suggestions from the ssNMR data. The 50 ns MD simulations are considered short compared to the time scale of protein folding conformational dynamics, so the sampled conformations should theoretically stay within a local energy well. Although the substrates and protein are mobile in solution, the overall geometry should not be significantly skewed from the crystal structures. Therefore, for all intermediates studied, we carefully checked the fluctuation of substrates and key residues by aligning the crystal structures and MD trajectories. Moreover, we also discuss the ways that different protonation states affect the conformation or catalysis of TRPS through changes in overall and local protein dynamics and in protein–water interactions.
Protonation states of E(Ain)
E(Ain) is the resting form of the enzyme; it occurs before PLP fuses with L-Ser in the β-catalytic reaction. The 15N chemical shift for the linking Lys ε-nitrogen from ssNMR reveals a protonated SB linkage, while 13C, 15N and 31P chemical shifts on PLP report that the PG, PO, and pyridine ring nitrogen are deprotonated.34 Our MD simulations show that the E(Ain):SB complex is locked into the binding pocket of the β-subunit. Figure 2(SB) shows the residues that make direct contact with E(Ain):SB. The PG forms multiple H-bonds with neighboring residues and is enclosed by His86, Thr190, and the loop Gly232, Gly233, Gly234, Ser235, and Asn236. Ser377 forms interactions with PN, as previously documented.34 Of note, based on pKa calculations performed with the MCCE program,36 His86 is assigned a doubly-protonated state with hydrogen atoms at both the ε- and δ-nitrogens. Although this residue can occasionally form H-bonds with the PG, the two protons at the nitrogen atoms of the imidazolyl ring can also form H-bonds to the Ser235 side chain and the Ser377 backbone simultaneously, allowing His86 to connect the α-helix and loop to create a well-formed cavity for TRPS β-site intermediates (see Supporting Information Fig. S2). In addition, the E(Ain) active site is filled with numerous water molecules. The crystal water molecules (PDB ID: 4HT3) Wat502 and Wat505 are highly stable during the 50 ns MD simulations. Wat502 bridges Ala237 and Cys230, and Wat505 stays localized between Cys230 and Asn375 to form H-bonds with both residues [Fig. 2(SB)].
Figure 2.

Protonation states of E(Ain). (left) The ligand (cyan) shows a proton at the SB nitrogen. The residues and water molecules forming interactions with E(Ain) are also shown. (middle) A proton was added at the pyridine nitrogen (PN). Motions of the ligand with two different protonation states, SB (cyan) and SB_PN (pink). (right) Comparison of a proton at the PO (yellow) and SB nitrogen (cyan). The distance between the PN and Ser377Oγ is shown in blue, and the distance between the phosphorous atom and Gly234N is shown in red. All snapshots are from 50-ns MD simulation.
To further test the structure of E(Ain):SB, we added one more proton at PN to create another protonation state designated E(Ain):SB_PN. Figure 2(SB_PN) shows the alignment of the two protonation states: E(Ain):SB and E(Ain):SB_PN. The largest difference between the two models is that the presence of two protons in E(Ain):SB_PN causes a shift in the position of the coenzyme in the binding pocket, resulting in the loss of interactions between the PG of E(Ain) and Gly232, Gly233, and Gly234. Also, the H-bond between the PN and Ser377 is missing (see the distance plots in Fig. 2). The shift of E(Ain):SB_PN in the MD simulations results from the rotation of the Lys87 side chain. The Lys residue has 5 rotatable side chain dihedral angles. The first and third dihedrals do not change during the simulation; however, the rotations of angles #2, #4, and #5 cause destabilization of the ligand (Supporting Information Fig. S3). In addition, the proton at the PN alters the motions of the coenzyme and changes the dynamics of nearby protein residues. The RMSDs of the protein backbone and side chains within 7 Å of the ligand were calculated (Supporting Information Fig. S4) and determined from MD simulations between 5 and 50 ns. The mean side chain RMSDs in the E(Ain):SB, E(Ain):SB_PN, and E(Ain):PO complexes were 1.87 ± 0.23, 2.51 ± 0.32 and 1.80 ± 0.15 Å, respectively, indicating that the protein residues near the binding cavity become more flexible when a proton is placed on the PN. Notably, our recent study showed that similar to other fold type II PLP enzymes, protonation at the PN position is precluded by interaction with a protonated Ser377 hydroxyl group,34 and our MD simulations agree well with this work.
We explored the effects of protonation at the PO by moving the proton from the SB nitrogen to the PO to create the state E(Ain):PO. Although the substrate and protein dynamics seem essentially unaffected by the switch of the proton position from the SB nitrogen to the PO, water molecules exhibit different behavior between E(Ain):SB and E(Ain):PO. Since E(Ain) shows open conformations of the β-subunit, the substrate in the β-site is well solvated by water molecules. Our MD simulations of E(Ain):SB show that the PG, PO, and Lys87-εN are all exposed to a number of water molecules (Supporting Information Fig. S5), which can bridge interactions between the SB nitrogen and PO, in agreement with observations from a recent study.34 RMSD calculations of water motions in the E(Ain):SB structure revealed that compared to bulk water, which relocates frequently, several crystallographic water molecules stay with the deprotonated PO for more than 30 ns. This suggests that the water molecules have a higher tendency to stay in the binding pocket when a proton is located on the SB nitrogen rather than the PO; indeed, simulations of E(Ain):PO show that the PO is surrounded by fewer water molecules when it possesses the proton. The mean number of water molecules found within 4 Å of the PO in the E(Ain):SB and E(Ain):PO simulations is 3.1 ±1.1 and 1.9 ±0.8, respectively (Supporting Information Fig. S6). The poor solvation for the E(Ain):PO complex may be due to the neutral PO; in contrast, a formal negative charge on the PO in the E(Ain):SB structure could attract more water molecules. The water molecules near the substrate and Lys87 may affect proton delivery from the SB nitrogen to the PO in the next catalytic steps, wherein E(Ain):SB converts to the E(A-A):PO intermediate.
Protonation states of E(A-A)
The stable E(A-A) intermediate is formed upon covalent binding of L-Ser to the PLP cofactor; several crystal structures are available showing the β-subunit in the closed conformation (we used PDB ID 4HN4). In contrast to E(Ain), which covalently binds Lys87, the E(A-A) intermediate does not form a covalent bond with the protein, and it is ready for fusing indole to L-Ser to produce L-Trp. Based on model compound studies,37 the 13C ssNMR spectra for E(A-A) indicate that the proton remains on the PO for a majority of the lifetime of the intermediate, though it is possible to transfer the proton to the SB nitrogen for short spans of time. The protein makes three direct contacts with the substrate-coenzyme complex [see Fig. 3(PO)]. First, the PG is locked in place through interactions with residues His86, Lys87, Thr190, Gly234, Ser235, and Asn236. The dihedral angles of the Lys87 side chain are highly rotatable, and the side chain can flip back and forth to form interactions with both the PG and Gly189. Second, the PN forms hydrogen-bonding interactions with the β-hydroxyl group of the Ser377 side chain. Third, CO atoms interact with the hydroxyl group of the Thr110 side chain and the backbone nitrogens of Gly111 and His115 (Fig. 4). In addition, we observed intra-molecular H-bonds between the SB nitrogen and PO. TRPS in complex with E(A-A) shows a closed conformation of the β-subunit, with no water molecules near the PN, PO, SB nitrogen, and COs. Although several highly mobile water molecules are located around the PG and form an interaction network during the MD simulations, in contrast to E(Ain), no structural (crystallographic) waters are observed.
Figure 3.

Protonation states of E(A-A). (PO) Presentation of E(A-A):PO binding to TRPS. The residues interacting with the phosphoryl group (PG), PN, and carboxylate oxygens (COs) of E(A-A):PO are shown in yellow, pink, and purple, respectively. (CO) The alignment of E(A-A):CO (yellow) and E(A-A):PO (cyan). Residues Lys87 and Gln114 are shown. The H-bond between Lys87 and the PO of E(A-A):CO is shown in green. (SB) The alignment of E(A-A):SB (pink) and E(A-A):PO (cyan). The Gln114 side chain forming H-bonds with the PO of E(A-A):SB is shown as a green dashed line. (PO_PN) The alignment of E(A-A):PO_PN (orange) and E(A-A):PO (cyan) is depicted. The Lys87 and Ser377 residues can form H-bonds with E(A-A):PO_PN. All snapshots are from 50-ns MD simulation.
Figure 4.

Percentage of H-bonds found between the CO atoms and the three residues, Thr110, Gly111 and His115. Percentage of H-bonds formed during 50-ns MD simulations of the TRPS in complex with E(A-A):PO, E(A-A):CO, E(A-A):SB and E(A-A):PO_PN.
The α-subunit produces indole, which is then transferred to the β-site through the tunnel followed by the series of chemical steps comprising Stage II of the β-reaction. To model the E(A-A) species with an indole positioned to form the E(Q)indole intermediate, the complex of BZI bound to E(A-A) was prepared. The X-ray crystal structure of this complex (PDB ID: 4HPX) shows BZI is bound in the same position indole would occupy in the E(A-A) complex for reaction with Cβ of the α-aminoacrylate intermedate.23 Once in the tunnel, BZI locates the entrance to the β-subunit active site, which is lined by the hydrophobic residues Glu109, His115, Leu166, Cys170, Leu188, Phe280 and Phe306 [Supporting Information Fig. S7(left)]. The Glu109 carboxyl group makes an H-bond to one BZI nitrogen; the indole ring of E(Aex2) binds in an essentially identical fashion.15 In this way, the residues around BZI create an optimal environment to stabilize the substrate in the binding pocket, which helps to explain earlier findings that the mutation of Glu109 could alter the substrate specificity and catalytic activity of the β-subunit.38 Although BZI was mobile during the simulation [Supporting Information Fig. S7(right)], the interactions between E(A-A):PO and TRPS mentioned previously remain intact. This indicates that the β-subunit active site can accommodate BZI while the coenzyme-substrate complex resides in the α-aminoacrylate form, which remains stable with a proton on the PO.
When the proton moves from the PO to the COs, denoted as E(A-A):CO, protein dynamics and conformations undergo changes that allow the β-site to open. The net charge of the carboxyl group changes from −1 to 0, which affects the H-bond network. Figure 4 shows the amount of H-bonding observed between the CO atoms and the residues Thr110, Gly111, and His115 during the 50 ns MD simulations. Compared to E(A-A):PO, and the E(A-A):SB and E(A-A):PO_PN complexes described in the following paragraph (which hold a −1 charge at CO atoms), fewer H-bonds are present between the COs and the protein for the E(A-A):CO structure. In particular, interactions between the COs and the side chain of Thr110 are missing. Although the structures of E(A-A):PO and E(A-A):CO seem similar upon alignment [Fig. 3(CO)], the weaker interactions around the COs of E(A-A):CO result in the destabilization of the residues around the substrate. The Lys87-ε-N now forms H-bonds with the PO, and Gln114 tends to move toward the PO instead of retaining a conformation exposed to solvent, which has not been observed in any X-ray structures. Furthermore, we also measured the distance between Arg141Cζ and Asp305Cγ, which defines the open and closed conformations of the β-subunit. The salt-bridge between Arg141 and Asp305 breaks when the proton transfers from the PO to the CO, indicative of an open β-site. This allows water molecules to move from bulk solution and join the cluster near the PO (Fig. 5). The theoretical formation of the E(A-A):CO structure in TRPS shows how changing a single proton location affects the local binding site and alters the overall protein dynamics and molecular motions.
Figure 5.

Comparison of water motions and protein conformations in the E(A-A):PO, E(A-A):CO and E(A-A):PO_PG complexes. Water molecules within 3.2 Å of E(A-A) substrates at 50 ns are shown in the bead representation. The red circle indicates that several water molecules cluster around the PO of E(A-A):CO and E(A-A):PO_PG. We also measured the distance between Arg141Cζ and Asp305Cγ, which defines the open and closed conformations of the β-site. The TRPS in complex with E(A-A):CO shows the open conformations occasionally, and the E(A-A):PO_PG complex shows the open conformations beginning at 12 ns in the simulation. A distance between Arg141Cζ and Asp305Cγ of less than 5.5 Å indicated the closed conformation and greater than 8.5 Å indicaed the open conformation. All snapshots are from 50-ns MD simulation.
Two other protonation states of the E(A-A) complex were also modeled: E(A-A):SB and E(A-A):PO_PN. The RMSD plot in Supporting Information Figure S8 shows that both of these structures have larger dynamic fluctuations compared to the E(A-A):PO structure. The PO of the E(A-A):SB complex holds a −1 charge, similar to E(A-A):CO, allowing the Gln114 side chain to hydrogen bond with the PO [Fig. 3(CO)] and causing a slight shift in ligand position. To retain H-bonds between the PN and the hydroxyl group of Ser377, the conformation of the E(A-A):PO_PN structure twist slightly, which allows the Lys87 side chain to rotate to the back side of the complex. This results in new interactions between the Lys87 side chain and the SB nitrogen [Fig. 3(PO_PN)], and a loss of interactions between the PG and Lys87. However, large movements of the Gln114 and Lys87 side chains are not evidenced in the available crystal structures, which further supports the dominance of the PO structure and the low populations of the E(A-A):SB and E(A-A):PO_PN complexes.
Protonation states of E(Q)indoline and E(Q)2AP
E(A-A) reacts too rapid with indole to give a quasi-stable E(Q)indole [Fig. 1(bottom)] for extended crystal studies. The indole analogue indoline, however, can be used in the reaction with E(A-A) to produce a long-lived, quasi-stable E(Q)indoline intermediate (PDB ID: 3PR2). The ssNMR results suggest that the proton of E(Q)indoline is capable of exchanging between the PO, CO, and SB nitrogen positions, and recent data suggests that the protonated PO form dominates the equilibrium. Like the E(A-A) structures, the E(Q)indoline:PO complex displays three major interactions with the enzyme: the PG interacts with His86, Lys87, Thr190, Gly234, Ser235, and Asn236; the PN interacts with Ser377; and the COs interact with Thr110, Gly111, Gln114, and His115. In fact, the E(A-A) and E(Q)indoline structures differ only in the binding of an indoline molecule to the serine-derived Cβ. The models also reveal an H-bond between the PO and SB nitrogen. Furthermore, the hydrophobic rings of indoline can interact well with the nonpolar functional groups of neighboring residues Glu109, His115, Leu166, Thr190, and Phe306, providing additional contacts between the coenzyme-substrate complex and the protein [Fig. 6(PO)] and resulting in a highly stable intermediate.
Figure 6.

Protonation states of E(Q)indoline. (PO) The residues interacting with the indoline ring of E(Q)indoline:PO are shown in the green bond representation. (CO) The alignment of E(Q)indoline:CO (yellow) and E(Q)indoline:PO (cyan). The good alignment of the residues Lys87, Gln114, and Ser377 is shown. (SB) The alignment of E(Q)indoline:SB (pink) and E(Q)indoline:PO (cyan). The residues Gln114 and Ser377 form H-bonds (red dash lines) with the ligand. (PO_PN) The alignment of E(Q)indoline:PO_PN (orange) and E(Q)indoline:PO (cyan). The hydroxyl group of Ser377 and Thr110 rotates away from E(Q)indoline:PO_PN. All snapshots are from 50-ns MD simulation.
When the proton shifts to the CO, E(Q)indoline:CO and E(Q)indoline:PO show good structural alignment [Fig. 6(CO)]. The key residues Lys87, Gln114, and Ser377 maintain similar conformations for both protonation states. The agreement and stability of both structures adds credence to ssNMR findings indicating that the proton may be in fast exchange between the PO and COs. In contrast to the changes that occur upon deprotonation of the PO in the E(A-A) complex, Gln114 does not appear to shift toward the PO, and the H-bonds between the COs of E(Q)indoline:CO and Thr110 remain stable, as in the E(Q)indoline:PO structure (Supporting Information Fig. S9). The differences between the E(Q)in-doline:CO and E(A-A):CO complexes may be due to distinct interactions between protein residues and indoline in the active site, providing better stabilization of the structure. Thus, for E(Q)indoline, an exchange of the proton from the PO to the COs has little overall effect on the dynamics and stability of the substrate-cofactor complex and the residues in the binding cavity.
The E(Q)indoline:SB form of the cofactor complex was also modeled, as it has been postulated to play a role in the exchange of the proton between the PO and CO positions. In this structure, the COs maintain their hydrogen bonds to Thr110, and Ser377 remains hydrogen-bonded to the PN. However, much like the E(A-A):CO and E(A-A):SB states, the side chain of Gln114 rotates to form interactions with the PO rather than remaining exposed to solvent [Fig. 6(SB)], promoting a slight twisting of the pyridine ring. These motions result in decreased stability in the binding pocket, which explains the lower population of the E(Q)indoline:SB form.
The traditional view of PLP enzymes holds that the PN should be protonated, so we investigated the effects of protonation at this site. Addition of a proton to the PN to create the E(Q)indoline:PO_PN complex results in the hydroxyl side chain of Ser377 rotating away from the PN and breaking the H-bond with the PN [Fig. 6(PO_PN)]. The PLP ring also moves away slightly from Ser377 due to the more crowded environment. This movement of the ring causes instability of the substrate-derived carboxyl group, as some of the H-bonds between the COs and Thr110 become lost (Supporting Information Fig. S9). Further instability results from the presence of water molecules between the COs and Thr110 at 40 to 50 ns during the MD simulations, which destroys the linkage between E(Q)indoline:PO_PN and Thr110 (Supporting Information Fig. S10). Thr110 is one residue of the COMM domain, and the missing interactions trigger the opening of the β-subunit of TRPS; hence, the salt-bridge between Arg141Cζ and Asp305Cζ cannot form, and water molecules move into the binding site (Supporting Information Fig. S11). The RMSD plot in Supporting Information Figure S12 indicates that the stability of coenzyme-substrate complexes decreases as shown: E(Q)indoline: PO >E(Q)indoline:CO > E(Q)indoline:SB > E(Q)indoline: PO_PN. Although the quinonoid form is most stable with a protonated PO, the fluctuations of the complexes containing a protonated CO or SB nitrogen are all within 1.0 Å, suggesting a similar geometry for each of the protonation states. However, the RMSD for E(Q)indoline:PO_PN increases significantly during the 50 ns simulation because the complex searches for a lower energy conformation. Our findings from MD simulations agree with ssNMR studies suggesting that the PN is likely unprotonated.
When 2AP is used as the incoming nucleophile in stage II of the β-reaction, it bonds to Cβ through its amino group to form another stable E(Q)indole analogue, E(Q)2AP (PDB ID: 4HPJ). The hydroxyl group forms an H-bond with Glu109, and the hydrophobic side chains of Leu166, Thr190 and Phe306 provide weak attractive forces to the ring [Fig. 7(PO)]. The E(Q)2AP complex is more tightly bound to the protein; the intermediate fails to turnover to an amino acid product. Simulations of this quino-noid intermediate with the protonation states E(Q)2AP:PO, E(Q)2AP:CO, and E(Q)2AP:SB show that the structures are all stable in the binding pocket, and the attractions between the protein and the PG, CO, and PN are all retained during the 50 ns simulations [Fig. 7(CO) and (SB)]. Gln114 can move toward the solvent or the substrate in the E(Q)2AP:CO and E(Q)2AP:SB simulations. The structures become unstable when a proton is added to the PN. The simulation of the E(Q)2AP:PO_PN complex shows missing H-bonds between the PN and Ser377 [Fig. 7(PO_PN)], and the resulting motions increase instability in the binding pocket. Overall, the motions of the E(Q)2AP analogue to TRPS are similar to the motions of the E(Q)indoline complexes in our study. Again, the suggested protonation states of E(Q)2AP from MD simulations agree with experimentally determined ssNMR chemical shifts.
Figure 7.

Protonation states of E(Q)2AP. (PO) The residues interacting with the 2-aminophenol ring of E(Q)2AP:PO are shown in a yellow bond representation. (CO) and (SB) The residue Gln114 is flexible during the 50 ns MD simulations. The side chain of Gln114 can move toward either the PO or solvent depending on protonation state. The conformations of Gln114 at 10, 20, 30, 40, and 50 ns are shown in different colors. (PO_PN) The key residues around E(Q)2AP:PO_PN are shown in yellow bond. The red circle indicates the missing H-bond between the PN and Ser377. All snapshots are from 50-ns MD simulation.
Roles of the pyridine nitrogen in PLP catalysis
The pyridine ring nitrogen is one of the functional groups critical to PLP catalysis, and an H-bond between the PN and enzymatic residues is essential for catalysis. Protonation of the PN in most PLP-dependent enzymes is traditionally proposed because it allows the coenzyme to adopt a zwitterionic structure, creating resonance-stabilized carbanionic intermediates. This stabilization is conventionally attributed to the ability of the protonated pyridine ring to act as an electron-sink.39,40 For example, early studies of aspartate aminotransferase confirmed that some enzymes use the cofactor with a fully protonated PN, which then forms a salt-bridge with Asp222 below the pyridine ring.41 However, recent studies suggest that some PLP enzymes, such as alanine racemase and O-acetylserine sulfhydrylase, use the PN in an unprotonated form. This remodulation of the PLP structure demonstrates that not all PLP enzymes require the protonated PN for stabilization of carbanionic intermediates.42–44 Our MD simulations confirm that although a proton never transfers to the PN in TRPS, the enzyme can use the β-hydroxyl group of Ser377 to form a strong H-bond with the PN, which still enables the formation of carbanionic intermediates. Attempts to bind a second proton to the PN cause a shift of the entire β-subunit, such as in the E(Ain) structure [Fig. 2(SB_PN)], or cause the PLP-substrate complex to twist in search of better interactions for Ser377, as in the E(A-A) and E(Q)indoline complexes [Fig. 3(PO_PN)]. It is clear that there is not enough space to accommodate a second proton in the area between the PN and Ser377.
Because the X-ray structures, ssNMR, and dynamic modeling studies preclude arrangements with a protonated PN, the negative charge density on the quinonoid intermediate must be accommodated through other weak and strong bonding interactions between the complex and the protein scaffolding at the site.42 One DFT computational study showed the distribution of atomic charges in PLP derivatives using both a protonated and unprotonated PN.45 However, consideration of the PLP-substrate complex and only a few nearby atoms is not enough to capture the full picture of protein–substrate-cofactor interactions, overall conformational changes, and dynamics of substrates binding to PLP enzymes. Therefore, to study whether the charge distribution on the PN plays a crucial role in controlling reaction specificity in PLP enzymes, we manually assigned a negative charge ranging from −0.4 to −0.9 at the PN of E(Q)indoline, with the total charges of the overall complex fixed at −4. Figure 8 shows the rotation of the Ser377 side chain as a function of charge at the PN. The H-bond between the PN and the hydroxyl group of Ser377 are missing with a charge −0.4. Although the H-bond is maintained initially with charges of −0.5, −0.6, and −0.7 on the PN, the side chain of Ser377 ultimately rotates away at 20–40 ns. As the negative charge on the PN is decreased to −0.75 and −0.8, the Ser377 side chain can rotate to the back side of the ring in the middle of the MD simulations; however, the crystallographic position of Ser377 was stable over the simulation time course. Structures generated for MD simulations best match the crystal structures when the PN charge is decreased to −0.9. Thus, the charge distribution of the pyridine ring is critical to the local stabilization of carbanionic intermediates, and protein conformations near the β-active site are perturbed with small changes to the PN. Notably, the E(Ain) and E(A-A) structures maintain the H-bond between Ser377 and the PN with charges of only −0.3 to −0.5 on the PN. The E(Q)indoline and E(Q)2AP forms display a higher negative charge on the PN, indicative of the charge delocalization required for carbanionic intermediates generated during catalysis.42
Figure 8.

The rotation of the Ser377 sidechain in response to changes of the charge on the PN. E(Q)indoline is presented in the bound representation. The MD snapshots of Ser377 are shown every 5 ns.
Roles of the phosphate group in PLP catalysis
The phosphate group has been a crucial inorganic modifier in a wide variety of protein–ligand systems, including PLP-dependent enzymes, which catalyze amino acid transformations.46 PG binding sites of most PLP enzymes are similar, with the oxygen atoms of the PG hydrogen bonding to a series of backbone Gly residues. The end result of this specificity is that chemical modifications of the 5′-phosphate group have a large impact on PLP binding.47 Although the deprotonated PG has been a paradigm in most systems of PLP catalysis, the way that the protonation state of this group modulates stability and catalysis remains unknown. Our study of the protonated (−1 charge) and deprotonated (−2 charge) PG in E(A-A) complexes show that the protonated structure maintains the same interactions as the deprotonated structure, except the H-bonds between the carboxylate group of L-Ser and Thr110/His115 are missing in the protonated complex (Fig. 4). These missing H-bonds are the result of movement of the Gln114 loop in E(A-A):PO_PG. The alignment of E(A-A):PO and E(A-A):PO_PG (Supporting Information Fig. S13) shows the loops containing Thr190 and Ser235 shift toward the substrate in an attempt to preserve the attractions between the PG and the protein backbone. The conformational changes of the loops around the PG may be attributed to the modification of the charge from −2 to −1, resulting in one fewer deprotonated oxygen atom available to interact with the protein. Although TRPS adjusts local geometries in the binding pocket to tolerate the protonation of the PG, the overall structure of the β-subunit gradually changes from the closed to the open conformation because the salt-bridge between Arg141Cζ and Asp305Cγ cannot be retained. The open β-subunit allows several water molecules to move into the binding pocket and cluster around the PO (Fig. 5). To confirm these results, we performed another MD simulation with a different random number seed and came to similar conclusions. In addition, simulations of the internal aldimine structures E(Ain):SB and E(Ain):SB_PG show that the dihedral angles of Lys87 rotate and the H-bond between the PN and Ser377 is missing when the PG is protonated (Supporting Information Fig. S14). Taken together, these results imply that a protonated PG alters the local protein structure in the binding site and also changes overall protein dynamics and water motions. The changes in structure that accompany protonation of the PG do not agree with the crystal structures, adding evidence to the idea that the PG carries a −2 charge throughout TRPS catalysis.23
Conclusions
We have used MD simulations to investigate the protein dynamics and structural stability involved in the PLP-dependent enzyme TRPS upon switching the location of a single proton to different functional groups on the PLP–substrate complex. These simulations illustrate the significance of how changing a single proton location interrupts the overall protein-substrate stability and restrains enzyme catalysis. The functional groups of the PLP cofactor, substrate, and substrate analogues play diverse roles in proton transfer. By studying the E(Ain), E(A-A), E(Q)indoline and E(Q)2AP complexes, the general features of each functional group, tabulated in Table I, can be summarized as follows: (a) In all TRPS intermediates, the unprotonated PG with a formal −2 charge serves as the main anchor for the cofactor, maintaining important protein-substrate interactions and allowing for a series of enzyme-catalyzed chemical transformations mediated by the functional groups of the PLP and substrates. (b) The unprotonated PN can form an H-bond with Ser377, which stabilizes the PLP ring structure by forcing it to maintain a locked conformation. (c) To stabilize car-banionic intermediates during catalysis, the electrons delocalize, changing the electrostatic and chemical environment of the ligand-substrate complex. (d) When the PO is unprotonated, the polar side chain of Gln114 moves toward the negatively charged oxygen, which affects the movements of the water molecules around the active site and induces the switch to the open conformation of the β-subunit for the E(Ain) form. (e) The COs typically form stable H-bonds with Thr110, Gly111, and His115. (f) Protonation of the PG, PN, or COs results in the absence of these H-bonds (except in the case of E(Q)indoline:CO and E(Q)2AP:CO), which alters the motions of the COMM domain and causes the opening of the β-subunit. By thoroughly mapping the conformational variations of TRPS due to changes in protonation state in the β-subunit active site, our study offers new insights into how this enzyme stabilizes the different intermediates formed at each catalytic step.
Table I.
Summary of MD Simulations in Each TRPS-Substrate Complex With a Single Proton Switching Between Different Functional Groups
| Protonation state intermediate | SB | PO | CO | SB_PN/PO_PN | SB_PG/PO_PG |
|---|---|---|---|---|---|
| E(Ain) | Forms stable H-bonds with TRPS. Waters are stable in the binding site. | Fewer waters around the PO, which affects the proton delivery. | H-bonds between PN and Ser377 are missing. Lys87 side chain rotates. | H-bonds between PN and Ser377 are missing. Lys87 side chain rotates. | |
| E(A-A) | Gln114 side chain tends to form H-bonds with PO. | PG, PN and CO form stable interactions with TRPS. | H-bonds between CO and Thr110 are missing. | H-bonds between PN and Ser377 are missing. | Thr190 and Ser235 loop moves. Salt-bridge (Arg141-Asp305) can- not be retained. |
| E(Q)indoline | Gln114 side chain occasionally forms H-bonds with PO. | PG, PN and CO form stable interactions with TRPS. | H-bonds between CO and Thr110 remain. Substrate is stable in the binding site. | H-bonds between PN and Ser377 are missing. | |
| E(Q)2AP | Gln114 side chain occasionally forms H-bonds with PO. | PG, PN and CO form stable interactions with TRPS. | H-bonds between CO and Thr110 are missing. Substrate is stable in the binding site. | H-bonds between PN and Ser377 are missing. |
Materials and Methods
Molecular systems
The crystal structures of the TRPS in complex with E(Ain), E(A-A), E(Q)indoline, and E(Q)2AP were obtained from the protein data bank (PDB) code 4HT3 with resolution 1.65 Å, code 4HN4 with resolution 1.64 Å, code 3PR2 with resolution 1.85 Å, and code 4HPJ with resolution 1.45 Å, respectively.23,32 The coordinates of the TRPS:E(A-A) complex including a benzimidazole (BZI) molecule was obtained from the PDB code 4HPX with resolution 1.30 Å.23 All systems studied include the ligand F9 bound in the α-active site and the MVC site in the β-subunit is occupied by Cs+ ion.
MD simulations
The standard Amber 14 package with graphics processing unit (GPU) acceleration was used for MD simulations.48–50 The protein and substrates were modeled with the Amber 99SB and general Amber force field, respectively.51,52 The atom charges of the intermediates were given by the vCharge model.53 We carefully minimized the systems through hydrogen atoms, side chains, and the entire protein complex for 500, 5000, and 50,000 steps, respectively. The counterion, Na+ based on the Coulombic potential, was placed where necessary to maintain the overall system as neutral. After solvating the system by the TIP3P water model to create the system in a rectangular box of 12 Å,54 we minimized the water and entire system for 10,000 and 20,000 steps, respectively, to correct small flaws. Water molecules were equilibrated for 40 ps. Then, all molecules, including the protein, substrates, cofactor, and solvent, were relaxed gradually by heating the system at 250, 275, and 300 K for 20, 20, and 160 ps, respectively. The particle mesh Ewald was turned on to consider long-range electrostatic interactions.55,56 The Langevin thermostat with a damping constant of 2 ps−1 was applied to maintain a temperature of 300 K, and the SHAKE algorithm was used to constrain hydrogen atoms during MD simulations.57 We collected the resulting trajectories every 1 ps with a time step of 2 fs in the isothermic-isobaric (NPT) ensemble, and the total simulation time for each protein–substrate system was 50 ns.
Analysis of simulation data
The root-mean-square deviation (RMSD), distance of atoms, and H-bond detection were calculated by the use of VMD.58 We defined an H-bond when the distance between a donor (D) and an acceptor (A) was <3.5 Å and the angle H-D•••A was <60°. The MD trajectories and snapshots were examined by VMD and PyMol program.58
Acknowledgments
Grant sponsor: US National Institutes of Health; Grant number: R01GM097569; Grant sponsor: NSF supercomputer centers.
C.A.C. and Y.M.H. congratulate Professor Ron Levy for his many accomplishments on the occasion of his 65th birthday. Thank you and happy birthday, Ron!
Abbreviations
- BZI
benzimidazole
- CO
carboxylate oxygen
- COMM domain
communication domain (β-subunit residues 102–189)
- DIT
dihydroiso-L-tryptophan
- E(Ain)
internal aldimine
- E(Q)2AP
2-aminophenol quinonoid
- E(Q)indoline
indoline quinonoid;enzyme-bound intermediates
- E(A-A)
aminoacrylate
- G3P
glyceraldehyde-3-phosphate
- GP
α-D,L-glycerol phosphate
- IGP
indole-3-glycerol phosphate
- MD
molecular dynamics
- PG
phosphoryl group
- PLP
pyridoxal 5′-phosphate
- PN
pyridine nitrogen
- PO
pyridoxyl phenolic oxygen
- SB
Schiff base
- ssNMR
solid-state NMR
- TRPS
tryptophan synthase
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Kirby AJ. Efficiency of proton transfer catalysis in models and enzymes. Acc Chem Res. 1997;30:290–296. [Google Scholar]
- 2.Hartwell E, Hodgson DRW, Kirby AJ. Exploring the limits of efficiency of proton-transfer catalysis in models and enzymes. J Am Chem Soc. 2000;122:9326–9327. [Google Scholar]
- 3.Schowen KB, Limbach HH, Denisov GS, Schowen RL. Hydrogen bonds and proton transfer in general-catalytic transition-state stabilization in enzyme catalysis. Biochim Biophys Acta Bioenerg. 2000;1458:43–62. doi: 10.1016/s0005-2728(00)00059-1. [DOI] [PubMed] [Google Scholar]
- 4.Hur O, Niks D, Casino P, Dunn MF. Proton transfers in the beta-reaction catalyzed by tryptophan synthase. Biochemistry. 2002;41:9991–10001. doi: 10.1021/bi025568u. [DOI] [PubMed] [Google Scholar]
- 5.Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu Rev Biophys Biomol Struct. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- 6.Alonso H, Cummins PL, Gready JE. Methyltetra-hydrofolate:corrinoid/iron-sulfur protein methyltransferase (MeTr): protonation state of the ligand and active-site residues. J Phys Chem B. 2009;113:14787–14796. doi: 10.1021/jp900181g. [DOI] [PubMed] [Google Scholar]
- 7.deDios AC, Oldfield E. Recent progress in understanding chemical shifts. Solid State Nucl Magn Reson. 1996;6:101–125. doi: 10.1016/0926-2040(95)01207-9. [DOI] [PubMed] [Google Scholar]
- 8.McDermott A, Polenova T. Solid state NMR: new tools for insight into enzyme function. Curr Opin Struct Biol. 2007;17:617–622. doi: 10.1016/j.sbi.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Oldfield E. Quantum chemical studies of protein structure. Philos Trans R Soc Lond B Biol Sci. 2005;360:1347–1361. doi: 10.1098/rstb.2003.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. 3-Dimensional structure of the tryptophan synthase alpha-2-beta-2 multienzyme complex from salmonella-typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 11.Bahar I, Jernigan RL. Cooperative fluctuations and subunit communication in tryptophan synthase. Biochemistry. 1999;38:3478–3490. doi: 10.1021/bi982697v. [DOI] [PubMed] [Google Scholar]
- 12.Dunn MF, Niks D, Ngo H, Barends TRM, Schlichting I. Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem Sci. 2008;33:254–264. doi: 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Walter MW. Structure-based design of agrochemicals. Nat Prod Rep. 2002;19:278–291. doi: 10.1039/b100919m. [DOI] [PubMed] [Google Scholar]
- 14.Dias MVB, Canduri F, da Silveira NJF, Czekster CM, Basso LA, Palma MS, Santos DS, de Azevedo WF. Molecular models of tryptophan synthase from mycobacterium tuberculosis complexed with inhibitors. Cell Biochem Biophys. 2006;44:375–384. doi: 10.1385/CBB:44:3:375. [DOI] [PubMed] [Google Scholar]
- 15.Rhee S, Parris KD, Hyde CC, Ahmed SA, Miles EW, Davies DR. Crystal structures of a mutant (beta K87T) tryptophan synthase alpha(2)beta(2) complex with ligands bound to the active sites of the alpha- and beta-subunits reveal ligand-induced conformational changes. Biochemistry. 1997;36:7664–7680. doi: 10.1021/bi9700429. [DOI] [PubMed] [Google Scholar]
- 16.Schneider TR, Gerhardt E, Lee M, Liang PH, Anderson KS, Schlichting I. Loop closure and intersubunit communication in tryptophan synthase. Biochemistry. 1998;37:5394–5406. doi: 10.1021/bi9728957. [DOI] [PubMed] [Google Scholar]
- 17.Sachpatzidis A, Dealwis C, Lubetsky JB, Liang PH, Anderson KS, Lolis E. Crystallographic studies of phosphonate-based alpha-reaction transition-state analogues complexed to tryptophan synthase. Biochemistry. 1999;38:12665–12674. doi: 10.1021/bi9907734. [DOI] [PubMed] [Google Scholar]
- 18.Pan P, Woehl E, Dunn MF. Protein architecture, dynamics and allostery in tryptophan synthase channeling. Trends Biochem Sci. 1997;22:22–27. doi: 10.1016/s0968-0004(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 19.Phillips RS, Miles EW, Holtermann G, Goody RS. Hydrostatic pressure affects the conformational equilibrium of salmonella typhimurium tryptophan synthase. Biochemistry. 2005;44:7921–7928. doi: 10.1021/bi050056b. [DOI] [PubMed] [Google Scholar]
- 20.Ferrari D, Niks D, Yang LH, Miles EW, Dunn MF. Allosteric communication in the tryptophan synthase bienzyme complex: roles of the beta-subunit aspartate 305 arginine 141 salt bridge. Biochemistry. 2003;42:7807–7818. doi: 10.1021/bi034291a. [DOI] [PubMed] [Google Scholar]
- 21.Barends TRM, Domratcheva T, Kulik V, Blumenstein L, Niks D, Dunn MF, Schlichting I. Structure and mechanistic implications of a tryptophan synthase quinonold intermediate. ChemBioChem. 2008;9:1024–1028. doi: 10.1002/cbic.200700703. [DOI] [PubMed] [Google Scholar]
- 22.Brzovic PS, Hyde CC, Miles EW, Dunn MF. Characterization of the functional role of a flexible loop in the alpha-subunit of tryptophan synthase from salmonella-typhimurium by rapid-scanning, stopped-flow spectroscopy and site-directed mutagenesis. Biochemistry. 1993;32:10404–10413. doi: 10.1021/bi00090a016. [DOI] [PubMed] [Google Scholar]
- 23.Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Allostery and substrate channeling in the tryptophan synthase bienzyme complex: evidence for two subunit conformations and four quaternary states. Biochemistry. 2013;52:6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spyrakis F, Raboni S, Cozzini P, Bettati S, Mozzarelli A. Allosteric communication between alpha and beta subunits of tryptophan synthase: modelling the open-closed transition of the alpha subunit. Biochim Biophys Acta Proteins Proteomics. 2006;1764:1102–1109. doi: 10.1016/j.bbapap.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Fatmi MQ, Ai R, Chang CeA. Synergistic regulation and ligand-induced conformational changes of tryptophan synthase. Biochemistry. 2009;48:9921–9931. doi: 10.1021/bi901358j. [DOI] [PubMed] [Google Scholar]
- 26.Huang XY, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 27.Dewar MJS, Storch DM. Alternative view of enzyme-reactions. Proc Natl Acad Sci U S A. 1985;82:2225–2229. doi: 10.1073/pnas.82.8.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houben KF, Dunn MF. Allosteric effects acting over a distance of 20–25 a in the escherichia-coli tryptophan synthase bienzyme complex increase ligand affinity and cause redistribution of covalent intermediates. Biochemistry. 1990;29:2421–2429. doi: 10.1021/bi00461a028. [DOI] [PubMed] [Google Scholar]
- 29.Drewe WF, Dunn MF. Detection and identification of intermediates in the reaction of L-serine with escherichia-coli tryptophan synthase via rapid-scanning ultraviolet visible spectroscopy. Biochemistry. 1985;24:3977–3987. doi: 10.1021/bi00336a027. [DOI] [PubMed] [Google Scholar]
- 30.Drewe WF, Dunn MF. Characterization of the reaction of L-serine and indole with escherichia-coli tryptophan synthase via rapid-scanning ultraviolet visible spectroscopy. Biochemistry. 1986;25:2494–2501. doi: 10.1021/bi00357a032. [DOI] [PubMed] [Google Scholar]
- 31.Ngo H, Kimmich N, Harris R, Niks D, Blumenstein L, Kulik V, Barends TR, Schlichting I, Dunn MF. Allosteric regulation of substrate channeling in tryptophan synthase: modulation of the L-serine reaction in stage I of the ss-reaction by alpha-site ligands. Biochemistry. 2007;46:7740–7753. doi: 10.1021/bi7003872. [DOI] [PubMed] [Google Scholar]
- 32.Lai J, Niks D, Wang Y, Domratcheva T, Barends TRM, Schwarz F, Olsen RA, Elliott DW, Fatmi MQ, Chang CeA, Schlichting I, Dunn MF, Mueller LJ. X-ray and NMR crystallography in an enzyme active site: the indoline quinonoid intermediate in tryptophan synthase. J Am Chem Soc. 2011;133:4–7. doi: 10.1021/ja106555c. [DOI] [PubMed] [Google Scholar]
- 33.Ngo H, Harris R, Kimmich N, Casino P, Niks D, Blumenstein L, Barends TR, Kulik V, Weyand M, Schlichting I, Dunn MF. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry. 2007;46:7713–7727. doi: 10.1021/bi700385f. [DOI] [PubMed] [Google Scholar]
- 34.Caulkins BG, Bastin B, Yang C, Neubauer TJ, Young RP, Hilario E, Huang YmM, Chang CeA, Fan L, Dunn MF, Marsella MJ, Mueller LJ. Protonation states of the tryptophan synthase internal aldimine active site from solid-state NMR spectroscopy: direct observation of the protonated schiff base linkage to pyridoxal-5′-phosphate. J Am Chem Soc. 2014;136:12824–12827. doi: 10.1021/ja506267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caulkins BG, Yang C, Hilario E, Fan L, Dunn MF, Mueller LJ. Catalytic roles of βLys87 in tryptophan synthase: 15 N solid state NMR studies. Biochim Biophys Acta. 2015;1854:1194–1199. doi: 10.1016/j.bbapap.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song Y, Mao J, Gunner MR. Mcce2: improving protein pK(a) calculations with extensive side chain rotamer sampling. J Comput Chem. 2009;30:2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oleary MH, Payne JR. C-13 NMR-spectroscopy of labeled pyridoxal 5′-phosphate – model studies, D-serine dehydratase, and L-glutamate decarboxylase. J Biol Chem. 1976;251:2248–2254. [PubMed] [Google Scholar]
- 38.Brzovic PS, Kayastha AM, Miles EW, Dunn MF. Substitution of glutamic-acid-109 by aspartic-acid alters the substrate specificity and catalytic activity of the beta-subunit in the tryptophan synthase bienzyme complex from salmonella-typhimurium. Biochemistry. 1992;31:1180–1190. doi: 10.1021/bi00119a030. [DOI] [PubMed] [Google Scholar]
- 39.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 40.Fogle EJ, Liu WS, Woon ST, Keller JW, Toney MD. Role of q52 in catalysis of decarboxylation and transamination in dialkylglycine decarboxylase. Biochemistry. 2005;44:16392–16404. doi: 10.1021/bi051475b. [DOI] [PubMed] [Google Scholar]
- 41.Sharif S, Fogle E, Toney MD, Denisov GS, Shenderovich IG, Buntkowsky G, Tolstoy PM, Huot MC, Limbach HH. NMR localization of protons in critical enzyme hydrogen bonds. J Am Chem Soc. 2007;129:9558–9559. doi: 10.1021/ja0728223. [DOI] [PubMed] [Google Scholar]
- 42.Griswold WR, Toney MD. Role of the pyridine nitrogen in pyridoxal 5′-phosphate catalysis: activity of three classes of PLP enzymes reconstituted with deaza-pyridoxal 5′-phosphate. J Am Chem Soc. 2011;133:14823–14830. doi: 10.1021/ja2061006. [DOI] [PubMed] [Google Scholar]
- 43.Shaw JP, Petsko GA, Ringe D. Determination of the structure of alanine racemase from bacillus stearo-thermophilus at 1.9-angstrom resolution. Biochemistry. 1997;36:1329–1342. doi: 10.1021/bi961856c. [DOI] [PubMed] [Google Scholar]
- 44.Tai CH, Cook PF. Pyridoxal 5′-phosphate dependent alpha,beta-elimination reactions: mechanism of O-acetylserine sulfhydrylase. Acc Chem Res. 2001;34:49–59. doi: 10.1021/ar990169l. [DOI] [PubMed] [Google Scholar]
- 45.Casasnovas R, Salva A, Frau J, Donoso J, Munoz F. Theoretical study on the distribution of atomic charges in the schiff bases of 3-hydroxypyridine-4-aldehyde and alanine. The effect of the protonation state of the pyridine and imine nitrogen atoms. Chem Phys. 2009;355:149–156. [Google Scholar]
- 46.Nardozzi JD, Lott K, Cingolani G. Phosphorylation meets nuclear import: a review. Cell Commu Signal. 2010;8:32. doi: 10.1186/1478-811X-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schnackerz KD, Andi B, Cook PF. P-31 NMR spectroscopy senses the microenvironment of the 5′-phosphate group of enzyme-bound pyridoxal 5′-phosphate. Biochim Biophys Acta Proteins Proteomics. 2011;(1814):1447–1458. doi: 10.1016/j.bbapap.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goetz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salomon-Ferrer R, Case DA, Walker RC. An overview of the amber biomolecular simulation package. WIREs Comput Mol Sci. 2013;3:198–210. [Google Scholar]
- 51.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins: Struct Funct Bioinf. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozpinar GA, Peukert W, Clark T. An improved generalized AMBER force field (GAFF) for urea. J Mol Model. 2010;16:1427–1440. doi: 10.1007/s00894-010-0650-7. [DOI] [PubMed] [Google Scholar]
- 53.Gilson MK, Gilson HSR, Potter MJ. Fast assignment of accurate partial atomic charges: an electronegativity equalization method that accounts for alternate resonance forms. J Chem Inf Comput Sci. 2003;43:1982–1997. doi: 10.1021/ci034148o. [DOI] [PubMed] [Google Scholar]
- 54.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 55.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 56.Salomon-Ferrer R, Goetz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 57.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-interaction of cartesian equations of motion of a system with constraints–molecular-dynamics of N-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 58.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]