

Highlights
-
•
We identify vinylsulfones as lead candidate inhibitors of Ebola virus and SARS-CoV.
-
•
K11777 inhibited Ebola virus and SARS-CoV entry in the sub-nanomolar range.
-
•
Potent inhibition correlated with the presence of a basic piperazine ring at P3.
-
•
Serine protease inhibitor and K11777 blocked coronavirus entry into caco-2 cells.
-
•
Camostat protected 6 out of ten mice from lethal infection with SARS-CoV.
Keywords: Vinylsulfones, Coronavirus, Filovirus, Cathepsin
Abstract
In order to gain entry into cells, diverse viruses, including Ebola virus, SARS-coronavirus and the emerging MERS-coronavirus, depend on activation of their envelope glycoproteins by host cell proteases. The respective enzymes are thus excellent targets for antiviral intervention. In cell culture, activation of Ebola virus, as well as SARS- and MERS-coronavirus can be accomplished by the endosomal cysteine proteases, cathepsin L (CTSL) and cathepsin B (CTSB). In addition, SARS- and MERS-coronavirus can use serine proteases localized at the cell surface, for their activation. However, it is currently unclear which protease(s) facilitate viral spread in the infected host. We report here that the cysteine protease inhibitor K11777, ((2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-methylpiperazine-1-carbonyl]amino}-3-phenylpropanamide) and closely-related vinylsulfones act as broad-spectrum antivirals by targeting cathepsin-mediated cell entry. K11777 is already in advanced stages of development for a number of parasitic diseases, such as Chagas disease, and has proven to be safe and effective in a range of animal models. K11777 inhibition of SARS-CoV and Ebola virus entry was observed in the sub-nanomolar range. In order to assess whether cysteine or serine proteases promote viral spread in the host, we compared the antiviral activity of an optimized K11777-derivative with that of camostat, an inhibitor of TMPRSS2 and related serine proteases. Employing a pathogenic animal model of SARS-CoV infection, we demonstrated that viral spread and pathogenesis of SARS-CoV is driven by serine rather than cysteine proteases and can be effectively prevented by camostat. Camostat has been clinically used to treat chronic pancreatitis, and thus represents an exciting potential therapeutic for respiratory coronavirus infections. Our results indicate that camostat, or similar serine protease inhibitors, might be an effective option for treatment of SARS and potentially MERS, while vinyl sulfone-based inhibitors are excellent lead candidates for Ebola virus therapeutics.
1. Introduction
Emerging viral diseases pose a unique risk to public health. Ebola virus, severe acute respiratory syndrome coronavirus (SARS-CoV) and members of the Henipavirus genus of paramyxoviruses are all highly pathogenic viruses that have arisen in the past 40 years and caused, or threaten to cause, major outbreaks. New viral threats continue to emerge, most recently demonstrated by a novel beta-coronavirus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), which was identified in 2012 (Zaki et al., 2012, Memish et al., 2013, de Groot et al., 2013). There are currently no approved vaccines or therapeutics for many of the highly pathogenic viruses potentially dependent on cathepsins, including Ebola virus, Nipah virus (NiV), MERS-CoV and SARS-CoV. Broad-spectrum antiviral drugs, with overlapping therapeutic indications, would facilitate rapid responses to new or changing pandemic threats, potentially even without precise identification of the agent. Targeting host factors involved in viral entry provides an excellent avenue for such drug development, due to the limited number of pathways involved (Zhou et al., 2011).
The glycoproteins of corona-, filo- and paramyxoviruses facilitate viral entry into target cells by binding to receptors and by driving fusion of viral and host cell membranes. However, the glycoproteins are synthesized as inactive precursors and depend on activation by host cell proteases to acquire a fusion active form. As a consequence, the respective enzymes are potential targets for broad-spectrum antiviral intervention. Cell culture studies demonstrated that endosomal cysteine proteases, in particular cathepsin B (CTSB) and/or L (CTSL), can activate the glycoproteins of filoviruses, SARS-CoV, other coronaviruses, and NiV and Hendra (HeV) viruses to facilitate entry into certain cell lines. In addition, activation of coronaviruses can also be accomplished by TMPRSS2, or other serine proteases located at the cell surface, or secreted into the extracellular space (Simmons et al., 2013). However, the respective roles of endosomal and cell surface proteases in viral spread in the infected host is unknown.
The development of protease inhibitors able to inhibit CTSL, CTSB and related proteases would be an excellent starting point for development of broad-spectrum antiviral therapies (Zhou et al., 2011). We describe here the discovery of K11777 and its related compounds, as broad-spectrum antivirals targeting endosomal proteases involved in viral entry. K11777, a cysteine protease inhibitor, blocked infection when viral entry did not require activating serine proteases, as is the case with ebolavirus (EBOV). K11777 also fully inhibited coronavirus infection, but only when target cell lines lacking activating serine proteases were used. If cells expressed cell-surface serine proteases known to activate coronaviruses, both K11777 and a serine protease inhibitor, such as camostat were required for full inhibition. Thus, both compounds were deployed to examine which activation pathway is predominant in vivo. Camostat displayed antiviral activity in a pathogenic animal model for SARS-CoV infection, indicating that serine protease inhibitors are suitable for treatment of SARS and potentially MERS. The predicted effect of K11777 and related cysteine protease inhibitors versus Ebola virus in vivo must await studies in approved biocontainment facilities.
2. Materials and methods
2.1. Libraries and commercial compounds
The cysteine protease inhibitor library screened in this work has been described elsewhere (Ang et al., 2011). Briefly, the library includes ∼2100 electrophilic cysteine protease inhibitors of various chemotype (glycine nitriles, ketobenzoxazoles, ketooxadiazoles, vinylsulfones, etc.), which were synthesized during the course of industrial drug discovery programs targeting human cathepsins (Palmer et al., 1995, Palmer et al., 2005, Palmer et al., 2006, Rydzewski et al., 2002). Camostat mesylate, leupeptin, bafilomycin A1, ammonium chloride, and chloroquine were purchased from Sigma–Aldrich.
2.2. Synthesis of vinylsulfone cysteine protease inhibitors
K11777 and novel P3 derivatives were synthesized according to the general approach described previously (Jaishankar et al., 2008) and as illustrated here (Scheme 1 ). The N-substituted piperazines were obtained from commercial sources or (for R = cyclopentyl and cyclopropylmethyl) were prepared by reductive amination of Boc-protected piperazine followed by treatment with HCl in dioxane (51–53% over two steps). We find that the final coupling of P3/P2 carboxylic acid to vinylsulfone amine is best accomplished via the mixed anhydride, as this minimized epimerization of the phenylalanine side chain. Final vinylsulfone analogs were >95% pure as judged by LC/MS analysis. Characterization data for final analogs is provided below.
Scheme 1.
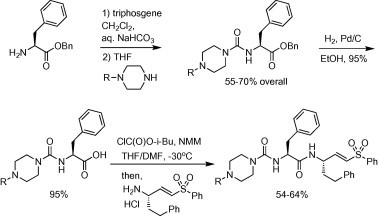
Synthesis of K11777 and P3-modified vinylsulfone analogs.
2.2.1. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-ethylpiperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256122)
1H NMR (400 MHz, CDCl3) δ 7.93–7.91 (m, 2H), 7.78–7.68 (m, 1H), 7.68–7.58 (m, 2H), 7.37–7.25 (m, 8H), 7.16–7.14 (m, 2H), 6.90 (dd, J = 4.8, 15.2 Hz, 1H), 6.35–6.31 (m, 2H), 4.67 (br. S., 1H), 4.52 (d, J = 6.8 Hz, 1H), 4.13 (br. S., 3H), 3.50 (br. S., 2H), 3.39 (br. S., 2H), 3.21–3.05 (m, 4H), 2.74–2.549 (m, 4H), 1.97–1.90 (m, 2H), 1.37 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.4, 156.6, 145.2, 140.1, 139.89, 136.4, 133.14, 130.14, 129.0, 128.9, 128.49, 128.29, 128.09, 127.3, 126.8, 125.9, 76.7, 76.4, 55.7, 54.2, 48.8, 47.8, 43.8, 37.8, 35.4, 31.4, 18.0; MS m/z = 589 [M+H]+.
2.2.2. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-(propan-2-yl)piperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256123)
1H NMR (400 MHz, CDCl3) δ 7.93–7.90 (m, 2H), 7.72–7.68 (m, 1H), 7.63–7.59 (m, 2H), 7.34–7.20 (m, 8H), 7.14–7.12 (m, 2H), 6.85 (dd, J = 4.8, 15.2 Hz, 1H), 6.69 (d, J = 8.2 Hz, 1H), 6.18 (dd, J = 1.7, 15.1 Hz, 1H), 5.05 (d, J = 7.3 Hz, 1H), 4.68 (m, 1H), 4.56 (d, J = 7.3 Hz, 1H), 3.45–3.28 (m, 4H), 3.10 (dd, J = 2.6, 7.3 Hz, 2H), 2.78–2.72 (m, 1H), 2.66–2.59 (m, 2H), 2.58–2.44 (m, 4H), 1.97–1.91 (m, 1H) 1.85–1.81 (m, 1H) 1.08 (m, 6H); MS m/z = 603 [M+H]+.
2.2.3. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-propylylpiperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256157)
1H NMR (400 MHz CDCl3) δ = 7.85–7.83 (m, 2H), 7.67–7.61 (m, 1H), 7.55 (t, J = 7.6 Hz, 2H), 7.25–7.16 (m, 8H), 7.08 (d, J = 7.0 Hz, 2H), 6.79 (d, J = 4.8 Hz, 1H), 6.83 (d, J = 4.8 Hz, 1H), 6.16 (dd, J = 1.6, 15.0 Hz, 1H), 5.56 (s, 1H), 5.19 (br. S., 1H), 4.62–4.60 (m, 1H), 4.47 (d, J = 7.5 Hz, 1H), 3.57–3.51 (m, 2H), 3.37–3.27 (m, 4H), 3.07 (d, J = 7.5 Hz, 2H), 2.65–2.48 (m, 2H), 2.37–2.22 (m, 6H), 1.93–1.72 (m, 3H), 1.52–1.39 (m, 4H), 0.93–0.85 (m, 3H); MS m/z = 603 [M+H]+.
2.2.4. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-phenylpiperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256158)
1H NMR (400 MHz, CDCl3) δ = 7.94–7.82 (m, 2H), 7.72–7.60 (m, 3H), 7.37–7.21 (m, 10H), 7.12 (d, J = 7.5 Hz, 2H), 7.00–6.96 (m, 3H), 6.87 (dd, J = 15.0 Hz, 4.4, 1H), 6.79 (d, J = 8.2 Hz, 1H), 6.19 (d, J = 15.0 Hz, 1H), 5.23 (d, J = 7.1 Hz, 1H), 4.69–4.59 (m, 2H), 3.57–3.49 (m, 4H), 3.17–311 (m, 6H), 2.66–2.57 (m, 2H), 295–1.78 (m, 2H); MS m/z = 637 [M+H]+.
2.2.5. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-(2-methoxyethyl)piperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256159)
1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 7.9 Hz, 2H), 7.64 (t, J = 7.5 Hz, 1H), 7.55 (t, J = 7.7 Hz, 2H), 7.27–7.15 (m, 8H), 7.06 (d, J = 7.1 Hz, 2H), 6.78 (dd, J = 4.9, 15.1 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H), 6.17 (d, J = 15.0 Hz, 1H), 5.74 (d, J = 6.8 Hz, 1H), 4.59 (d, J = 4.2 Hz, 1H), 4.40 (q, J = 7.6 Hz, 1H), 3.71–3.69 (m, 3H), 3.49 (br. S., 3H), 3.32 (s, 3H), 3.16–3.14 (m, 2H), 3.06–2.8 (m, 4H), 2.65–2.51 (m, 4H), 1.89–1.79 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 172.6, 156.7, 146.1, 140.4, 139.8, 137., 133.6, 130.3, 129.3, 129.1, 128.5, 128.5, 128.3, 127.4, 127.0, 126.2, 77.4, 76.6, 66.5, 58.8, 57.2, 56.4, 52.1 49.2, 41.1, 35.22, 31.8; MS m/z = 619 [M+H]+.
2.2.6. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-tert-butylpiperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256160)
1H NMR (400 MHz, CDCl3) δ = 7.84–7.82 (m, 2H), 7.64–7.61 (m, 1H), 7.58–7.52 (m, 2H), 7.27–7.14 (m, 8H), 7.08–7.03 (m, 2H), 6.87 (br. S., 1H), 6.79 (dd, J = 4.8, 15.2 Hz, 1H), 6.22 (d, J = 15.0 Hz, 1H), 5.99 (br. S., 1H), 4.61–4.59 (m, 1H), 4.41 (d, J = 7.5 Hz, 1H), 4.07–3.96 (m, 2H), 3.49–3.36 (m, 4H), 3.12–2.98 (m, 2H), 2.68–2.45 (m, 2H), 2.68–2.45 (m, 4H), 1.88–1.80 (m, 2H), 1.34 (s, 9H); MS m/z = 617 [M+H]+.
2.2.7. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-cyclopentylpiperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256161)
1H NMR (400 MHz, CHLOROFORM-d) δ = 7.84–7.81 (m, 2H), 7.65–7.61 (m, 1H), 7.56–7.52 (m, 2H), 7.28–7.14 (m, 8H), 7.09–7.07 (m, 2H), 7.01 (br. S, 1H), 6.80 (dd, J = 4.7, 15.1 Hz, 1H), 6.24–6.20 (m, 1H), 6.06 (br. S., 1H), 4.61–4.59 (m, 1H), 4.42 (q, J = 7.5 Hz, 1H), 3.99 (t, J = 16.1 Hz, 2H), 3.48–3.33 (m, 4H), 3.13–2.97 (m, 3H), 2.64–2.52 (m, 4H), 2.42 (br. S., 1H), 1.97–1.78 (m, 8H); MS m/z = 629 [M+H]+.
2.2.8. (2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-(cyclopropylmethyl)piperazine-1-carbonyl]amino}-3-phenylpropanamide (SMDC-256162)
1H NMR (400 MHz, CDCl3) δ 7.83–7.81 (m, 2H), 7.65–7.61 (m, 1H), 7.55–7. 51 (m, 2H), 7.25–7.15 (m, 8H), 7.06–7.04 (m, 3H), 6.80 (dd, J = 4.8, 15.0 Hz, 1H), 6.23 (dd, J = 1.6, 15.0 Hz, 1H), 6.12 (br. D., J = 6.8 Hz, 1H), 4.60–4.59 (m, 1H), 4.42 (d, J = 7.5 Hz, 1H), 4.02 (br. S., 2H), 3.53 (br. S., 2H), 3.34 (br. S., 2H), 3.11–2.97 (m, 2H), 2.80 (d, J = 7.1 Hz, 2H), 2.68–2.48 (m, 4H), 1.87–1.79 (m, 2H), 1.04–0.97 (m, 1H), 0.73–0.68 (m, 2H), 0.31 (m, 2H); MS m/z = 615 [M+H]+.
2.3. Cell lines and reagents
Human Embryonic Kidney 293 cells, 293T cells, clone 17 (293T/17), and Vero cells were obtained from American Type Culture Collection (ATCC), while the Huh7.5 cell line was a gift from Apath LLC. All cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen) supplemented with 10% FBS and Penicillin and Streptomycin (10 U/ml). 293T/17 stably expressing ACE2 (293T/ACE2) were established by transfecting 293T/17 cells with pcDNA6 (Invitrogen) encoding the ACE2 gene and selecting for stable transformants using blasticidin S (2.5 μg/ml). 293 stably expressing human CD13 [also called aminopeptidase N (APN)] (293/CD13) were established by transfecting cells with pcDNA3 (Invitrogen) encoding the CD13 gene and selecting for stable transformants using geneticin 418 (2.0 mg/ml). Expression of CD13 was measured with flow cytometric analysis.
2.4. Plasmids and gene constructs
Lentiviral pseudotypes were generated from two plasmids, one encoding the envelope and the second an envelope-deficient HIV reporter construct – either pNL4-3 Luc-R-E- (pNL-luc) or pNL4-3.REN.R-E- (Zhou et al., 2011, Connor et al., 1995). Plasmids encoding spike (S) proteins from human coronaviruses SARS-CoV, NL63 and 229E, MERS-CoV, glycoproteins (GP) from filoviruses EBOV (formerly known as ebolavirus Zaire), Sudan ebolavirus (SUDV), Tai Forest ebolavirus (TAFV), Reston ebolavirus (RESTV), and Marburg (MARV), as well as NiV F and G, Lassa virus GP, vesicular stomatitis virus (VSV) G protein, chikungunya virus (CHIKV) E1/E2, and MLV envelope, have been described (Zhou et al., 2011, Zhou et al., 2012, Simmons et al., 2005, Simmons et al., 2004, Simmons et al., 2002, Salvador et al., 2013, Salvador et al., 2009). Bundibugyo ebolavirus (BEBOV) GP was a gift from Edward Wright (University of Westminster). HCV E1E2 was synthesized (Genscript, CA) while Junin virus G protein was a gift from Sean Amberg (Siga Technologies, OR). Plasmids encoding cellular type II transmembrane serine proteases (TTSP) TMPRSS2 were previously described (Glowacka et al., 2011).
2.5. Pseudovirion production and titration
Lentiviral pseudovirions were produced essentially as previously described (Zhou et al., 2010). Briefly, 293T/17 cells were transfected with up to 30 μg of viral envelope encoding plasmid and 10 μg of pNL4-3 reporter backbone per 10-cm dish by calcium phosphate transfection. The next day, expression was induced with sodium butyrate (10 mM) for 6 h before washing once. Forty hours after transfection, supernatant was filtered through a 0.45 μm-pore-size filter and frozen at −80 °C. Virus was titrated essentially as it would be used in the screening assay. If required, virions were purified and concentrated by ultracentrifugation (28,000 rpm in a SW28 rotor, Beckman) over a 20% sucrose cushion, resuspended in Hank’s balanced salt solution (HBSS) and stored at −80 °C as aliquots. Pseudoviruses were normalized for equal infectivity by transduction of target cells with serially diluted stock followed 48 h later by determination of luciferase activity in cell lysates according to the manufacturer’s instruction (Promega). VSV-based pseudotypes bearing Junin virus G were produced essentially as described (Steffen et al., 2013) by transfecting 293T cells with 16 μg of Junin G plasmid and then infecting the cells with recombinant VSVΔG-GFP(VSV-G). Progeny VSVΔG-GFP(Junin-G) virus was then collected, titrated and used for inhibition studies. In the case of NiV, VSVΔG-GFP(NiV F/G) viruses were produced via calcium phosphate transfection of 293T cells with 10 ug of NiV F and 10 μg of NiV G. Transfected cells were left for 16 h before an initial medium change; then infected with recombinant VSVΔG-GFP(VSV-G) (MOI 0.1–0.3) after five additional hours. Media alone or compound of interest were then added at the desired concentration and cells were incubated overnight before supernatant was harvested and filtered. To assay for inhibition, production of entry-competent virus was examined. Target cells were pre-plated at 25,000 cells/50 μl in 96 well plates and allowed to attach overnight. 50 μl of undiluted VSVΔG-GFP NiV F/G made in the presence or absence of inhibitor was added. Cells were incubated at 37 °C with 5% CO2 for two days, and then washed and fixed with 2% paraformaldehyde and GFP fluorescence determined by flow cytometry using a Becton Dickinson LSRII cytometer and FlowJo software. 100% infection was determined with samples infected with pseudovirons produced from cells with no compound exposure.
2.6. Screening assays
High-throughput screens for viral entry inhibitors were performed in 384-well plates using the dual-envelope pseudotype (DEP) assay (Zhou et al., 2011). Briefly, compounds and controls were diluted in DMEM with 10% FBS to 50 μM (5% DMSO) and 10 μl were transferred to 384-well white tissue culture plates (NUNC) using a Biomek FX-P (Beckman-Coulter). A mixture of the target virus [e.g., (HIV-luc(SARS-CoV S)) and the control virus [HIV-ren(Lassa GP) or HIV-ren(MLV Env)] was made, with the concentration and ratio derived empirically to give similar robust levels of reporter expression. 10 μl of reporter virus mix was added to each well using a Matrix Well-Mate (Thermo Scientific). 30 μl of cells (170,000 cells per milliliter) were then added to all wells. Plates were incubated for two days at 37 °C/5% CO2 and firefly and renilla luciferase reporter expression was determined using the Dual-Glo luciferase assay substrate (Promega).
2.7. Dose response curves
Assays for dose response curves were performed in 96-well white tissue culture plates (NUNC). Target cells were pretreated with test compounds or inhibitors serially diluted in medium, followed by either a single virus or a two reporter virus mixture, depending on the purpose of the assay. The env/reporter combinations were reversed in order to demonstrate inhibition was not directed at the backbone or reporter enzyme rather than entry. Plates were incubated for two days at 37 °C/5% CO2 and firefly and renilla luciferase reporter expression was determined using the Dual-Glo luciferase substrate (Promega), or detection of firefly luciferase reporter expression using the Bright-Glo™ luciferase substrate (Promega). The infectivity for pseudotyped VSVs with NiV F/G was analyzed by measuring the number of GFP expressing cells by flow cytometric analysis.
2.8. Dual inhibition study
Either Caco2 or 293-CD13 cells transiently expressing TMPRSS2 were pretreated with serially diluted K11777, a combination of serially diluted K11777 and camostat mesylate at 1 or 10 μM or a combination of serially diluted camostat mesylate and K11777 at 2.5 μM for 60 min at 37 °C and then incubated with infectivity-normalized pseudoviruses in the presence of the inhibitors. The cells were then cultured at 37 °C/5% CO2 for two days and luciferase expression was measured.
2.9. Live SARS-CoV assays and cytotoxicity
Antiviral replication with Urbani and Toronto-2 strains of live SARS-CoV, as well as cytotoxicity of selected compounds was investigated using three in vitro assays, cytopathic effect (CPE) inhibition assay, neutral red (NR) uptake assay, and virus yield reduction assay as described in Kumaki et al. (2011).
For cell viability assays, cells were seeded in 96-well black tissue culture plates (Costar) and treated with compounds with final concentration of 1% DMSO. The quantity of the ATP present in metabolically active cells was determined with CellTiter-Glo® luminescent cell viability assay kits (Promega, Madison, WI).
2.10. Camostat and SMDC256160 in mice
SMDC256160 (50 mg/kg), camostat (30 mg/kg) alone, SMDC256160 (50 mg/kg) combined with camostat (30 mg/kg), or negative control (water) were administrated into 6–8 week old female BALB/c mice by oral gavage twice a day for 9 days beginning 10 h prior to virus exposure. Ten mice were assigned to each group. The Texas Biomedical Research Institute’s institutional (Texas Biomed) animal care and use committee approved all animal protocols. Live virus assays were performed at the ABSL-4 facility at Texas Biomed using a mouse adapted strain of SARS-CoV (MA15) kindly provided by Ralph Baric (University of North Carolina). Mice were infected by administering 10,000 pfu of virus by intranasal instillation.
2.11. Data analysis
Statistical calculations were performed in Excel (Microsoft, Seattle, WA). Compounds from the primary screens were considered inhibitory when the luciferase readings of SARS-CoV, but not the internal control pseudotyped viruses, fell below the pre-defined cut-off, mean-3 × SD (m-3SD). IC50 (50% inhibitory concentration) and CC50 (50% cell cytotoxic concentration) values were calculated using non-linear regression analysis based on the sigmoidal dose response equation using PRISM 6 (GraphPad Software Inc) (applied to the percent inhibition and concentration data. A selectivity index (SI) was calculated using the formula SI = CC50/IC50.
3. Results
3.1. Discovery of the broad-spectrum antiviral K11777
We recently developed an internally-controlled dual virus HTS assay for identification of inhibitors of viral entry (Zhou et al., 2011). Using SARS-CoV entry assays, we screened a library of ∼2100 cysteine protease inhibitors with confirmed activity against human cathepsins. Unsurprisingly, a large number of hits were identified. Upon validation of the hits, the most robust activity was observed for K11777 ((2S)-N-[(1E,3S)-1-(benzenesulfonyl)-5-phenylpent-1-en-3-yl]-2-{[(E)-4-methylpiperazine-1-carbonyl]amino}-3-phenylpropanamide) (Fig. 1 a), a compound known to inhibit cruzain, a cathepsin-L like protease from the protozoan parasite Trypanosoma cruzi (Engel et al., 1998). In addition, K11777 inhibits a variety of cysteine proteases, including human cathepsins (Choy et al., 2013) and cathepsin-like proteases from several other parasites (Ndao et al., 2013, Abdulla et al., 2007).
Fig. 1.
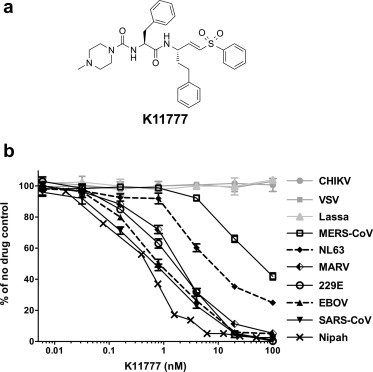
Identification of protease inhibitor K11777 as a broad-spectrum antiviral drug candidate. (a) Structure of K11777; N-methyl-piperazine-phenylalanyl-homophenylalanyl-vinylsulfone phenyl. (b) Dose response curves of compound K11777 against pseudoviruses with a variety of different viral envelopes. Data shown as mean ± SD of triplicate measurements. Representative experiments of at least three experiments are shown.
To determine whether K11777 can inhibit entry driven by other viral envelope proteins, HIV-based pseudotypes bearing spikes from coronaviruses (SARS-CoV, HCoV-229E, NL63, MERS-CoV) or glycoproteins from filoviruses (EBOV, SUDV, TAFV, RESTV, BEBOV and MARV) were examined together with control pseudotypes. We also tested the ability of K11777 to prevent activation and hence infectivity during production of VSV-based pseudotypes (Salvador et al., 2009) bearing NiV F and G. K11777 was active against all the major enveloped viruses previously known to require cathepsin-mediated proteolysis, including a variety of coronaviruses and filoviruses, especially EBOV (Fig. 1b; Table 1 ). K11777 inhibited SARS-CoV pseudovirus entry with an IC50 of 0.68 nM (Fig. 1b, Table 1) while no toxicity was observed, CC50 > 10 μM (data not shown). MERS-CoV and NL63 envelope required higher concentrations of K11777 for inhibition, likely due to less reliance on CTSL (Gierer et al., 2013, Hofmann et al., 2006). Nevertheless, the IC50s were very low: 46 nM for MERS-CoV and <7 nM for NL63. In contrast, 100 nM K11777 did not inhibit infection mediated by envelope glycoproteins from an alphavirus (CHIKV), a rhabdovirus (VSV), a flavivirus (HCV), the retroviruses MLV-A and XMRV or two arenaviruses, Lassa and Junin virus.
Table 1.
K11777 inhibits entry of a variety of enveloped viruses.
| Pseudoviruses | Family | Genome type | Cells | IC50 (nM)a |
|---|---|---|---|---|
| SARS-CoV | Coronaviridae | ssRNA(+) | 293T-ACE2 | 0.68 ± 0.09 |
| HCoV-229E | Coronaviridae | ssRNA(+) | 293T-CD13 | 1.48 ± 0.13 |
| NL63 | Coronaviridae | ssRNA(+) | 293T-ACE2 | 6.78 ± 0.24 |
| MERS-CoV | Coronaviridae | ssRNA(+) | Vero | 46.12 ± 6.63 |
| Zaire ebolavirus (EBOV) | Filoviridae | ssRNA(−) | 293T | 0.87 ± 0.06 |
| Sudan ebolavirus (SUDV) | Filoviridae | ssRNA(−) | 293T | 1.14 ± 0.07 |
| Tai Forest ebolavirus (TAFV) | Filoviridae | ssRNA(−) | 293T | 2.26 ± 0.10 |
| Reston ebolavirus (RESTV) | Filoviridae | ssRNA(−) | 293T | 3.37 ± 0.29 |
| Bundibugyo ebolavirus (BEBOV) | Filoviridae | ssRNA(−) | 293T | 5.91 ± 0.50 |
| Marburg (MARV) | Filoviridae | ssRNA(−) | 293T | 1.90 ± 0.16 |
| Nipah | Paramyxoviridae | ssRNA(−) | 293T | 0.42 ± 0.03 |
| Chikungunya | Togaviridae | ssRNA(+) | 293T | >100 |
| Vesicular stomatitis | Rhabdoviridae | ssRNA(−) | 293T | >100 |
| Amphotropic murine leukemia | Retroviridae | ssRNA(−)RT | 293T | >100 |
| Xenotropic murine leukemia | Retroviridae | ssRNA(−)RT | 293T | >100 |
| Lassa | Arenaviridae | ssRNA(−) | 293T | >100 |
| Junin | Arenaviridae | ssRNA(−) | Vero | >100 |
| HCV (H77 envelope) genotype 1a | Flaviviridae | ssRNA(+) | Huh7.5 | >100 |
IC50 (inhibitory concentration) values are the concentrations required to inhibit the infectivity of the pseudotyped viruses on cells by 50%, which were determined from dose response curves. All envelopes apart from Nipah and Junin were used to make HIV-based pseudotypes. Target cells (293T, 293T expressing ACE2 or CD13, or Vero cells) were then pretreated with serial dilutions of K11777 and exposed to virus. VSV-based pseudotypes were made by transfecting cells with Nipah F and G plasmids, or Junin envelope, and transducing with VSVΔG(GFP)-G. Progeny virus was then collected and titered on target cells. A non-linear regression analysis based on the Sigmoidal dose response equation was applied to the percent inhibition and concentration data. Data is shown as means of triplicate measurements ± standard deviation. Values are representative of at least two independent experiments.
3.2. Alternative proteases for entry
Coronaviruses including SARS-CoV, human coronavirus 229E (hCoV-229E) and MERS-CoV use two distinct pathways for cell entry: (i) the endosomal pathway, in which spike activation is facilitated by the pH-dependent endosomal protease CTSL; or (ii) entry at the plasma membrane, which relies on spike activation by secreted or surface proteases – such as trypsin and type II transmembrane serine proteases HAT (human airway trypsin-like protease) or TMPRSS2 (Gierer et al., 2013, Bertram et al., 2011, Bertram et al., 2013). The serine protease inhibitor camostat mesylate (camostat) inhibits the enzymatic activity of TMPRSS2 and other cell-surface proteases involved in coronavirus activation (Kawase et al., 2012). We therefore assessed whether K11777 displays antiviral activity in TMPRSS2 expressing cells. For this, we incubated target cells with camostat, K11777, or a combination of K11777 and camostat and then infected with pseudoviruses bearing 229E-S. K11777 alone demonstrated up to ∼70% inhibition of 229E-S-mediated transduction. Simultaneous treatment with camostat and K11777 increased inhibition to ∼90% (Fig. 2 a, left panel). Similar inhibition patterns were obtained using the human intestinal epithelial cell line Caco-2, which express endogenous TMPRSS2 and cathepsins (Fig. 2b). In contrast, K11777 alone fully blocked Ebola pseudovirus infection, while camostat had no impact on viral infection (Fig. 2a, middle panel). Finally, treatment of cells with K11777, camostat or both, had no impact on VSV-G driven viral entry (Fig. 2a, right panel), which is known to be independent of cysteine and serine protease activity. These results indicate that both serine and cysteine proteases can activate 229E-S for viral entry, as expected, while EBOV-GP exclusively relies on cysteine proteases for activation.
Fig. 2.
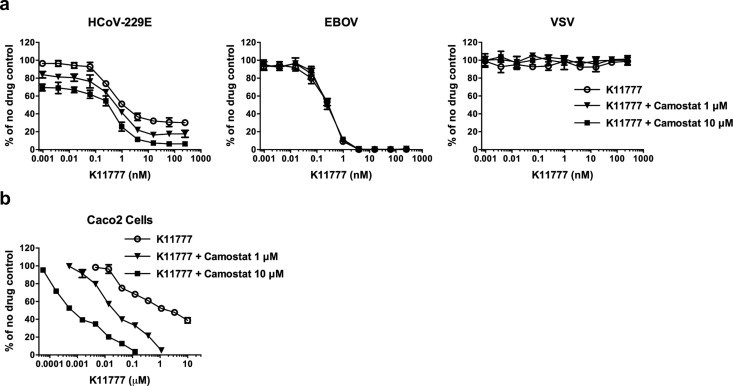
Inhibition of serine and/or cysteine proteases in cells transfected with, or endogenously expressing, TMPRSS2. 293T-CD13 cells transiently expressing TMPRSS2 (a) or Caco2 cells (b) were pretreated with serially diluted compound K11777, or a combination of serially diluted K11777 and camostat at two different concentrations (1 or 10 μM), followed by incubation with infectivity-normalized pseudoviruses in the presence of the inhibitors. The cells were then cultured at 37 °C/5% CO2 for two days and luciferase expression was measured. (a) Simultaneous treatment with both K11777 and camostat for 229E-S, EBOV or VSV-G pseudovirus infection. (b) Enhanced inhibition by a combination of K11777 and camostat for 229E-S mediated viral entry using Caco2 cells.
3.3. In vitro antiviral activity of new vinylsulfone analogs
We next synthesized a series of K11777 analogs to further explore the antiviral activity of vinylsulfone-class protease inhibitors (Table 2 ). Given that the piperazine ring in K11777 is basic (pKa ∼7.8 for the conjugate acid) we considered that the compound might accumulate in the acidic (lysosomal and endosomal) compartments where target proteases such as CTSL and CTSB are abundant. To explore this notion and to more generally evaluate structure–activity trends, we synthesized new vinylsulfone analogs in which the substituent on the piperazine ring nitrogen atom was modified systematically. While the majority of these analogs (Table 2) retain a basic piperazine ring, the N-phenyl analog SMDC-256158 is only weakly basic (pKa ∼3.42 for the conjugate acid) and thus will be neutral at physiological pH and would not be expected to exhibit lysosomotropic behavior. Nearly all of the new analogs possessed potency comparable or superior to K11777 against SARS-CoV and EBOV (Table 2), the most potent analogs being SMDC256122 (SARS-CoV IC50 = 0.04 nM; EBOV IC50 = 0.12 nM), SMDC256159 (SARS-CoV IC50 = 0.07 nM; EBOV IC50 = 0.16 nM) and SMDC256160 (SARS-CoV IC50 = 0.08 nM; EBOV IC50 = 0.11 nM).
Table 2.
Structure and antiviral activity of K11777 analogs modified at the P3 position.
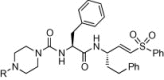 | |||||
|---|---|---|---|---|---|
| Compound | P3 substituent R= |
MW | pKaa | HIV-luc (SARS-CoV S)b IC50 (nM) |
HIV-luc (EBOV GP) IC50 (nM) |
| K11777 | Me | 575 | 7.02 | 0.32 ± 0.02 | 0.36 ± 0.02 |
| SMDC-256122 | Et | 589 | 7.29 | 0.04 ± 0.01 | 0.12 ± 0.01 |
| SMDC-256123 | i-Pr | 603 | 7.57 | 0.11 ± 0.01 | 0.25 ± 0.07 |
| SMDC-256157 | n-Pr | 603 | 7.59 | 0.24 ± 0.03 | 0.42 ± 0.03 |
| SMDC-256158 | Ph | 637 | 3.42 | 2.49 ± 0.34 | 2.69 ± 0.43 |
| SMDC-256159 | —CH2CH2OCH3 | 619 | 6.82 | 0.07 ± 0.02 | 0.16 ± 0.02 |
| SMDC-256160 | t-Bu | 617 | 7.87 | 0.08 ± 0.01 | 0.11 ± 0.03 |
| SMDC-256161 | Cyclopentyl | 629 | 8.01 | 0.25 ± 0.16 | 0.18 ± 0.01 |
| SMDC-256162 | Cyclopropylmethyl | 615 | 7.73 | 0.16 ± 0.03 | 0.10 ± 0.01 |
Calculated in MarvinSketch 5.5.0.1 from ChemAxon Ltd.
IC50 (inhibitory concentration) values of SARS-CoV or EBOV are the concentrations required to inhibit the infectivity of SARS-CoV or EBOV pseudotyped viruses on 293T-ACE2 cells by 50%, which were determined from dose response curves. A non-linear regression analysis based on the Sigmoidal dose response equation was applied to the percent inhibition and concentration data. Data is shown as means of quadruplicate measurements ± standard deviation. Values are representative of at least three independent experiments.
Of particular note from the structure–activity data is that the weakly basic analog SMDC-256158 was 10–100-fold less potent than the other basic and protonatable vinylsulfone analogs (Table 2). The reduced potency of SMDC-256158 is likely not related to the size of the phenyl substituent, since even larger, biaryl P3 substituents are known to be well tolerated in cathepsin-L like proteases such as cruzain (Beaulieu et al., 2010). Also consistent with this interpretation, we find that other bulky tert-butyl and cyclopentyl groups are tolerated in analogs like SMDC-256160 and SMDC-256161. Therefore, the most likely explanation is that as a weak base and the only analog expected not to be protonated at physiological pH, SMDC-256158 does not accumulate in the lysosome to the same extent that the other, more basic, analogs do. Conversely, K11777 and the other basic analogs accumulate in acidic endosomal compartments where target cysteine proteases such as CTSL and CTSB are located.
To further verify the antiviral effects of three of the most efficient drug candidates, inhibition assays were carried out with two strains (Urbani and Toronto-2) of replication competent SARS-CoV, and using two separate readouts of replication (summarized in Table 3 ). IC50 values ranged from <0.05 to 0.52 μM for K11777, <0.48 to <2.26 μM for SMDC256159, and <0.05 to 0.32 μM for SMDC256160. Furthermore, 90% reduction in viral yields of 0.35–1.04 μM (K11777), <0.48–7.1 μM (SMDC256159) and 0.49–12.2 μM (SMDC256160) were observed. We also performed studies with authentic hCoV-229E and determined the IC50 for all three compounds to be approximately 0.2 μM (data not shown). For the three compounds CC50’s were all above 25 μM. The selective indices (SI, CC50/IC50) ranged from 94.5 (SMDC256159 inhibition against the Toronto-2 strain) to over 1000. Thus, these compounds were identified as ideal tools to determine whether cysteine or serine proteases promote SARS-CoV spread in the host.
Table 3.
Inhibition of live SARS-CoV replication in Vero 76 cells.
| Compound | Virus strain | CPE inhibition |
Neutral red (NR) assay |
Virus yield reduction | ||||
|---|---|---|---|---|---|---|---|---|
| IC50a (μM) | CC50b (μM) | SIc | IC50a (μM) | CC50b (μM) | SIc | IC90d (μM) | ||
| K11777 | Urbani | <0.05 ± 0 | >105.6 ± 59.3 | >2112 | 0.52 ± 0.17 | >100.3 ± 64.2 | >192.9 | 0.35 ± 0.35 |
| Toronto-2 | <0.05 ± 0 | 85.2 ± 0 | >1704 | 0.35 ± 0.17 | 52.7 ± 7.8 | 150.6 | 1.04 ± 1.22 | |
| SMDC256159 | Urbani | 0.65 ± 0.81 | >109.2 ± 49.8 | >168 | <2.26 ± 3.56 | >91.6 ± 65.6 | >40.5 | 7.1 ± 7.6 |
| Toronto-2 | <0.48e | 85.6 | >178.3 | 0.65 | 61.4 | 94.5 | <0. 48 | |
| SMDC256160 | Urbani | <0.08 ± 0.05 | 50.6 ± 26.7 | >632.5 | <0.13 ± 0.03 | >81.8 ± 58.5 | >629.2 | 0.49 ± 0 |
| Toronto-2 | <0.05 | 27.6 | >552 | 0.32 | 30.8 | 96.3 | 12.2 | |
50% virus inhibitory concentration.
50% cell cytotoxic concentration of drug.
Selective index: SI = CC50/IC50.
90% virus inhibitory concentration.
No SDs were reported when n ⩽ 2.
3.4. Evaluating the efficacy of SMDC256160 in a lethal SARS-CoV BALB/c mouse model
The pharmacokinetics and bioavailability of SMDC256159 and SMDC256160 in male and female Sprague–Dawley rats were determined following a single i.v. or p.o. dose administration (data not shown) and demonstrated similar profiles to K11777 (Jacobsen et al., 2000).
In initial experiments, the antiviral efficacy of low-dose (1–10 mg/kg) SMDC256160 was examined in a lethal SARS-CoV mouse model (Day et al., 2009). While there was a trend toward protection, there was no statistically significant reduction in mortality or disease severity (data not shown). Experiments were therefore repeated at higher doses of cysteine protease inhibitor (50 mg/kg), either alone or in combination with the serine protease inhibitor, camostat (30 mg/kg) (Fig. 3 ). SMDC-256160 alone was no more effective than vehicle treated controls (Fig. 3). In contrast, camostat was effective in protecting mice against death due to a lethal infection by SARS-CoV, with a survival rate of ∼60%. Combining both classes of inhibitors did not significantly improve survival versus camostat alone. Thus, SARS-CoV depends on serine protease activity for viral spread in vivo.
Fig. 3.
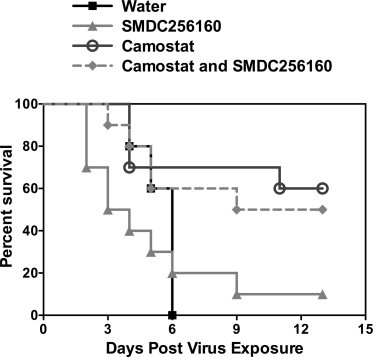
Effects of per os administered SMDC256160 and/or camostat on survival of BALB/c mice infected with a lethal SARS-CoV. Ten mice per group were dosed twice a day by oral gavage with SMDC256160 and/or camostat or diluent alone (sterile water) for 9 days beginning 10 h prior to infection with 10,000 pfu of mouse-adapted SARS-CoV.
4. Discussion
Viral entry is a multi-step process and an attractive target for antivirals (Zhou and Simmons, 2012). The fact that disparate pathogenic viruses such as SARS-CoV, EBOV and NiV all utilize a common host factor for entry – CTSL – suggested that inhibitors of cathepsins might have broad applicability. Cysteine proteases have proved to be druggable targets and their inhibitors are generally of low toxicity.
We screened a library of drug-like compounds with established activity against CTSL and CTSB for activity against SARS-CoV and filoviruses, including EBOV. We describe here the confirmation that protease inhibitors, such as K11777 and related compounds, are broad-spectrum antiviral drug candidates targeting viral entry. A number of additional vinylsulfone analogs were synthesized, some of which exhibited enhanced potency compared to K11777. Most notably, potent antiviral activity was correlated with the presence of a basic piperazine ring at the P3 position, a finding that is consistent with accumulation in endosomal (acidic) compartments where the target cysteine proteases required for viral entry are located. The vinylsulfones described herein were broadly active against viral entry for three viral families: the corona-, filo- and paramyxoviruses, and are very well tolerated in vivo (Barr et al., 2005).
The notion that coronaviruses, including SARS-CoV, use both a cathepsin-dependent endosomal pathway and a direct cell-surface serine protease-mediated pathway for entry (Simmons et al., 2013) is supported by our finding that the combination of K11777 and camostat was superior to either compound alone. In contrast, EBOV infection was effectively inhibited by K11777, but not by camostat. While unidentified additional proteases have been reported to mediate infection by other filoviruses, such as MARV (Gnirss et al., 2012) and SUDV (Misasi et al., 2012), efficient inhibition by the vinylsulfone analogs suggests that the unidentified proteases are cysteine proteases related to CTSB and L. Activation of NiV and HeV appears to be fully dependent on CTSL and/or CTSB (Pager et al., 2006, Diederich et al., 2008, Diederich et al., 2012). Thus, vinylsulfones are promising antiviral lead compounds for further optimization as potent inhibitors of these two important groups of pathogenic emerging viruses, including EBOV.
Previous reports showed that compound K11777 and analogs have satisfactory safety and pharmacokinetic profiles in rodents, dogs and primates (Abdulla et al., 2007). The fact that K11777, as a vinylsulfone, is an irreversible and not highly selective cysteine protease inhibitor does not appear to be a liability, at least if it is used as a short course antiviral. Indeed, in the case of filoviruses, the lack of target selectivity is likely a boon – increasing effectiveness by also inhibiting secondary proteases (Gnirss et al., 2012, Misasi et al., 2012).
The availability of a novel, highly potent and largely non-toxic cysteine protease inhibitor, SMDC256160, afforded the opportunity to assess whether the activity of cysteine or serine proteases is required for viral spread in vivo. For this, a mouse model for lethal SARS-CoV infection was employed. Notably, only inhibition of serine proteases mitigated SARS-CoV pathogenesis in vivo. Thus, future development of anti-coronavirus therapeutics should focus on inhibiting serine rather than cysteine proteases, with camostat being an excellent starting candidate. Indeed, in Japan camostat is used clinically, particularly to treat chronic pancreatitis (Ikeda et al., 1988, Sai et al., 2010), with a reasonable safety profile (Fiopan® Tablets, 2009).
5. Conclusions
Our results showed that targeting viral entry, and more specifically, the endosomal proteolysis step of entry, is an attractive strategy to discover new antiviral agents – particularly for filoviruses, like EBOV, and some paramyxoviruses. Although endosomal and cell-surface proteases can facilitate coronavirus entry in cultured cells, only the activity of serine proteases is required for viral spread in the infected host. Nevertheless, the highly potent cysteine protease inhibitors identified here might be excellent starting points for the development of highly effective inhibitors of Ebola virus and Paramyxovirus entry, and constitute excellent research tools for dissecting the molecular mechanisms of viral entry.
Acknowledgements
This work was supported by grants R01AI074986 and R21AI107165 from the National Institute of Allergy and Infectious Diseases (to G.S.) and funding from the Sandler Foundation (to A.R.R.). Preliminary animal studies were supported by NIAID contracts HHSN266200600011C (SRI International) and HHSN272201000039I, PI. John Morrey, PhD (to D.B.).
References
- Abdulla M.H., Lim K.C., Sajid M., McKerrow J.H., Caffrey C.R. Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 2007;4:e14. doi: 10.1371/journal.pmed.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang K.K., Ratnam J., Gut J., Legac J., Hansell E. Mining a cathepsin inhibitor library for new antiparasitic drug leads. PLoS Negl. Trop. Dis. 2011;5:e1023. doi: 10.1371/journal.pntd.0001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr S.C., Warner K.L., Kornreic B.G., Piscitelli J., Wolfe A. A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob. Agents Chemother. 2005;49:5160–5161. doi: 10.1128/AAC.49.12.5160-5161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu C., Isabel E., Fortier A., Masse F., Mellon C. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg. Med. Chem. Lett. 2010;20:7444–7449. doi: 10.1016/j.bmcl.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Bertram S., Glowacka I., Muller M.A., Lavender H., Gnirss K. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011;85:13363–13372. doi: 10.1128/JVI.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram S., Dijkman R., Habjan M., Heurich A., Gierer S. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 2013;87:6150–6160. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy J.W., Bryant C., Calvet C.M., Doyle P.S., Gunatilleke S.S. Chemical-biological characterization of a cruzain inhibitor reveals a second target and a mammalian off-target. Beilstein J. Org. Chem. 2013;9:15–25. doi: 10.3762/bjoc.9.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R.I., Chen B.K., Choe S., Landau N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Day C.W., Baric R., Cai S.X., Frieman M., Kumaki Y. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395:210–222. doi: 10.1016/j.virol.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot R.J., Baker S.C., Baric R.S., Brown C.S., Drosten C. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederich S., Thiel L., Maisner A. Role of endocytosis and cathepsin-mediated activation in Nipah virus entry. Virology. 2008;375:391–400. doi: 10.1016/j.virol.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederich S., Sauerhering L., Weis M., Altmeppen H., Schaschke N. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J. Virol. 2012;86:3736–3745. doi: 10.1128/JVI.06628-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel J.C., Doyle P.S., Hsieh I., McKerrow J.H. Cysteine protease inhibitors cure an experimental Trypanosoma cruzi infection. J. Exp. Med. 1998;188:725–734. doi: 10.1084/jem.188.4.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierer S., Bertram S., Kaup F., Wrensch F., Heurich A. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 2013;87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowacka I., Bertram S., Muller M.A., Allen P., Soilleux E. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011;85:4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirss K., Kuhl A., Karsten C., Glowacka I., Bertram S. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology. 2012;424:3–10. doi: 10.1016/j.virol.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H., Simmons G., Rennekamp A.J., Chaipan C., Gramberg T. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J. Virol. 2006;80:8639–8652. doi: 10.1128/JVI.00560-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S., Manabe M., Muramatsu T., Takamori K., Ogawa H. Protease inhibitor therapy for recessive dystrophic epidermolysis bullosa. In vitro effect and clinical trial with camostat mesylate. J. Am. Acad. Dermatol. 1988;18:1246–1252. doi: 10.1016/s0190-9622(88)70130-9. [DOI] [PubMed] [Google Scholar]
- Jacobsen W., Christians U., Benet L.Z. In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab. Dispos. 2000;28:1343–1351. [PubMed] [Google Scholar]
- Jaishankar P., Hansell E., Zhao D.M., Doyle P.S., McKerrow J.H. Potency and selectivity of P2/P3-modified inhibitors of cysteine proteases from trypanosomes. Bioorg. Med. Chem. Lett. 2008;18:624–628. doi: 10.1016/j.bmcl.2007.11.070. [DOI] [PubMed] [Google Scholar]
- Kawase M., Shirato K., van der Hoek L., Taguchi F., Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012;86:6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y., Wandersee M.K., Smith A.J., Zhou Y., Simmons G. Inhibition of severe acute respiratory syndrome coronavirus replication in a lethal SARS-CoV BALB/c mouse model by stinging nettle lectin, Urtica dioica agglutinin. Antiviral Res. 2011;90:22–32. doi: 10.1016/j.antiviral.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memish Z.A., Zumla A.I., Al-Hakeem R.F., Al-Rabeeah A.A., Stephens G.M. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 2013;368:2487–2494. doi: 10.1056/NEJMoa1303729. [DOI] [PubMed] [Google Scholar]
- Misasi J., Chandran K., Yang J.Y., Considine B., Filone C.M. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 2012;86:3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndao M., Nath-Chowdhury M., Sajid M., Marcus V., Mashiyama S.T. A cysteine protease inhibitor rescues mice from a lethal Cryptosporidium parvum infection. Antimicrob. Agents Chemother. 2013;57:6063–6073. doi: 10.1128/AAC.00734-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pager C.T., Craft W.W., Jr., Patch J., Dutch R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology. 2006;346:251–257. doi: 10.1016/j.virol.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer J.T., Rasnick D., Klaus J.L., Bromme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995;38:3193–3196. doi: 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- Palmer J.T., Bryant C., Wang D.X., Davis D.E., Setti E.L. Design and synthesis of tri-ring P3 benzamide-containing aminonitriles as potent, selective, orally effective inhibitors of cathepsin K. J. Med. Chem. 2005;48:7520–7534. doi: 10.1021/jm058198r. [DOI] [PubMed] [Google Scholar]
- Palmer J.T., Hirschbein B.L., Cheung H., McCarter J., Janc J.W. Keto-1,3,4-oxadiazoles as cathepsin K inhibitors. Bioorg. Med. Chem. Lett. 2006;16:2909–2914. doi: 10.1016/j.bmcl.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Rydzewski R.M., Bryant C., Oballa R., Wesolowski G., Rodan S.B. Peptidic 1-cyanopyrrolidines: synthesis and SAR of a series of potent, selective cathepsin inhibitors. Bioorg. Med. Chem. 2002;10:3277–3284. doi: 10.1016/s0968-0896(02)00173-6. [DOI] [PubMed] [Google Scholar]
- Sai J.K., Suyama M., Kubokawa Y., Matsumura Y., Inami K. Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J. Gastroenterol. 2010;45:335–341. doi: 10.1007/s00535-009-0148-1. [DOI] [PubMed] [Google Scholar]
- Salvador B., Zhou Y., Michault A., Muench M.O., Simmons G. Characterization of Chikungunya pseudotyped viruses: identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology. 2009;393:33–41. doi: 10.1016/j.virol.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador B., Sexton N.R., Carrion R., Jr., Nunneley J., Patterson J.L. Filoviruses utilize glycosaminoglycans for their attachment to target cells. J. Virol. 2013;87:3295–3304. doi: 10.1128/JVI.01621-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Wool-Lewis R.J., Baribaud F., Netter R.C., Bates P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002;76:2518–2528. doi: 10.1128/jvi.76.5.2518-2528.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U.S.A. 2004;101:4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Zmora P., Gierer S., Heurich A., Pohlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 2013;100:605–614. doi: 10.1016/j.antiviral.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen I., Liss N.M., Schneider B.S., Fair J.N., Chiu C.Y. Characterization of the Bas-Congo virus glycoprotein and its function in pseudotyped viruses. J. Virol. 2013;87:9558–9568. doi: 10.1128/JVI.01183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiopan® Tablets [package insert]. Ono Pharmaceutical Co., Ltd, Chuo-ku, Osaka, Japan; August 2009.
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Simmons G. Development of novel entry inhibitors targeting emerging viruses. Expert Rev. Anti Infect. Ther. 2012;10:1129–1138. doi: 10.1586/eri.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Lu K., Pfefferle S., Bertram S., Glowacka I. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010;84:8753–8764. doi: 10.1128/JVI.00554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Agudelo J., Lu K., Goetz D.H., Hansell E. Inhibitors of SARS-CoV entry – identification using an internally-controlled dual envelope pseudovirion assay. Antiviral Res. 2011;92:187–194. doi: 10.1016/j.antiviral.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Steffen I., Montalvo L., Lee T.H., Zemel R. Development and application of a high-throughput microneutralization assay: lack of xenotropic murine leukemia virus-related virus and/or murine leukemia virus detection in blood donors. Transfusion. 2012;52:332–342. doi: 10.1111/j.1537-2995.2011.03519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]