

Abstract
Background
This study was designed to investigate the effects of different doses of levetiracetam on aquaporin 4 (AQP4) expression in rats after fluid percussion injury.
Material/Methods
Sprague-Dawley rats were randomly divided into 4 groups: sham operation group, traumatic brain injury group, low-dose levetiracetam group, and high-dose levetiracetam group. Brain edema models were established by fluid percussion injury, and intervened by the administration of levetiracetam. Samples from the 4 groups were collected at 2, 6, 12, and 24 h, and at 3 and 7 days after injury. Histological observation was performed using hematoxylin-eosin staining and immunohistochemical staining. AQP4 and AQP4 mRNA expression was detected using Western blot assay and RT-PCR. Brain water content was measured by the dry-wet method.
Results
Compared with the traumatic brain injury group, brain water content, AQP4 expression, and AQP4 mRNA expression were lower in the levetiracetam groups at each time point and the differences were statistically significant (P<0.05). The intervention effects of high-dose levetiracetam were more apparent.
Conclusions
Levetiracetam can lessen brain edema from fluid percussion injury by down-regulating AQP4 and AQP4 mRNA expression. There is a dose-effect relationship in the preventive effect of levetiracetam within a certain extent.
MeSH Keywords: Aquaporin 4, Brain Edema, Real-Time Polymerase Chain Reaction
Background
Brain edema is a major pathological change following traumatic brain injury. Mild brain edema causes neurological dysfunction and severe brain edema is life-threatening; brain edema is a major determinant of patient prognosis [1–5]. Aquaporin 4 (AQP4) is a major membrane protein that mediates 2-way transmembrane transport of water molecules in the brain tissue, and plays a key role in producing traumatic brain edema [6–10].
Levetiracetam derived from pyrrolidone is an analogue of piracetam and a novel orally active antiepileptic drug used in adjunctive therapy of primary generalized tonic-clonic seizures (partial seizures and myoclonic seizures). Levetiracetam achieves antiepileptic effects by binding to synaptic vesicle proteins of neurons in the central nervous system. Levetiracetam has a high capacity to bind to synaptic vesicle proteins, which can inhibit abnormal discharge of neurons and exert antiepileptic effects. Levetiracetam is characterized by high bioavailability, linear curve, low protein binding rate, no hepatic enzyme induction, ideal pharmacokinetic properties, and good tolerability and safety. In addition to the antiepileptic effect, levetiracetam also has been shown to resist apoptosis, inhibit the proliferation of glial cells, and mitigate brain edema [11–13], but there is lack of molecular biological evidence.
This study sought to investigate the effects of levetiracetam on brain edema and AQP4 expression so as to provide theoretical evidence for the effects of levetiracetam on lessening traumatic brain edema in a rat model of brain edema induced by fluid percussion injury.
Material and Methods
Experimental animals
We obtained 192 healthy specific-pathogen-free male Sprague-Dawley rats aged 6–9 weeks and weighing 210–270 g from the Experimental Animal Center of Hebei Medical University in China (certificate no. SCXK(Ji)2008-1-003). The rats were housed at room temperature (20–25°C), with a 12-h light/dark cycle (8:00–12:00), and were allowed free access to food and water. All procedures were approved by the Animal Care Committee of Hebei Medical University and were in accordance with the guidelines of the National Institutes of Health (Bethesda, MD, USA) on the care and use of animals.
Main reagents and instruments
We used levetiracetam oral liquid (300 ml: 30 g, Approval no. GYZZJ20150002; NextPharma SAS, France), rabbit anti-mouse AQP4 polyclonal antibody, biotin-labeled goat anti-rabbit IgG, diaminobenzidine color kit and enhanced chemiluminescence kit (Santa Cruz Biotechnology, USA), RNA extraction reagent (Invitrogen), ribonuclease inhibitor (RNasin), dNTP, reverse transcriptase (M-MLV), random primers, the Taq DNA polymerase and agarose (Promega, USA), a quantitative fluorescent reagent kit (Hot Start Fluorescent PCR Core Reagent Kits, SYBR Green I) (BBI Company), a fluid percussion injury device (NatureGene Corp., USA), and the 7300 real-time PCR system (ABI, USA).
Model establishment
The rats were intraperitoneally anesthetized with 10% chloral hydrate (0.3 g/kg), and fixed on a stereotaxic apparatus. Craniectomy was performed on the right hemisphere at 3 mm posterior to the anterior fontanelle and 3 mm lateral to the median line. The skull was resected 5 mm in diameter, and the rat dura was kept intact. The injury hub, consisting a female Luer-Lok fitting, was affixed to the craniectomy site with cyanoacrylate adhesives and dental cement, which was connected to the transfer tube of the fluid percussion injury device via a male Luer-Lok. Animal models of edema were established by moderate fluid percussion pulse (21±1ms duration) on the right parietal cortex at 0.2±0.01MPa. A single moderate fluid percussion pulse of injury was caused by releasing of the pendulum. The sinusoidal pressure curve was captured by digital-storage oscilloscope through pressure transducer. Fluid percussion injury was graded as mild (0.01–0.10 MPa), moderate (0.15–0.21 MPa), or severe (0.31–0.37 MPa) according to the strength of pressure. A pressure of 0.2 MPa was generated when the release angle of the pendulum was set at 21.5 degrees [14–16]. The SO group did not receive fluid percussion and only the bone window was exposed.
Grouping and administration of drugs
The rats were randomly divided into a sham operation group (SO group, n=48), a traumatic brain injury group (TBI group, n=48), a low-dose levetiracetam group (LL group, n=48), and a high-dose levetiracetam group (LH group, n=48). TBI, LL, and LH groups received moderate fluid percussion. Rats in the LL and LH groups were intragastrically administered levetiracetam oral liquid 100 mg/kg/d and 300 mg/kg/d, once a day, at 15 min after injury. Rats in the SO and TBI groups were intragastrically administered an equivalent volume of physiological saline.
The SO group, TBI group, LL group, and LH group were each divided into 6 subgroups according to time after injury (2, 6, 12, and 24 h and 3 and 7 days).
At 2, 6, 12, and 24 h and 3 and 7 days after model establishment, 8 rats in each group were intraperitoneally injected with 10% chloral hydrate (0.3 g/kg), perfused with 4% paraformaldehyde in the heart, and decapitated. The skull was opened and the brain tissue was obtained. Brain water content was measured. Brain tissue received hematoxylin-eosin staining, immunohistochemical staining, Western blot assay, and RT-PCR.
Brain water content as detected by the dry-wet method
Brain water content was measured using the dry-wet method. At 2, 6, 12, and 24 and 3 and 7 days after model establishment, 8 rats in each group were sacrificed. Approximately 200±50 mg of brain tissue obtained from the anterior border in the percussion area was dried with filter paper, and weighed with an electronic analytical balance with 0.0001-g accuracy to obtain wet weight (W). The tissue was dried in a 110°C dryer for 24 h and weighed to obtain dry weight (D). In accordance with Elliot formula [17], brain water content (%) was calculated by (W–D)/W×100%.
Hematoxylin-eosin staining and immunohistochemical staining
At 2, 6, 12, and 24 h and 3 and 7 days after model establishment, 8 rats in each group were sacrificed. An approximately 4-mm-thick piece of brain tissue in the percussion area was fixed in 4% paraformaldehyde at 4°C for 24 h and embedded in paraffin, and then sliced into 4-μm sections. Paraffin sections were dewaxed, hydrated, and stained with hematoxylin and eosin. The extent and scope of balloon cells were observed in the edema area with a light microscope.
Horseradish peroxidase-labeled streptavidin (S-P method) was used in immunohistochemical staining. Primary antibody and secondary antibody were rabbit anti-rat AQP4 polyclonal antibody (1:100) and biotin-labeled goat anti-rabbit IgG (1:1,000), respectively. Samples were visualized with a diaminobenzidine color kit. Goat serum and PBS were used as negative controls instead of primary antibody. The presence of brown coarse or fine particles on the cell membrane was considered as a positive reaction.
AQP4 protein expression as detected by western blot assay
At 2, 6, 12, and 24 h and 3 and 7 days after model establishment, 8 rats in each group were sacrificed. A 200-mg piece of brain tissue from the posterior border in the percussion area was cut into pieces on ice. Membrane proteins in each group were extracted with a Thermo Scientific Pierce Protein Extraction Kit, and quantified with a Thermo Scientific Pierce BCA Protein Quantification Kit. Forty μg of proteins were loaded and separated on 10% separating gel and 5% stacking gel for sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The proteins were electrically transferred to nitrocellulose membranes. The membranes were blocked with 5% defatted milk powder at room temperature for 2 h, incubated with blocking solution-diluted rabbit anti-rat AQP4 polyclonal antibody (1:200) at 4°C overnight, with 5% defatted milk powder-diluted goat anti-rabbit IgG (1:6000) at room temperature for 2 h, and visualized with an enhanced chemiluminescence kit. The internal reference was β-actin. Integral optical density (IOD) values of target band and internal reference were analyzed with a gel image analysis system (Gel-Pro Analyzer Version 3.0). The ratio of IOD values of target band and internal reference represented relative expression levels of protein.
AQP4 mRNA expression as detected by RT-PCR
At 2, 6, 12, and 24 h and 3 and 7 days after model establishment, total RNA was extracted from brain tissues of each group using a Trizol RNA Isolation extraction kit. RNA purity was measured using ultraviolet spectrophotometry. OD values at 260 nm and 280 nm (OD260 and OD280) were determined using the ultraviolet spectrophotometry. The ratio of OD260 to OD280 was set between 1.8 and 2.0. cDNA was synthesized with a M-MLV reverse transcription kit. PCR amplification was conducted taking cDNA as a template. With NCBI Genebank and Primer Premier 5.0 software, primer sequences were designed and synthesized by Sangon Biotech, Shanghai, China. β-actin was considered as an internal reference. Primer sequences were as follows:
AQP4 primer:
Upstream 5′-GAATTCAGCACCTGTGATAG-3′;
Downstream 5′-CGCTGAGCGGCACAGAGATG-3′;
β-actin primer:
Upstream 5′-CTAGGCACCAGCGTGTGATG-3′;
Downstream 5′-GAACGTCTCACACATGATCTGG-3′
Quantitative fluorescent PCR was performed with a quantitative fluorescent reagent kit (SYBR Green I) using the 7300 real-time PCR system. Thermal cycling parameters were: 96°C for 4 min, followed by 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s. Fluorescence signal was collected at 72°C for 30 s in each cycle. After amplification, β-actin served as an internal reference gene. Compared with the control group, relative quantitative value of the target gene was obtained and used for statistical analysis.
Statistical analysis
Results were analyzed using SPSS17.0 statistical software and expressed as mean ±SD. Means among multiple groups were compared using 2-way ANOVA. The comparison between groups was conducted using Tukey’s post hoc comparisons. A value of P<0.05 was considered statistically significant.
Results
Histological observation
As shown in Figures 1 and 2, in the SO group, the space around neurons was widened, and cellular edema was ballooning and became worsen at 2–12 h after injury. Neuronal swelling reached a peak and presented vacuolated cells at 24 h. Swelling lasted for 3 days. At 24 h, swelling was relieved; gliosis and inflammatory cell infiltration appeared. At 7 days, cellular edema mostly disappeared, and a scar-like change was visible in the percussion area. Compared with the SO group, cellular edema was lessened at various time points in the LL and LH groups; the number of balloon cells was remarkably reduced and the space around cells became narrowed. Compared with the LL group, cellular edema was mitigated at 12 and 24 h in the LH group.
Figure 1.

The HE staining of brain tissue in different groups after 24 h. (A) SO group, (B) TBI group, (C) LL group, (D) LH group. There was no significant pathological lesion in brain tissue slice of SO group. The swollen cells were successively alleviated in TBI, LL, and LH groups. Scale bar: 100 μm (×400).
Figure 2.

The expression of AQP4 in different groups after 24 h by immunohistochemistry staining. (A) SO group, (B) TBI group, (C) LL group, (D) LH group. The positive cells were consecutively decreased in TBI, LL, and LH groups. Scale bar: 100 μm (×400).
Determination of brain water content
Brain water content was slightly lower in the LL and LH groups than in the TBI group at 2 h, but no statistical significance was detected (P>0.05). At 6 h, brain water content was significantly lower in the LL and LH groups than in the TBI group (79.77±1.12, 76.83±1.67, 74.88±2.43; P<0.05), and no statistically significant difference was detectable between the LL and LH groups (P>0.05). At 12 h, brain water content was increased in the 3 groups (81.62±1.39, 77.70±1.56, 75.69±2.00), and a statistically significant difference was observed among the 3 groups (P<0.05). At 24 h, brain water content reached the peak (82.41±1.66, 78.49±1.59, 75.94±1.47), and a statistically significant difference was observed among the 3 groups (P<0.05). At 3 days, brain water content began to decrease (81.41±1.41, 77.40±1.70, 75.47±1.33), and a statistically significant difference was observed among the 3 groups (P<0.05). At 7 days, brain water content was noticeably reduced in each group (77.80±0.75, 75.51±2.37, 74.81±0.78); brain water content was slightly lower in the LL and LH groups than in the TBI group (P<0.05), but no significant difference in brain water content was found between the LL and LH groups (P>0.05; Table 1, Figure 3).
Table 1.
Brain water content at different time points in each group (χ̄±S%, n=8).
| Time | Brain water content (%) | |||
|---|---|---|---|---|
| SO | TBI | LL | LH | |
| 2 h | 72.47±0.83 | 75.76±1.12a | 75.03±1.53a | 74.55±0.83a |
| 6 h | 72.18±0.72 | 79.77±1.12a | 76.83±1.67a,b | 74.88±2.43a,b |
| 12 h | 72.12±0.63 | 81.62±1.39a | 77.70±1.56a,b | 75.69±2.00a,b,c |
| 24 h | 72.24±1.00 | 82.41±1.66a | 78.49±1.59a,b | 75.94±1.47a,b,c |
| 3 d | 72.44±0.66 | 81.41±1.41a | 77.40±1.70a,b | 75.47±1.33a,b |
| 7 d | 72.1.3±0.77 | 77.80±0.75a | 75.51±2.37a,b | 74.81±0.78a,b |
P<0.05, vs. SO group;
P<0.05, vs. TBI group;
P<0.05, vs. LL group at the same time point.
Figure 3.
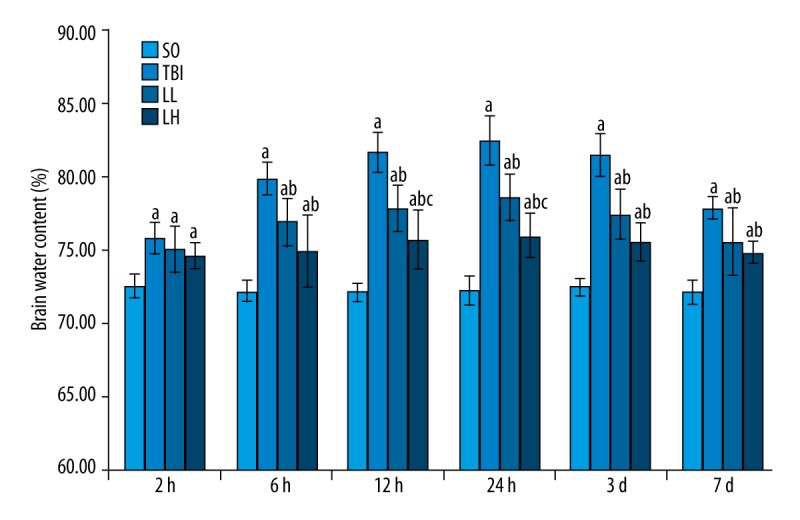
Levetiracetam effects on brain water content in rats with brain edema in each group. Each column represents the mean (%) ±SD. Results were analyzed by 2-way ANOVA followed by Tukey’s post hoc comparisons (each group, n=8). a P<0.05, vs. SO group; b P<0.05, vs. TBI group; c P<0.05, vs. LL group at the same time point.
Determination of AQP4 using Western blot assay
AQP4 expression showed an ascendant trend in the TBI, LL, and LH groups compared with the SO group (P<0.05). AQP4 expression was lower in the LL and LH groups than in the TBI group at various time points (except at 2 h), and the decrease was apparent at 12 and 24 h and 3 days (P<0.05). AQP4 expression was lower in the LH group than in the LL group at 12 and 24 h (P<0.05; Table 2, Figures 4).
Table 2.
AQP4 expression at different time points in each group (χ̄±S%, n=8).
| Time | AQP4 expression | |||
|---|---|---|---|---|
| SO | TBI | LL | LH | |
| 2 h | 0.085±0.011 | 0.141±0.028a | 0.130±0.008a | 0.119±0.084a |
| 6 h | 0.085±0.011 | 0.164±0.020a | 0.134±0.021a,b | 0.128±0.023a,b |
| 12 h | 0.086±0.013 | 0.236±0.017a | 0.198±0.019a,b | 0.146±0.026a,b,c |
| 24 h | 1.069±0.081 | 1.636±0.055a | 1.480±0.062a,b | 1.345±0.090a,b,c |
| 3 d | 0.095±0.011 | 0.206±0.021a | 0.164±0.025a,b | 0.149±0.014a,b |
| 7 d | 0.095±0.013 | 0.163±0.014a | 0.131±0.006a,b | 0.128±0.005a,b |
P<0.05, vs. SO group;
P<0.05, vs. TBI group;
P<0.05, vs. LL group at the same time point.
Figure 4.
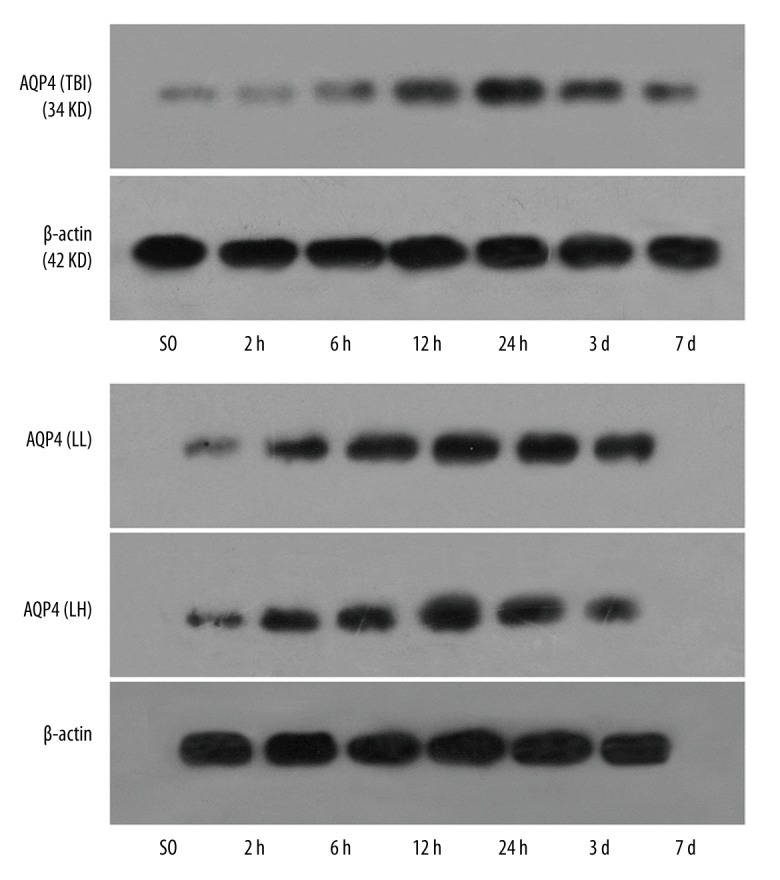
The expression of AQP4 protein in the different groups by Western blot assay with β-actin as housekeeping gene.
Detection of AQP4 mRNA expression using RT-PCR
AQP4 mRNA expression was significantly greater in the TBI, LL, and LH groups than in the SO group (P<0.05). AQP4 mRNA expression was significantly lower in the LL and LH groups than in the TBI group at various time points (except 2 h) (P<0.05). Significant differences in AQP4 mRNA expression were detectable between the LL and LH groups at 12 and 24 h (P<0.05; Table 3, Figures 5, 6).
Table 3.
AQP4 mRNA expression at different time points in each group (χ̄±S%, n=8).
| Time | AQP4 mRNA expression | |||
|---|---|---|---|---|
| SO | TBI | LL | LH | |
| 2 h | 0.968±0.063 | 1.185±0.052a | 1.135±0.023a | 1.108±0.036a |
| 6 h | 0.093±0.078 | 1.325±0.061a | 1.199±0.057a,b | 1.118±0.080a,b |
| 12 h | 0.976±0.053 | 1.473±0.087a | 1.365±0.085a,b | 1.243±0.047a,b,c |
| 24 h | 1.069±0.081 | 1.636±0.055a | 1.480±0.062a,b | 1.345±0.090a,b,c |
| 3 d | 0.980±0.077 | 1.441±0.080a | 1.226±0.056a,b | 1.145±0.043a,b |
| 7 d | 1.063±0.077 | 1.203±0.050a | 1.175±0.053a,b | 1.110±0.042a,b |
P<0.05, vs. SO group;
P<0.05, vs. TBI group;
P<0.05, vs. LL group at the same time point.
Figure 5.
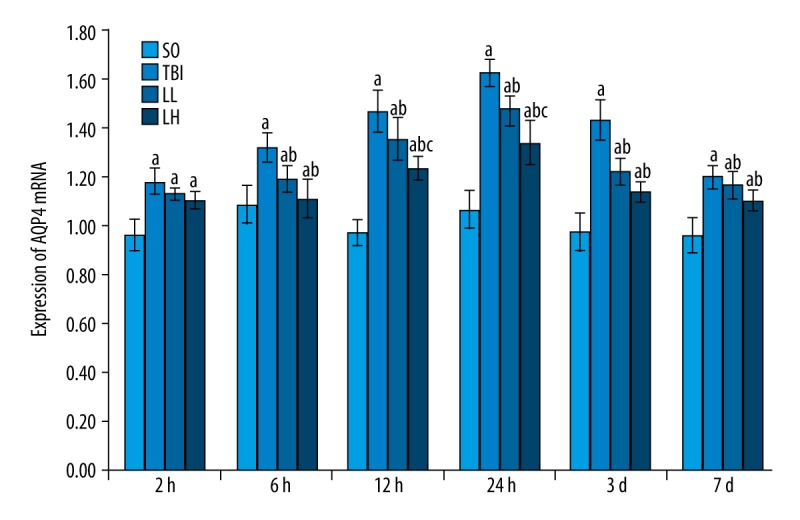
Levetiracetam effects on AQP4 mRNA expression in rats of each group. RT-PCR was used to measure AQP4 mRNA expression levels. The data are presented as mean±SD and were analyzed by 2-way ANOVA followed by Tukey’s post hoc comparisons (each group, n=8). a P<0.05, vs. SO group; b P<0.05, vs. TBI group; c P<0.05, vs. LL group at the same time point.
Figure 6.
The amplification curve of AQP4 cDNA (RT-PCR).
Discussion
AQP4 is expressed mainly in astrocytes and ependymal cells. AQP4 expression is abundant on the subarachnoid and perivascular astrocyte surface and glial limiting membrane formed by the astrocyte end-feet-surrounded capillary wall; this distribution is called a polarized distribution. This distribution feature indicates that AQP4 plays a key role in maintaining water balance in the brain, and is an important structural basis of water regulation and transport among glial cells, cerebrospinal fluid, and blood vessels [18–22]. AQP4 expression was positively correlated with brain water content after fluid percussion injury on the swollen area, which indicated that AQP4 participated in the formation of brain edema. The trend of changes in AQP4 expression was basically consistent with the changes in brain water content, which suggest that traumatic brain injury activated AQP4 on the astrocyte end-feet; the water molecule entered astrocytes and produced brain edema [23–26]. Rao et al. demonstrated that AQP expression was evidently up-regulated in the swollen area following fluid percussion injury, and brain edema could be obviously lessened by down-regulating AQP4 expression with siRNA silencing or cycloheximide [27]. Protein kinase C agonist (activated protein kinase C causes AQP4 phosphorylation and the loss of biological activity) phorbol ester, or dibutyryl not only apparently reduced brain water content, but also noticeably inhibited AQP4 expression. These results show that phorbol ester or dibutyryl relieved brain edema by suppressing AQP4 expression, and verified that the up-regulation of AQP4 expression was involved in the occurrence of traumatic brain edema [28,29]. In a rat model of lateral fluid percussion-induced severe traumatic brain injury, decompression craniectomy could diminish AQP4 expression and mitigate brain edema [30]. In a controlled cortical impact model, rapid microsurgical skull reconstruction could increase AQP4 expression and aggravate brain injury [31]. In the present study, the alterations in brain edema induced by fluid percussion were consistent with the trends of AQP4 and AQP4 mRNA expression, which confirmed that the up-regulation of AQP4 expression was the main cause of traumatic brain edema.
Levetiracetam can be rapidly absorbed orally, and has anti-epileptic and neuroprotective effects ]32,33]. In a controlled cortical impact model, levetiracetam could relieve inflammatory reaction and promote the recovery of neurological function [34]. In a rat model of closed craniocerebral injury and subarachnoid hemorrhage, levetiracetam could protect neurons and lessen cerebral vasospasm [35]. This study confirmed that levetiracetam could not alter the trend of brain edema, but could noticeably diminish the degree of brain edema. Moreover, high-dose levetiracetam could more evidently reduce the degree of brain edema, in a dose-effect relationship. A positive correlation between AQP4 and AQP4 mRNA expression suggested that the changes in AQP4 mRNA expression determined the changes in AQP4 expression. This indicates that the increase in AQP4 protein expression might be induced by the increase in AQP4 mRNA synthesis. The trend of the inhibitory effect of levetiracetam on AQP4 and AQP4 mRNA expression was consistent with the trend of the effect of levetiracetam on decreasing brain water content. Taken together, levetiracetam mitigated fluid percussion-induced brain edema, possibly by down-regulating AQP4 mRNA expression and suppressing AQP4 expression. These findings provide experimental evidence for clinical use of levetiracetam in the treatment of craniocerebral injury.
Conclusions
This study showed that levetiracetam could effectively reduce brain water content, lessen brain edema, and protect brain tissue by down-regulating AQP4 mRNA expression. The precise mechanism underlying the down-regulation of AQP4 mRNA expression deserves further investigations.
Footnotes
Source of support: This work was supported by the Natural Science Foundation Project of Hebei Province (no. H2014307060) and the Department of Science and Technology of Hebei Province (no. 13277767D)
Conflict of interest
None.
References
- 1.Sviri GE. Massive cerebral swelling immediately after cranioplasty, a fatal and unpredictable complication: report of 4 cases. J Neurosurg. 2015 doi: 10.3171/2014.11.JNS141152. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2.Margulies S, Anderson G, Atif F, Badaut J, et al. Combination therapies for traumatic brain injury: Retrospective considerations. J Neurotrauma. 2016;33(1):101–12. doi: 10.1089/neu.2014.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filippidis AS, Liang X, Wang W, et al. Real-time monitoring of changes in brain extracellular sodium and potassium concentrations and intracranial pressure after selective vasopressin-1a receptor inhibition following focal traumatic brain injury in rats. J Neurotrauma. 2014;31:1258–67. doi: 10.1089/neu.2013.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marmarou CR, Liang X, Abidi NH, et al. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP, V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014;1581:89–102. doi: 10.1016/j.brainres.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed F, Plantman S, Cernak I, Agoston DV. The temporal pattern of changes in serum biomarker levels reveals complex and dynamically changing pathologies after exposure to a single low-intensity blast in mice. Front Neurol. 2015;6:114. doi: 10.3389/fneur.2015.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Badaut J, Fukuda AM, Jullienne A, Petry KG. Aquaporin and brain diseases. Biochim Biophys Acta. 2014;1840:1554–65. doi: 10.1016/j.bbagen.2013.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lv Q, Fan X, Xu G, et al. Intranasal delivery of nerve growth factor attenuates aquaporins-4-induced edema following traumatic brain injury in rats. Brain Res. 2013;1493:80–89. doi: 10.1016/j.brainres.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 8.Lo Pizzo M, Schiera G, Di Liegro I, et al. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol Sci. 2013;34:1309–14. doi: 10.1007/s10072-012-1233-4. [DOI] [PubMed] [Google Scholar]
- 9.Jullienne A, Badaut J. Molecular contributions to neurovascular unit dysfunctions after brain injuries: Lessons for target-specific drug development. Future Neurol. 2013;8:677–89. doi: 10.2217/fnl.13.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shenaq M, Kassem H, Peng C, et al. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1α after traumatic brain injury (TBI) J Neurol Sci. 2012;323:134–40. doi: 10.1016/j.jns.2012.08.036. [DOI] [PubMed] [Google Scholar]
- 11.Benge JF, Phenis RA, Bernett A, et al. Neurobehavioral effects of levetiracetam in patients with traumatic brain injury. Front Neurol. 2013;4:195. doi: 10.3389/fneur.2013.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shetty AK. Prospects of levetiracetam as a neuroprotective drug against status epilepticus, traumatic brain injury, and stroke. Front Neurol. 2013;4:172. doi: 10.3389/fneur.2013.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szaflarski JP, Nazzal Y, Dreer LE. Post-traumatic epilepsy: Current and emerging treatment options. Neuropsychiatr Dis Treat. 2014;10:1469–77. doi: 10.2147/NDT.S50421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon CE, Lighthall JW, Anderson TE. Physiologic, histopathologic, and cineradiographic characterization of a new fluid-percussion model of experimental brain injury in the rat. J Neurotrauma. 1988;5:91–104. doi: 10.1089/neu.1988.5.91. [DOI] [PubMed] [Google Scholar]
- 15.Dixon CE, Lyeth BG, Povlishock JT, et al. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–19. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- 16.McIntosh TK, Vink R, Noble L, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–44. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 17.Elliott KA, Jasper H. Measurement of experimentally induced brain swelling and shrinkage. Am J Physiol. 1949;157:122–29. doi: 10.1152/ajplegacy.1949.157.1.122. [DOI] [PubMed] [Google Scholar]
- 18.Laird MD, Shields JS, Sukumari-Ramesh S, et al. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iliff JJ, Chen MJ, Plog BA, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180–93. doi: 10.1523/JNEUROSCI.3020-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dardiotis E, Paterakis K, Tsivgoulis G, et al. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J Neurotrauma. 2014;31:1920–26. doi: 10.1089/neu.2014.3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cartagena CM, Phillips KL, Tortella FC, et al. Temporal alterations in aquaporin and transcription factor HIF1α expression following penetrating ballistic-like brain injury (PBBI) Mol Cell Neurosci. 2014;60:81–87. doi: 10.1016/j.mcn.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Kapoor S, Kim SM, Farook JM, et al. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J Neurosci. 2013;33:17398–403. doi: 10.1523/JNEUROSCI.2756-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Rodriguez AB, Acaz-Fonseca E, Viveros MP, Garcia-Segura LM. Changes in cannabinoid receptors, aquaporin 4 and vimentin expression after traumatic brain injury in adolescent male mice. Association with edema and neurological deficit. PLoS One. 2015;10:e0128782. doi: 10.1371/journal.pone.0128782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitchen P, Day RE, Taylor LH, et al. Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J Biol Chem. 2015;290:16873–81. doi: 10.1074/jbc.M115.646034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu W, Tian R, Hao S, et al. A pre-injury high ethanol intake in rats promotes brain edema following traumatic brain injury. Br J Neurosurg. 2014;28:739–45. doi: 10.3109/02688697.2014.915007. [DOI] [PubMed] [Google Scholar]
- 26.Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J Cereb Blood Flow Metab. 2013;33:1621–32. doi: 10.1038/jcbfm.2013.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao KV, Reddy PV, Curtis KM, Norenberg MD. Aquaporin-4 expression in cultured astrocytes after fluid percussion injury. J Neurotrauma. 2011;28:371–81. doi: 10.1089/neu.2010.1705. [DOI] [PubMed] [Google Scholar]
- 28.Amorini AM, Dunbar JG, Marmarou A. Modulation of aquaporin-4 water transport in a model of TBI. Acta Neurochir Suppl. 2003;86:261–63. doi: 10.1007/978-3-7091-0651-8_56. [DOI] [PubMed] [Google Scholar]
- 29.Karasu A, Aras Y, Sabancı PA, et al. The effects of protein kinase C activator phorbol dibutyrate on traumatic brain edema and aquaporin-4 expression. Ulus Travma Acil Cerrahi Derg. 2010;16:390–94. [PubMed] [Google Scholar]
- 30.Tomura S, Nawashiro H, Otani N, et al. Effect of decompressive craniectomy on aquaporin-4 expression after lateral fluid percussion injury in rats. J Neurotrauma. 2011;28:237–43. doi: 10.1089/neu.2010.1443. [DOI] [PubMed] [Google Scholar]
- 31.Glover LE, Tajiri N, Lau T, et al. Immediate, but not delayed, microsurgical skull reconstruction exacerbates brain damage in experimental traumatic brain injury model. PLoS One. 2012;7:e33646. doi: 10.1371/journal.pone.0033646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou H, Hurwitz M, Fowler L, Wagner AK. Abbreviated levetiracetam treatment effects on behavioural and histological outcomes after experimental TBI. Brain Inj. 2015;29:78–85. doi: 10.3109/02699052.2014.955528. [DOI] [PubMed] [Google Scholar]
- 33.Ramakrishnan V, Dahlin R, Hariri O, et al. Anti-epileptic prophylaxis in traumatic brain injury: A retrospective analysis of patients undergoing craniotomy versus decompressive craniectomy. Surg Neurol Int. 2015;6:8. doi: 10.4103/2152-7806.149613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou H, Brayer SW, Hurwitz M, et al. Neuroprotective, neuroplastic, and neurobehavioral effects of daily treatment with levetiracetam in experimental traumatic brain injury. Neurorehabil Neural Repair. 2013;27:878–88. doi: 10.1177/1545968313491007. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Gao J, Lassiter TF, et al. Levetiracetam is neuroprotective in murine models of closed head injury and subarachnoid hemorrhage. Neurocrit Care. 2006;5:71–78. doi: 10.1385/NCC:5:1:71. [DOI] [PubMed] [Google Scholar]