

Abstract
The present study aimed to investigate the mechanism by which Aurora kinase A (AURKA) promotes cell migration and invasion in head and neck squamous cell carcinoma (HNSCC). Transwell assays were performed to investigate the cell migration and invasion abilities of AURKA, whilst western blotting was used to analyze the protein expression in FaDu and Hep2 cells, each treated with pharmacological inhibitors. Following the inhibition of AURKA, Akt and focal adhesion kinase (FAK), the migration and invasion of the FaDu and Hep2 cells decreased. The expression of phosphorylated (p)-AURKA and p-FAK (Y397) was observed to decrease following FaDu and Hep2 cell treatment with VX-680, a small molecular inhibitor of AURKA. The expression of p-Akt and p-FAK (Y397) ceased following treatment with the Akt inhibitor triciribine. The expression of p-FAK (Y397) decreased, however, p-Akt expression did not change following treatment with the FAK inhibitor TAE226. In conclusion, AURKA activates FAK through the AURKA/Akt/FAK signaling pathway, promoting the migration and invasion of HNSCC cells, which may subsequently provide a novel approach for the treatment of HNSCC.
Keywords: Aurora kinase A, head and neck squamous cell carcinoma, Akt, FAK
Introduction
Head and neck squamous cell carcinoma (HNSCC) is one of the most prevalent types of human cancer, with ~500,000 novel cases being diagnosed worldwide every year (1). HNSCC is the sixth leading cause of cancer-associated mortality (2). Although the molecular pathogenesis of HNSCC is not yet fully understood, it has been reported that aberrant cell division is one of the main mechanisms of carcinogenesis (3). Undergoing chromosome condensation, nuclear envelope breakdown, separation, bipolar-spindle assemblage, chromosome segregation and cytokinesis, the cells separate and replicated genetic material is split into two daughter cells (3). A complex regulatory network of kinase and phosphatase regulate this process aiding the accuracy of cell division.
There are three homologues of Aurora kinase (A, B and C) (4). Aurora kinase A (AURKA) has been implicated in numerous types of cancer, including colonic, breast, liver, gastric, uterine, ovarian, non-small cell lung, pancreatic and esophageal cancer (5–10). Furthermore, a correlation has been observed between the overexpression of AURKA mRNA and tumor progression and shortened survival in patients with HNSCC (11). Despite AURKA mRNA and protein being frequently overexpressed in various types of cancer, they are not always correlated with the gene amplification (12). The overexpression of AURKA has also been reported in gastric (13), breast (14) and ovarian (15) cancer. Therefore, besides gene amplification, other mechanisms, including transcriptional activation (16), suppression of protein degradation (17) and activation of certain signaling pathways (18), may also serve essential roles.
Focal adhesion kinase (FAK), a non-receptor tyrosine kinase, is a key regulator of cell proliferation, migration and invasion, and is involved in the development and progression of cancer. It has previously been established that FAK serves a vital role in the mediation of signal transduction pathways, involved in cell attachment, migration, invasion, proliferation and survival, which are crucial for cancer development and metastasis (19,20). Numerous studies have described increased expression of FAK protein in a variety of human cancers, including sarcomas, astrocytomas, and carcinomas of the breast, colon, thyroid, prostate, oral cavity, liver, stomach and ovary (20). Furthermore, Canel et al (21) demonstrated that FAK expression may be employed as an effective index for cervical lymph node metastases in patients with laryngeal squamous cell carcinoma. Additionally, Akt may be activated as result of AURKA overexpression. It has been demonstrated that an AURKA inhibitor may overcome AURKA-induced chemoresistance in various types of cancer (22).
The present study aimed to validate the hypothesis that AURKA activates FAK through the AURKA/Akt/FAK signaling pathway, and subsequently promotes the cell migration and invasion of HNSCC cells. The current study may provide a basis for the future development of inhibitors of the AURAK/Akt/FAK signaling pathway, with the aim to alleviate and eventually treat HNSCCs.
Materials and methods
Cell lines and cell culture
Human HNSCC cell lines (FaDu and Hep2) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS) and 1 µg/ml penicillin/streptomycin.
Furthermore, the FaDu and Hep2 cells were treated with 75 nM of the AURKA inhibitor, VX-680 (Selleck Chemicals, Houston, TX, USA), for 24 and 48 h (23), with 100 µM of the FAK inhibitor, TAE226 (Selleck Chemicals), for 12 and 24 h (24), or with 5 µM of the Akt inhibitor, triciribine (Selleck Chemicals), for 6 and 12 h (25), respectively. Subsequent to the treatment of each inhibitor, for the aforementioned specific times, the cells were harvested for the following experiments.
Transwell migration and invasion assay
The cell migration capability was determined using the previously described methods (26). A total of 600 µl medium containing 20% FBS was added to the lower chamber, whilst a total of 3×104 cells in 150 µl serum-free medium were added to the upper chamber. The Transwell chambers (8 µm; 24-well format; Corning Incorporated, Corning, NY, USA) were incubated at 37°C overnight. The cells were treated with the various inhibitors for the indicated times. Following the scraping of non-migrating cells from the upper surface of the membrane with cotton swabs, crystal violet was used to stain the cells that had migrated to and invaded the bottom chamber. The cells were then counted under a microscope (3 fields at random with ×100 magnification; U-ULS100HG; Olympus Optical Co. Ltd., Tokyo, Japan). For the invasion assay, the insert membrane was coated with diluted Matrigel Basement Membrane Matrix (BD Biosciences, Franklin Lakes, NJ), and the assay was conducted in a similar manner to the aforementioned assay.
Western blot analysis
Following the treatment of the cells with the various inhibitors, the cells were then dissolved using Pierce radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, Inc.) containing proteinase inhibitors (2.5 µg/ml leupeptin, 1 µg/ml aprotinin and 1 mM phenylmethanesulfonyl fluoride). Subsequently, the total protein within the lysates was quantified using a protein assay kit obtained from Bio-Rad Laboratories Inc. (Hercules, CA, USA). Furthermore, 50 µg protein was lysed and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The membrane was blotted with antibodies against phosphorylated (p)-AURKA (dilution, 1:3,000; catalog no. 3079P), AURKA (dilution, 1:1,000; catalog no. 3092S), p-Fak (Y397; dilution, 1:1000; catalog no. 3283), p-Fak (Y925; dilution, 1:1,000; catalog no. 3284P), Fak (dilution, 1:1,000; catalog no. 3285P), p-Akt473 (dilution, 1:1,000; catalog no. 9271) and Akt (dilution, 1:1,000; catalog no. 9272), all monoclonal Anti-rabbit IgG antibodies, obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA), and p-Fak (Y861; dilution, 1:1,000; monoclonal, Anti-rabbit IgG; catalog no. ab81293) obtained from Abcam (Cambridge, UK). The antibodies were added for 1 h at room temperature, and were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h. During the procedure, the glyceraldehyde 3-phosphate dehydrogenase level was regarded as the loading control.
Statistical analysis
The data from all experiments were analyzed with GraphPad Prism software (version 6; GraphPad Software, Inc., La Jolla, CA, USA) and shown as the mean±standard deviation. The Student's t-test was performed to assess the difference between the experimental and control groups. Values of P<0.05, indicated by *, were considered to indicate a statistically significant difference and values of P<0.01, indicated by **, were considered to indicate a highly statistically significant difference.
Results
Migration of FaDu and Hep2 cells decreases following treatment with AURKA, Akt and FAK inhibitors
Transwell migration assays were performed in order to investigate the role of AURKA in the migration of FaDu and Hep2 cells. Initially, when compared with the control, VX-680 decreased the migration of the FaDu cells to 66.6% at 24 h (P<0.01) and to 62.0% at 48 h (P<0.01) (Fig. 1A). Meanwhile, in the Hep2 cells, VX-680 decreased migration to 57.6% at 24 h (P<0.01) and 66.5% at 48 h (P<0.01) (Fig. 1B). Secondly, when compared with the control, TAE226 was observed to decrease the migration of the FaDu cells to 58.3% at 12 h (P<0.01) and to 60.3% at 24 h (P<0.01), respectively (Fig. 2A). Regarding the Hep2 cells, TAE226 was observed to decrease migration to 72.0% at 12 h (P<0.01) and to 66.1% at 24 h (P<0.01) (Fig. 2B). Lastly, triciribine decreased the migration of the FaDu cells to 79.3% at 6 h (P>0.05) and to 75.6% at 12 h (P>0.05; Fig. 3A), whereas in the Hep2 cells, triciribine decreased the migration to 71.8% at 6 h (P>0.05) and 68.8% at 12 h (P<0.05) (Fig. 3B).
Figure 1.
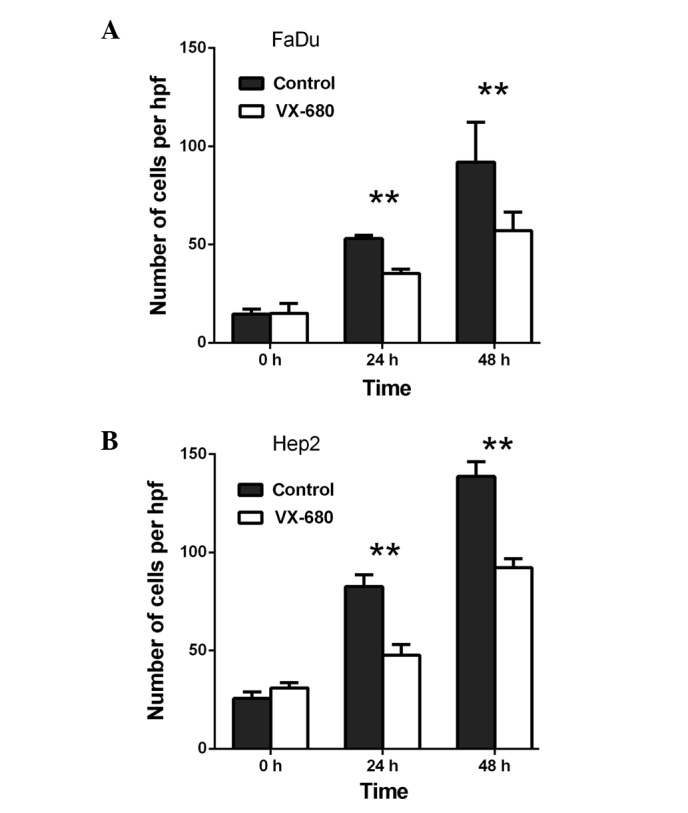
Effect of VX-680 on the migration of (A) FaDu and (B) Hep2 cells. The cells were treated with 75 nM VX-680 for different periods of time. **P<0.01 vs. control. Hpf, high-power field.
Figure 2.

Effect of TAE226 on the migration of (A) FaDu and (B) Hep2 cells. The cells were treated with 100 µM TAE226 for different periods of time. **P<0.01 vs. control. Hpf, high-power field.
Figure 3.

Effect of triciribine on the migration of (A) FaDu and (B) Hep2 cells. The cells were treated with 5 µM triciribine for different periods of time. *P<0.05 vs. control. Hpf, high-power field.
Capability of FaDu and Hep2 cell invasion is decreased following the treatment with AURKA, Akt and FAK inhibitors
Transwell invasion assays were performed to investigate the role of AURKA in the invasion of the FaDu and Hep2 cells. As presented in Fig. 4, VX-680 decreased the invasion of the FaDu cells to 33.3% at 24 h (P<0.01) and to 60.4% at 48 h (P<0.05) (Fig. 4A), whereas VX-680 was observed to decrease the invasion of the Hep2 cells to 51.2% at 24 h (P<0.05) and to 67.0% at 48 h (P<0.05) (Fig. 4B). Secondly, TAE226 was observed to decrease the invasion of the FaDu cells to 52.1% at 24 h (P<0.05; Fig. 5A), whereas in the Hep2 cells, TAE226 decreased invasion to 49.6% at 12 h (P<0.05) and to 58.5% at 24 h (P<0.05) (Fig. 5B). Lastly, triciribine decreased the invasion of the FaDu cells to 58.8% at 6 h (P<0.05) and to 86.7% at 12 h (P<0.05) (Fig. 6A), whilst in the Hep2 cells, triciribine decreased invasion to 61.3% at 6 h (P<0.05) and to 67.1% at 12 h (P<0.05) (Fig. 6B).
Figure 4.

Effect of VX-680 on the invasion of (A) FaDu and (B) Hep2 cells. The cells were treated with 75 nM VX-680 for different periods of time. *P<0.05 and **P<0.01 vs. control. Hpf, high-power field.
Figure 5.

Effect of TAE226 on the invasion of (A) FaDu and (B) Hep2 cells. The cells were treated with 100 µM TAE226 for different periods of time. *P<0.05 vs. control. Hpf, high-power field.
Figure 6.

Effect of triciribine on the cell invasion of (A) FaDu cells and (B) Hep2 cells. The cells were treated with 5 µM triciribine for different times *P<0.05 vs. control. Hpf, high-power field.
Suppression of AURKA inhibits p-FAKY397 phosphorylation
The FaDu and Hep2 cells were treated with 75 nM VX-680, with western blot analysis then used with the p-FAK antibody to examine the effect of AURKA suppression on Y397 phosphorylation. Following the downregulation of AURKA, p-AURKA and p-FAK Y397 expression decreased (P<0.05); however, the expression of p-FAK Y861 and p-FAK Y925 did not change (Fig. 7). The results also demonstrated that the treatment with VX-680 at 24 h did not significantly decrease p-FAK Y397 phosphorylation (P<0.05), but did completely block Y397-phosphorylation in the Hep2 cells at 48 h (P<0.01; Fig. 7). The expression of p-Akt was also slightly decreased (P<0.01 in FaDu cells; P<0.05 in Hep2 cells; Fig. 7).
Figure 7.

(A) Effects of downregulation of AURKA on FAK and Akt phosphorylation in FaDu and Hep2 cells. p-FAK Y397 and p-Akt were extremely decreased. (B) The expression levels of p-AURKA, p-Akt, p-FAK(Y861), p-FAK(Y925) and p-FAK(Y397) in FaDu cells were normalized to GAPDH. (C) The expression levels of p-AURKA, p-Akt, p-FAK(Y861), p-FAK(Y925) and p-FAK(Y397) in Hep2 cells were normalized to GAPDH. The bar represents the mean ± standard deviation of results from three separate experiments. *P<0.05; **P<0.01. AURKA, Aurora kinase A; p, phosphorylated; Akt, protein kinase B; FAK, focal adhesion kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
AURKA promotes migration and invasion through the Akt-FAK signaling pathway
Following the inhibition of Akt, the expression of p-Akt (P<0.01) and p-FAK (Y397) were decreased (P<0.05; Fig. 8). By contrast, following the inhibition of FAK, the expression of p-FAK (Y397) decreased, but p-Akt expression did not change (P<0.05 in FaDu cells; P<0.01 in Hep2 cells; Fig. 9). Thus, the downregulation of AURKA suppressed p-FAK via the inhibition of p-Akt.
Figure 8.

(A) Effect of downregulation of Akt in FaDu and Hep2 cells. p-FAK Y397 was extremely decreased. (B) The expression levels of p-Akt and p-FAK(Y397) in FaDu cells were normalized to GAPDH. (C) The expression levels of p-Akt and p-FAK(Y397) in Hep2 cells were normalized to GAPDH. The bar represents the mean ± standard deviation of results from three separate experiments. *P<0.05; **P<0.01. p, phosphorylated; Akt, protein kinase B; FAK, focal adhesion kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Figure 9.

(A) Effect of downregulation of FAK in FaDu and Hep2 cells. p-FAK Y397 was extremely decreased, but p-Akt was not altered. (B) The expression levels of p-Akt and p-FAK(Y397) in FaDu cells were normalized to GAPDH. (C) The expression levels of p-Akt and p-FAK(Y397) in Hep2 cells were normalized to GAPDH. The bar represents the mean ± standard deviation of results from three separate experiments. *P<0.05; **P<0.01. p, phosphorylated; Akt, protein kinase B; FAK, focal adhesion kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
The role of AURKA as an oncogene has been supported by a number of previous studies (3,27). Zhang et al demonstrated that the suppression of AURKA expression inhibits the growth and invasiveness of laryngeal squamous cell carcinoma cells, in vitro and in vivo (28). AURKA has been observed to positively regulate the G2 to M phase of the cell cycle, and activation of AURKA in late G2 is inhibited by DNA damage (3). Furthermore, high expression levels of AURKA have been associated with late clinical stages and regional metastasis in HNSCC (29). With research progression, a greater number of trials have been performed to investigate the treatment of HNSCC with Aurora kinase inhibitors (30). Li et al demonstrated that Akt promotes cell survival through its ability to phosphorylate and activate several pro-apoptotic targets. Subsequent consequences of this include the phosphorylation of Fak and the activation of signal transduction pathways, ultimately contributing to AURKA-mediated tumorigenesis (31). However, the pathway by which the migration and invasion of HNSCC was enhanced was not determined. Chemotherapy, radiotherapy and surgery have made great advances, however, the rate of morbidity remains high in HNSCC (2). In the present study, it was demonstrated that AURKA upregulated FAK via the activation of the AURKA/Akt/FAK signaling pathway. This subsequently led to the promotion of cell migration and invasion in HNSCC, indicating a strong association with the overexpression of AURKA and the AURKA/Akt/FAK signaling pathway.
The current study provided evidence that Aurora kinase inhibitors, which are implicated in the AURKA/Akt/FAK signaling pathway, should not only be considered in clinical trials for the treatment of HNSCC patients, but should also be tested in combination with other therapeutic drugs. During the process of future clinical trials, it may be beneficial for pre-clinical investigators to examine the various doses and schedules. Rational emergency treatment and advisable drug pharmacodynamics should be taken into account in such trials with volunteers. Finally, further investigation may determine the potential of using aurora kinase inhibitors as a target therapy for the treatment of other types of cancer.
In conclusion, a better understanding of the role that AURKA may serve in recurrence and metastasis, alongside the association between AURKA and Akt pathways in HNSCC, provides a novel insight and rationale for the possibility of further combined molecular targeting therapy in HNSCC and other types of cancer.
Acknowledgements
This study was supported by a grant from the Research Project of Shanghai Science and Technology Commission (no. 12ZR1418700).
References
- 1.Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127–135. doi: 10.1038/nrc1549. [DOI] [PubMed] [Google Scholar]
- 3.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 4.Stenoien DL, Sen S, Mancini MA, Brinkley BR. Dynamic association of a tumor amplified kinase, Aurora-A, with the centrosome and mitotic spindle. Cell Motil Cytoskeleton. 2003;55:134–146. doi: 10.1002/cm.10120. [DOI] [PubMed] [Google Scholar]
- 5.Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, Kirkner GJ, Goel A, Fuchs CS, Ogino S. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia. 2009;11:418–425. doi: 10.1593/neo.09154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitzen JJ, de Jonge MJ, Verweij J. Aurora-A kinase inhibitors. Crit Rev Oncol Hematol. 2010;73:99–110. doi: 10.1016/j.critrevonc.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Milam MR, Gu J, Yang H, Celestino J, Wu W, Horwitz IB, Lacour RA, Westin SN, Gershenson DM, Wu X, Lu KH. STK15 F31I polymorphism is associated with increased uterine cancer risk: A pilot study. Gynecol Oncol. 2007;107:71–74. doi: 10.1016/j.ygyno.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa E, Takenaka K, Katakura H, Adachi M, Otake Y, Toda Y, Kotani H, Manabe T, Wada H, Tanaka F. Perimembrane Aurora-A expression is a significant prognostic factor in correlation with proliferative activity in non-small-cell lung cancer (NSCLC) Ann Surg Oncol. 2008;15:547–554. doi: 10.1245/s10434-007-9653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang R, Wang JH, Chu XY, Geng HC, Chen LB. Expression of STK15 mRNA in hepatocellular carcinoma and its prognostic significance. Clin Biochem. 2009;42:641–647. doi: 10.1016/j.clinbiochem.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 10.Yang SB, Zhou XB, Zhu HX, Quan LP, Bai JF, He J, Gao YN, Cheng SJ, Xu NZ. Amplification and overexpression of Aurora-A in esophageal squamous cell carcinoma. Oncol Rep. 2007;17:1083–1088. [PubMed] [Google Scholar]
- 11.Zhang H, Chen X, Jin Y, Liu B, Zhou L. Overexpression of Aurora-A promotes laryngeal cancer progression by enhancing invasive ability and chromosomal instability. Eur Arch Otorhinolaryngol. 2012;269:607–614. doi: 10.1007/s00405-011-1629-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang LH, Xiang J, Yan M, Zhang Y, Zhao Y, Yue CF, Xu J, Zheng FM, Chen JN, Kang Z, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010;70:9118–9128. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- 13.Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, Shimomura K, Nakamura Y, Inazawa J, Abe T, Yamagishi H. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br J Cancer. 2001;84:824–831. doi: 10.1054/bjoc.2000.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 15.Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9:1420–1426. [PubMed] [Google Scholar]
- 16.Weaver KL, Alves-Guerra MC, Jin K, Wang Z, Han X, et al. NACK is an integral component of the Notch transcriptional activation complex and is critical for development and tumorigenesis. Cancer Res. 2014;74:4741–4751. doi: 10.1158/0008-5472.CAN-14-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Archewa P, Pata S, Chotjumlong P, Supanchart C, Krisanaprakornkit S, Iamaroon A. Akt2 and p-Akt overexpression in oral cancer cells is due to a reduced rate of protein degradation. J Investig Clin Dent. Epub ahead of print. 2015 doi: 10.1111/jicd.12194. [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 19.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22:359–374. doi: 10.1023/A:1023725029589. [DOI] [PubMed] [Google Scholar]
- 20.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 21.Canel M, Secades P, Rodrigo JP, Cabanillas R, Herrero A, Suarez C, Chiara MD. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res. 2006;12:3272–3279. doi: 10.1158/1078-0432.CCR-05-1583. [DOI] [PubMed] [Google Scholar]
- 22.Yang F, Guo X, Yang G, Rosen DG, Liu J. AURKA and BRCA2 expression highly correlate with prognosis of endometrioid ovarian carcinoma. Mod Pathol. 2011;24:836–845. doi: 10.1038/modpathol.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guan Z, Wang XR, Zhu XF, Huang XF, Xu J, Wang LH, Wan XB, Long ZJ, Liu JN, Feng GK, et al. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res. 2007;67:10436–10444. doi: 10.1158/0008-5472.CAN-07-1379. [DOI] [PubMed] [Google Scholar]
- 24.Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, Magis A, Ostrov D, Cance WG, Golubovskaya VM. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8:2435–2443. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 26.Feng R, Chen X, Yu Y, Su L, Yu B, Li J, Cai Q, Yan M, Liu B, Zhu Z. miR-126 functions as a tumour suppressor in human gastric cancer. Cancer Lett. 2010;298:50–63. doi: 10.1016/j.canlet.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link. TRENDS in Cell Biology. 2005;15:241–250. doi: 10.1016/j.tcb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Chen X, Liu B, Zhou L. Effects of stable knockdown of Aurora kinase A on proliferation, migration, chromosomal instability, and expression of focal adhesion kinase and matrix metalloproteinase-2 in HEp-2 cells. Mol Cell Biochem. 2011;357:95–106. doi: 10.1007/s11010-011-0879-1. [DOI] [PubMed] [Google Scholar]
- 29.Reiter R, Gais P, Jütting U, Steuer-Vogt MK, Pickhard A, Bink K, Rauser S, Lassmann S, Höfler H, Werner M, Walch A. Aurora kinase A messenger RNA overexpression is correlated with tumor progression and shortened survival in head and neck squamous cell carcinoma. Clin Cancer Res. 2006;12:5136–5141. doi: 10.1158/1078-0432.CCR-05-1650. [DOI] [PubMed] [Google Scholar]
- 30.Naruganahalli KS, Lakshmanan M, Dastidar SG, Ray A. Therapeutic potential of Aurora kinase inhibitors in cancer. Curr Opin Investig Drugs. 2006;7:1044–1051. [PubMed] [Google Scholar]
- 31.Li DW, Sun YJ, Sun ZF, Dong P. Involvement of focal adhesion kinase in cellular proliferation, apoptosis and prognosis of laryngeal squamous cell carcinoma. J Laryngol Otol. 2012;126:1127–1133. doi: 10.1017/S0022215112001971. [DOI] [PubMed] [Google Scholar]