

The crystal structure of an N-terminally truncated form of leucine zipper II (LZII) of JIP3 alone shows an unexpected antiparallel arrangement. This study draws attention to the fact that LZII of JIP3/JIP4 is a versatile structural motif, modifications of which can impact partner recognition and thus biological functions.
Keywords: JIP3, JIP4, leucine zipper II, crystal structure, molecular dynamics, antiparallel coiled coil, buried asparagine
Abstract
JIP3 and JIP4, two highly related scaffolding proteins for MAP kinases, are binding partners for two molecular motors as well as for the small G protein ARF6. The leucine zipper II (LZII) region of JIP3/4 is the binding site for these three partners. Previously, the crystal structure of ARF6 bound to JIP4 revealed LZII in a parallel coiled-coil arrangement. Here, the crystal structure of an N-terminally truncated form of LZII of JIP3 alone shows an unexpected antiparallel arrangement. Using molecular dynamics and modelling, the stability of this antiparallel LZII arrangement, as well as its specificity for ARF6, were investigated. This study highlights that N-terminal truncation of LZII can change its coiled-coil orientation without affecting its overall stability. Further, a conserved buried asparagine residue was pinpointed as a possible structural determinant for this dramatic structural rearrangement. Thus, LZII of JIP3/4 is a versatile structural motif, modifications of which can impact partner recognition and thus biological function.
1. Introduction
JIP3 and JIP4 (JNK-interacting proteins 3 and 4) were first identified as scaffold proteins for JNK and p38 mitogen-activated protein kinase (MAPK) signalling modules (Ito et al., 1999 ▸; Kelkar et al., 2000 ▸; Lee et al., 2002 ▸). JIP3 [also known as JSAP1 in mammals, Sunday Driver (SYD) in Drosophila and UNC-16 in Caenorhabditis elegans] and JIP4 (splice variants known as JLP and SPAG9 in mammals) are two close homologues. Both JIP3 and JIP4 are soluble cytoplasmic proteins (Kelkar et al., 2000 ▸, 2005 ▸). While JIP3 is mainly expressed in brain, JIP4 exhibits a ubiquitous pattern of expression (Ito et al., 1999 ▸; Kelkar et al., 2000 ▸, 2005 ▸; Lee et al., 2002 ▸), with its splice variant SPAG9 (sperm-associated antigen 9) being expressed exclusively in testis (Shankar et al., 1998 ▸; Jagadish et al., 2005 ▸).
JIP3 and JIP4 are large multi-domain proteins encompassing several highly conserved regions, among which are two coiled-coil regions (known as leucine zipper I and II). Leucine zipper II (LZII) of JIP3/JIP4 is the binding site for two microtubule-based molecular motors: kinesin 1 (Jeppesen & Hoerber, 2012 ▸) and the dynein–dynactin complex (Roberts et al., 2013 ▸). The recruitment of JIP3/JIP4 by kinesin 1 (Bowman et al., 2000 ▸; Nguyen et al., 2005 ▸; Montagnac et al., 2009 ▸) and the dynein–dynactin complex (Cavalli et al., 2005 ▸; Montagnac et al., 2009 ▸) allows their transport, as well as that of their binding partners, in two opposite directions throughout the cell. LZII of JIP3/JIP4 is also the binding site for the small G protein ARF6, which regulates the organization of the actin cytoskeleton and membrane trafficking (D’Souza-Schorey & Chavrier, 2006 ▸; Myers & Casanova, 2008 ▸). The specific interaction of JIP3/JIP4 with ARF6, together with kinesin 1 and the dynactin–dynein complex, controls the trafficking of endocytic vesicles during cytokinesis (Montagnac et al., 2009 ▸) and regulates endosomal tubules for MT1-MMP exocytosis in cancer invasion (Marchesin et al., 2015 ▸).
In order to better understand the mode of recognition of JIP3/4 by ARF6 and kinesin 1 at the molecular level, we previously determined the crystal structure of JIP4-LZII bound to ARF6 (PDB entry 2w83; Isabet et al., 2009 ▸). We are now investigating the recruitment of JIP3/4 by the kinesin 1 light chain (KLC). To increase our chance of obtaining crystals of this complex, we tested various fragments of JIP3/4-LZII and KLC that interact. In the course of our attempts, and despite the presence of KLC, an N-terminally truncated form of LZII of JIP3 crystallized alone. To date, no structural data for the unbound LZII of JIP3/4 are available, and because such information is required to visualize the structural impact of the interaction of partners such as ARF6, we determined its three-dimensional structure at 2.06 Å resolution. Unexpectedly, the structure of this truncated JIP3-LZII revealed an antiparallel coiled-coil arrangement that differs from the parallel coiled-coil arrangement observed for JIP4-LZII with a complete N-terminus bound to ARF6 (Isabet et al., 2009 ▸). In order to evaluate the stability of this antiparallel coiled-coil orientation, as well as the structural determinants forming the basis of this critical structural rearrangement, molecular-dynamics calculations were performed on parallel and antiparallel models of JIP3 and JIP4. In addition, we investigated the ability of this antiparallel JIP3-LZII to recognize its partner, ARF6. Together, the results highlight that N-terminal truncation of LZII can dramatically impact its overall structure as well as its ability to recognize its binding partners.
2. Materials and methods
2.1. Primary-sequence analysis
To perform primary-sequence analysis, human JIP3 and JIP4 (NCBI accession Nos. NP_055948.2 and NP_001123999.1, respectively) were used. Sequence alignment of human JIP3 and JIP4 was performed using ClustalW (Combet et al., 2000 ▸). Coiled-coil primary-sequence characteristics were analysed using Paircoil2 (McDonnell et al., 2006 ▸). The phylogenetic study was performed using the full-length sequences of JIP3 and JIP4 homologues retrieved using BLAST searches (Altschul et al., 1997 ▸) of the nr database and aligned together using the MAFFT E-INS-i algorithm (Katoh & Standley, 2013 ▸). Alignments were trimmed to remove regions with more than 50% of gaps in a column and a phylogenetic tree was calculated using the PhyML program using standard parameters (LG substitution model with four substitution-rate categories; Guindon et al., 2010 ▸).
2.2. Plasmid construction, protein production and purification
cDNA encoding a fragment of LZII of human JIP3 that consists of heptad repeats 3–9 plus the first four residues of heptad repeat 10 (residues 433–486; JIP3-LZII_3-10) was cloned into pGST-Parallel-1 (a modified plasmid based on pGEX-4T-1; GE Healthcare) using the EcoRI/XhoI restriction sites. JIP3-LZII_3-10 was produced in Escherichia coli Rosetta cells as a GST-fusion protein. Note that the GST tag can be removed using TEV protease, leaving a GAMDPEF sequence before residue 433 of JIP3-LZII_3-10. Cells were collected after induction with 0.3 mM IPTG for 4 h at 30°C. Frozen bacteria were suspended in 50 mM Tris–HCl pH 8.0 containing 250 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1% Triton X-100, 5 mM MgCl2, 1 mM PMSF, 2 µg ml−1 aprotinin, 2 µg ml−1 leupeptin, 0.7 mg ml−1 lysozyme and 0.02 mg ml−1 DNAse. The lysate was incubated for 1 h at 4°C and was then disrupted by sonication. The lysate was ultracentrifuged at 40 000 rev min−1 for 30 min at 4°C and the supernatant was incubated at 4°C with Glutathione Sepharose 4B beads (Amersham Biosciences) for 2 h. After washing, purified TPR domain of kinesin 1 KLC1 was added to the beads containing JIP3-LZII_3-10 in a 1:2 molar ratio and incubated for 2 h at 4°C. The GST tag of JIP3-LZII_3-10 was cleaved by the addition of TEV protease [1:50(w:w) overnight at 4°C], and the flowthrough containing the complex was collected at a concentration of 3.1 mg ml−1.
2.3. Protein crystallization, data collection and structure determination
Crystals of the JIP3-LZII_3-10 fragment were grown in sitting drops containing equal volumes of protein solution and reservoir solution by the vapour-diffusion method at 290 K. The crystallization solution consisted of 1.6 M amonium sulfate, 100 mM citrate pH 5.5, 2% PEG 400. The protein solution contained a mixture of JIP3-LZII_3-10 and KLC1-TPR fragments at 3.1 mg ml−1. Despite the presence of the KLC1-TPR fragment in the protein solution, the crystals contained only the JIP3-LZII_3-10 fragment. Crystals were transferred briefly to a cryoprotectant composed of reservoir solution supplemented with 20% ethylene glycol and cooled in liquid nitrogen. Diffraction data were collected at 100 K on the PROXIMA 1 beamline at the SOLEIL synchrotron. The crystals diffracted to 2.06 Å resolution and belonged to space group C2 with three molecules (A, B and C) in the asymmetric unit. The X-ray data were integrated and scaled using XDS (Kabsch, 2010 ▸). The structure was determined by molecular replacement with Phaser (McCoy et al., 2007 ▸) using as a search model molecule C (JIP4) from the ARF6–JIP4-LZII complex structure (PDB entry 2w83; Isabet et al., 2009 ▸) (i) with heptad repeats 1 and 2 removed and (ii) with the residues that differ between JIP4 and JIP3 mutated to the JIP3 sequence. Three molecules were found in the asymmetric unit. Heptad repeats 9 and 10, which were missing from the search model, were then manually built. Refinement was then carried out using PHENIX (Adams et al., 2010 ▸) and graphical model building was performed using Coot (Emsley & Cowtan, 2004 ▸). It should be noted that the N-terminal residues (GAMDPEF) belonging to the linker between the GST tag and JIP3-LZII_3-10 were modelled in electron density and numbered as residues 426–432. Data-collection and refinement statistics are presented in Table 1 ▸. MolProbity was used for model validation (Chen et al., 2010 ▸). Atomic coordinates and structure factors have been deposited in the PDB as entry 4pxj. The figures were produced using PyMOL (DeLano, 2002 ▸).
Table 1. Data-collection and refinement statistics for JIP3-LZII_3-10.
Values in parentheses are for the highest resolution shell.
| PDB code | 4pxj |
| Data collection | |
| Beamline | PROXIMA 1, SOLEIL |
| Wavelength (Å) | 0.9788 |
| Temperature (K) | 100 |
| Detector | Pilatus 6M |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 137.83, b = 63.04, c = 50.22, β = 97.95 |
| Resolution (Å) | 38.70–2.06 (2.13–2.06) |
| Unique reflections | 26641 |
| R merge † (%) | 4.1 (29.5) |
| Mean I/σ(I) | 24.20 (5.48) |
| Completeness (%) | 99.9 (99.7) |
| Multiplicity | 6.7 (6.7) |
| Refinement | |
| No. of reflections | 26642 |
| R work/R free ‡ (%) | 17.9/20.5 |
| No. of atoms | |
| Total | 1588 |
| Water | 207 |
| B factor (Å2) | |
| Overall | 43.9 |
| Water | 51.1 |
| R.m.s.d. from ideal | |
| Bond lengths (Å) | 0.003 |
| Bond angles (°) | 0.62 |
| Ramachandran plot§ | |
| Favoured (%) | 99.40 |
| Allowed (%) | 0.60 |
| Outliers (%) | 0.00 |
R
merge = 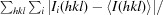
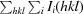 , where 〈I(hkl)〉 is the mean intensity of a set of equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of a set of equivalent reflections.
R
work = 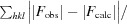
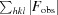 for the 95% of the reflection data used in refinement. F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
free is equivalent to R
work except that it was calculated for a randomly chosen 5% test set excluded from refinement.
for the 95% of the reflection data used in refinement. F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively. R
free is equivalent to R
work except that it was calculated for a randomly chosen 5% test set excluded from refinement.
Ramachandran analysis was performed using MolProbity (Chen et al., 2010 ▸).
2.4. Modelling
For molecular-dynamics calculations, antiparallel JIP4-LZII_3-10 (JIP4-apLZII_3-10) and parallel JIP3-LZII_1-8 (JIP3-pLZII_1-8) models were computed. For the JIP4-apLZII_3-10 model, the antiparallel JIP3-LZII_3-10 crystal structure (this study) was used as an initial model and all residues that differed were graphically mutated to the JIP4 sequence using PyMOL (DeLano, 2002 ▸). For the JIP3-pLZII_1-8 model, the parallel JIP4-LZII_1-8 crystal structure (PDB entry 2w83; Isabet et al., 2009 ▸) was used as an initial model (the two ARF6 molecules were removed). All residues that differed between JIP4 and JIP3 were graphically mutated to the JIP3 sequence. For both models, the rotamer of each mutated residue was carefully chosen in order to prevent steric hindrance with the rest of the structure. Finally, energy-minimization cycles were performed on both models before molecular-dynamics calculations. For structural analysis, two models of the antiparallel JIP3-LZII_3-10–ARF6 complex were computed. Model 1 and model 2 result from the superimposition of the JIP3-apLZII_3-10 crystal structure on chain C and chain D, respectively, of the JIP4-pLZII_1-8 homodimer of the JIP4–ARF6 complex structure (PDB entry 2w83; Isabet et al., 2009 ▸). Of note, because the interaction of the two ARF6 molecules with the JIP3/4 homodimer is virtually identical, models 1 and 2 were computed with only one ARF6 molecule.
2.5. Molecular dynamics
All molecular-dynamics (MD) calculations were performed using the Amber 12 molecular-dynamics package (Case et al., 2012 ▸) and the FF12SB force field. The LEaP software was employed to add H atoms to the proteins, solvate the systems with water molecules and add counter-ions. Conventional protonation states at neutral pH were considered for all side chains, which is adequate given the high solvent exposure of the proteins. The exact number of water molecules and counter-ions can be found in Supplementary Table S1. All models were subjected to a four-stage minimization using the SANDER module of Amber 12. Harmonic potentials (force constant of 50 kcal mol−1 Å−2) were used to restrain the positions of selected atoms in the systems. In the first stage (500 steps), all atoms except those from water molecules were restrained. In the second stage (800 steps), the restraints on the H atoms were released as well. In the third stage (2500 steps), only the backbone chain atoms were restrained. In the fourth stage (6000 steps), all of the system was free and this process ended in a full energy minimization. A three-stage equilibration MD protocol was performed using the PMEMD (Particle Mesh Ewald Molecular Dynamics) module of Amber 12. In the first simulation (50 ps) the temperature was raised from 0 to 300 K at constant volume, followed by a density equilibration (50 ps in the NPT ensemble with weak restraints of 2 kcal mol−1 Å−2 on the backbone atoms) and by 500 ns in the NPT ensemble (300 K, 100 kPa). The Langevin thermostat and Berendsen barostat (Berendsen et al., 1984 ▸) were used in all simulations. Subsequent production simulations in the NTP ensemble were carried out, with a total length of 500 ns for JIP3/4 models, recording microstates every 10 ps. All MD simulations used the SHAKE algorithm to fix the bond length between H atoms and heavy atoms. The time step was 2 fs. The CCPTRAJ (Roe & Cheatham, 2013 ▸) module of Amber 12 was used to analyse the MD results and to calculate the root-mean-square fluctuation (r.m.s.f.) and the root-mean-square deviation (r.m.s.d.) of the Cα atoms in relation to the initial structure.
3. Results
3.1. Leucine zipper II of JIP3 and JIP4
Human JIP3 and JIP4 (1336 and 1311 residues, respectively) are close homologues that share 56% sequence identity, with four regions that are highly conserved (Fig. 1 ▸ a). In the N-terminal part of the molecule, JIP3 and JIP4 possess two conserved coiled-coil regions consisting of heptad repeats with apolar residues at the a and d positions. Because these two regions exhibit a number of leucine residues at position d, which is a feature of the leucine-zipper motif, they have been called leucine zipper I (LZI) and leucine zipper II (LZII), respectively. Primary-sequence analysis using Paircoil2 (McDonnell et al., 2006 ▸) revealed that LZII consists of 11 heptad repeats (LZII_1-11) corresponding to residues 420–496 of JIP3 and residues 396–472 of JIP4 (Fig. 1 ▸ b). LZII of JIP3 and JIP4 exhibits the characteristic leucine residue at position d except in heptad repeats 7, 10 and 11. Sequence alignment of the LZII_1-11 fragment of the JIP3 and JIP4 homologues reveals that 52 residues are identical (67.5% identity), 18 residues are strongly similar, two residues are weakly similar and five residues are different (Fig. 1 ▸ b). Note that the last heptad repeat, heptad 11, is the least conserved part of LZII of JIP3 and JIP4.
Figure 1.

Organization of JIP3 and JIP4. (a) Scheme of full-length JIP3/4 domain organization. (b) Sequence alignment of the LZII region (heptad repeats 1–11) of JIP3 and JIP4. Position d of each heptad repeat is indicated in red. In the JIP4 sequence, residues identical to those in JIP3 are indicated by a dash.
A multiple sequence alignment of full-length homologues of JIP3 and JIP4 shows that the LZII region is conserved in evolution in all species. JIP-like homologues can be found in several clades of metazoans, including insects and worms. In invertebrates, a single homologue of JIP3/4 is found, including UNC-16 in Caenorhabditis elegans, which was shown to share similar functions with JIP3 and JIP4 (Byrd et al., 2001 ▸; Sakamoto et al., 2005 ▸; Brown et al., 2009 ▸). The duplication giving rise to the JIP3 and JIP4 subfamilies can be positioned at the origin of the vertebrate clade since a single version of the protein is found in the species most closely related to vertebrates such as lancelets (Branchiostoma floridae) and tunicates (Ciona intestinalis) (Supplementary Fig. S1a). Interestingly, LZII of JIP3 orthologues is flanked by two insertions of around ten residues each compared with JIP4 (Supplementary Fig. S1b). These insertions are not observed in the JIP-like homologues in invertebrates, suggesting that they are specific to vertebrate JIP3.
3.2. Crystal structure of the JIP3-LZII_3-10 fragment
The structure of an N-terminally truncated form of LZII of human JIP3 encompassing heptad repeats 3–9 plus the first four residues from heptad repeat 10 (hereafter referred to as JIP3-LZII_3-10; residues 433–486; Fig. 2 ▸ a) was determined at 2.06 Å resolution (PDB entry 4pxj; statistics in Table 1 ▸) with three molecules (A, B and C) in the asymmetric unit. Molecules A and B associate together to form a homodimer, while molecule C associates with its crystallographic symmetry-related molecule (molecule C*) by the crystal twofold axis. No significant structural difference is observed between dimers AB and CC*, with their entire Cα traces showing an r.m.s.d. of 0.77 Å (calculated over 103 residues from the dimer). Careful examination of the crystal packing, which exhibits a high solvent content of around 78%, rules out a possible crystallization artefact.
Figure 2.

Three-dimensional structure of the antiparallel JIP3-LZII_3-10 fragment. (a) Scheme of the quaternary organization of the antiparallel JIP3-LZII_3-10 homodimer. The primary sequence of JIP3-LZII_3-10 is reported above. (b) Three-dimensional structure of the antiparallel JIP3-LZII_3-10 fragment (two orthogonal views). Leucine residues at position d are indicated in red in (a) and are shown as sticks in (b). Heptad repeats are highlighted using a rainbow colour code. (c) A simulated-annealing composite OMIT map contoured at 1σ is shown for a region overlapping three heptad repeats of the antiparallel JIP3-LZII_3-10 homodimer structure. The simulated-annealing composite OMIT map was calculated on the entire homodimer using PHENIX (Adams et al., 2010 ▸).
The overall structure of the JIP3-LZII_3-10 fragment consists of two unbroken α-helices that wind around each other in a 79.5 Å long and straight coiled coil (Fig. 2 ▸ b). The total surface area buried by both helices of the JIP3-LZII_3-10 homodimer is 2305 Å2. The JIP3-LZII_3-10 structure exhibits an antiparallel arrangement with heptad 3 facing heptad 9′/10′, heptad 4 facing heptad 8′/9′, heptad 5 facing heptad 7′/8′, heptad 6 facing heptad 6′/7′, heptad 7 facing heptad 5′/6′, heptad 8 facing heptad 4′/5′ and heptad 9 facing heptad 3′/4′ from the opposite helix (where a prime indicates a heptad/residue that resides in a different helix). The two helices of JIP3-LZII_3-10 interact together through the characteristic coiled-coil side-chain packing known as knobs-into-holes interactions, with an apolar residue (‘knob’) from one helix buried in a ‘hole’ formed by four hydrophobic residues from the opposite helix. Fig. 2 ▸(c) shows the details of the interaction between the two helices of the antiparallel JIP3-LZII_3-10 structure. Also, a composite simulated-annealing OMIT map reveals the positions of the carbonyl groups of the main chain for each helix (Fig. 2 ▸ c).
3.3. Structural comparison of JIP3-apLZII_3-10 and JIP4-pLZII_1-8
Previously, we determined the crystal structure of LZII of JIP4 bound to ARF6 (PDB entry 2w83; Isabet et al., 2009 ▸). The JIP4-LZII fragment used for crystallization consisted of heptad repeats 1–9 plus the first four residues from heptad repeat 10 (hereafter called the JIP4-LZII_1-10 fragment; residues 392–462; Supplementary Fig. S2a). Of note, since no electron density was observed for heptad repeat 9 and beyond, only heptad repeats 1–8 were fully modelled; therefore, we will refer to this as the JIP4-LZII_1-8 structure hereafter. The JIP4-LZII_1-8 structure consists of two unbroken α-helices that wind around each other in a 84.5 Å long and straight coiled-coil structure (Supplementary Fig. S2b). Interestingly, while the JIP3-LZII_3-10 structure shows an antiparallel coiled-coil arrangement (JIP3-apLZII_3-10; Fig. 2 ▸), the JIP4-LZII_1-8 structure revealed a parallel coiled-coil arrangement (JIP4-pLZII_1-8; Supplementary Fig. S2). As a consequence, the dyad symmetry of the dimer is perpendicular to the long axis for the JIP3-apLZII_3-10 structure, while it follows the long axis for the JIP4-pLZII_1-8 structure. Superposition of the JIP3-apLZII_3-10 and JIP4-pLZII_1-8 structures on one helix of the coiled coil (using residues from heptad repeats 3–8) clearly shows a shift of the opposite helix by half a helix turn (Fig. 3 ▸ a). Thus, while the interface of the two dimers shares characteristic coiled-coil knobs-into-holes interactions, the electrostatic properties of the homodimer surface differ significantly between JIP3-apLZII_3-10 and JIP4-pLZII_1-8 (Fig. 3 ▸ b).
Figure 3.

Structural comparison of JIP3-apLZII_3-10 and JIP4-pLZII_1-8. (a) Superposition of the JIP3-apLZII_3-10 structure (the same rainbow colour code is used as in Fig. 2 ▸) and the JIP4-pLZII_1-8 structure (grey; PDB entry 2w83). (b) A solvent-accessible surface representation of the JIP3-apLZII_3-10 and JIP4-pLZII_1-8 structures shown in two orthogonal views. Hydrophobic residues are shown in yellow and polar residues in white, while negative and positive residues are indicated in red and blue, respectively.
3.4. Molecular-dynamics assessment of the stability of the JIP3-apLZII_3-10 structure
In order to evaluate the stability of the antiparallel arrangement of LZII of JIP3 in solution, we performed molecular-dynamics (MD) calculations on the JIP3-apLZII_3-10 structure, as well as on the JIP4-pLZII_1-8 structure (PDB entry 2w83; Isabet et al., 2009 ▸) for comparison. The r.m.s.d. values are around 2.0 and 2.1 Å on average with small fluctuations (standard deviation of 0.3 Å), which is indicative of good structural preservation for both models (Figs. 4 ▸ a and 4 ▸ b). Note that unstable, strained proteins (or incorrectly determined structures) start unfolding within a few nanoseconds, which did not happen in these cases. The observation of the model during the long MD simulations did not reveal any major changes in the folding for both the parallel and the antiparallel arrangements of LZII. We cannot rule out that unfolding/dissociation of the models may take place on a larger time scale owing to eventual high barriers in the free-energy hypersurface. This would be a very uncommon scenario, in particular in very small peptides that are very exposed to the solvent and without any intricate wrapping of the two chains. These computational data reveal that the antiparallel arrangement of the JIP3-LZII_3-10 structure is intrinsically stable in solution.
Figure 4.

Molecular dynamics on parallel and antiparallel arrangements of JIP3/4-LZII models. Graphical representations of the r.m.s.d. over time for JIP3/4-LZII models are shown. R.m.s.d. values calculated along the simulation time are compared with the initial structure of the system for (a) the JIP3-apLZII_3-10 structure, (b) the JIP4-pLZII_1-8 structure, (c) the JIP3-pLZII_1-8 model and (d) the JIP4-apLZII_3-10 model.
3.5. Impact of JIP3/4 primary-sequence variations on LZII orientation
LZII of JIP3 was crystallized in an antiparallel arrangement, while that of JIP4 was in a parallel arrangement. Thus, we wondered whether primary-sequence variations between these two close homologues (Fig. 1 ▸ b) could be structural determinants for this structural rearrangement. To investigate this point, we modelled (i) JIP4-LZII_3-10 with an antiparallel arrangement (referred to as the JIP4-apLZII_3-10 model) and (ii) JIP3-LZII_1-8 with a parallel arrangement (referred to as the JIP3-pLZII_1-8 model). Concerning the antiparallel arrangement, comparison of the JIP4-apLZII_3-10 model and the JIP3-apLZII_3-10 structure highlighted two different residues in the core of the JIP4 dimer, namely Val441 (Ala465 in JIP3) and Asn452 (Ile476 in JIP3). Both Val441 and Asp452 fit well in the core of the antiparallel coiled coil and should not disturb the characteristic knobs-into-holes interactions of JIP4-apLZII_3-10. Concerning the parallel arrangement, comparison of the JIP3-pLZII_1-8 model and the JIP4-pLZII_1-8 structure highlights one difference in the core of the JIP3 dimer, namely Ala465 (Val441 in JIP4), which should not induce steric hindrance at the interface of the coiled coil. This structural analysis suggests that primary-sequence variations between JIP3 and JIP4 should not prevent the JIP3-LZII_1-8 fragment from assembling into a parallel arrangement or the JIP4-LZII_3-10 fragment assembling into an antiparallel arrangement. However, we cannot exclude that these primary-sequence variations favour one or the other of these coiled-coil orientations.
Thus, we performed MD calculations on the JIP3-pLZII_1-8 and JIP4-apLZII_3-10 in silico models to evaluate their stability compared with the JIP3-apLZII_3-10 and JIP4-pLZII_1-8 X-ray structures (Figs. 4 ▸ a and 4 ▸ b). The r.m.s.d. values for these two models are around 2.1 and 2.2 Å, respectively, on average, with small fluctuations (standard deviation of 0.4 Å), which is indicative of good structural preservation (Figs. 4 ▸ c and 4 ▸ d). Together, these computational data suggest that both parallel and antiparallel arrangements of LZII are intrinsically stable in solution regardless of the primary-sequence variations between JIP3 and JIP4.
3.6. Modelling of ARF6 recognition by the antiparallel JIP3-LZII
In order to investigate whether the JIP3/4-LZII binding partner ARF6 can recognize the antiparallel JIP3-LZII, we took advantage of the JIP4-pLZII–ARF6 complex structure that we had determined previously (PDB entry 2w83; Isabet et al., 2009 ▸). Note that ARF6 binds to heptad repeats 3, 4 and 5 of JIP4-pLZII (Supplementary Fig. S2a), which are present in the N-terminally truncated JIP3-LZII_3-10 fragment. Thus, we computed two putative models of the antiparallel JIP3-LZII bound to ARF6. The first model (model 1) results from the structural superimposition of JIP3-apLZII_3-10 onto chain C of JIP4-pLZII_1-8 that contacts switch I and switch II of ARF6 (Fig. 5 ▸ a). The second model (model 2) results from the superimposition of the JIP3-apLZII_3-10 onto chain D of JIP4-pLZII_1-8 that contacts the interswitch tip and the switch II end of ARF6 (Fig. 5 ▸ b).
Figure 5.

Modelling of ARF6 bound to the antiparallel JIP3-LZII_3-10 fragment. Superposition of the ARF6–JIP4-pLZII_1-8 complex (PDB entry 2w83; JIP4 is shown in grey) with two ARF6–JIP3-apLZII_3-10 models: (a) model 1 (JIP3 in orange) and (b) model 2 (JIP3 in yellow). Superposition was performed on ARF6, which is indicated in blue with its switch regions in light blue. Below the overall view, a detailed view of the interface is indicated. Residues of ARF6, JIP3 and JIP4 involved in the interface are shown in stick representation and are labelled.
Structural analysis of these two models suggests that ARF6 would not recognize and bind to the antiparallel JIP3-LZII. Indeed, in both models the helix of JIP3-apLZII that is superposed on that of JIP4-pLZII exhibits identical interactions with ARF6, but the second helix with reverse orientation exhibits dramatic differences. Overall, the second helix (i) faces different heptad repeats (heptad repeats 7, 8 and 9 and 8, 9 and 10, respectively, for models 1 and 2) towards ARF6, (ii) exhibits a half helix-turn shift of its backbone and (iii) directs side chains in the opposite direction (Fig. 5 ▸). In model 1, Thr428 and Asp432 from JIP4-pLZII are replaced by Ala468 and Ala464, respectively, in JIP3-apLZII, which can no longer make hydrogen bonds to and van der Waals contacts with ARF6 (Fig. 5 ▸ a). Furthermore, Asn418 and Ile421 from JIP4-pLZII are replaced by Glu478 and Arg475, respectively, in JIP3-apLZII, while the negatively charged Asp425 is replaced by the positively charged Lys471 (Fig. 5 ▸ a). These last three differences should impact the electrostatic interactions at the interface with ARF6. In model 2, the hydrophobic Leu413 and Leu420 from JIP4-pLZII are substituted by equivalent residues in JIP3-apLZII (Leu483 and Ile476, respectively; Fig. 5 ▸ b). However, these latter are slightly shifted away from ARF6, which might decrease the affinity of JIP3-apLZII for ARF6. More importantly, two other hydrophobic residues from JIP4-pLZII, Ile415 and Val416, are substituted by negatively charged glutamates in JIP3-apLZII (Glu481 and Glu480, respectively; Fig. 5 ▸ b). Also, another charge reversion is observed at the edge of the interface, with Lys423 being substituted by a glutamate residue (Glu473 in JIP3). Together, the presence of these three glutamate residues facing ARF6 might dramatically change the electrostatic properties of the interface. Finally, these two JIP3-apLZII–ARF6 models exhibit differences that would generate steric hindrances, charge repulsions and a loss of hydrophobic and hydrophilic contacts at the interface.
4. Discussion
Unexpectedly, the crystal structure of an N-terminally truncated form of leucine zipper II (LZII) of JIP3 reveals an antiparallel coiled-coil arrangement. Molecular-dynamics (MD) calculations support the stability of the antiparallel arrangement of this truncated JIP3 fragment. Previously, we determined the crystal structure of ARF6 bound to JIP4-LZII with a complete N-terminus in a parallel coiled-coil arrangement (Isabet et al., 2009 ▸). The association of the ARF6–JIP4-pLZII complex observed in the crystal was validated using site-directed mutagenesis and affinity-binding experiments in solution (Isabet et al., 2009 ▸). Note that the JIP4 residues involved in ARF6 binding are conserved in JIP3, suggesting that it recognizes ARF6 in the same way. We cannot exclude that LZII of JIP3/4 alone adopts an antiparallel arrangement and the presence of ARF6 (or other binding partners) induces a dramatic structural rearrangement of LZII leading to the formation of a stable complex between ARF6 and the parallel LZII of JIP3/4. However, in the absence of additional experimental evidence, we consider here that the proper orientation of LZII of both JIP3 and JIP4 is parallel. Modelling and MD calculations suggest that primary-sequence variations between JIP3 and JIP4 are not structural determinants for the different coiled-coil orientation observed in the JIP3-LZII_3-10 structure. Instead, we hypothesize that the N-terminal truncation of heptad repeats 1 and 2 of LZII may be critical for this structural rearrangement. The two first heptad repeats, as a trigger sequence, can induce a strong initial interaction with themselves and favour the proper parallel arrangement observed in the JIP4-LZII_1-10 structure (bound to ARF6; Isabet et al., 2009 ▸). Note that in both the JIP3-LZII_3-10 and JIP4-LZII_1-10 fragments heptad repeat 11 and the last part of heptad repeat 10 are missing, which reasonably excludes this C-terminal part of LZII from being a structural determinant for this structural rearrangement. Interestingly, an asparagine residue is spotted at position a of heptad 2 of both JIP3 and JIP4 (Fig. 1 ▸ b). Note that this asparagine is strictly conserved in evolution in all species (Supplementary Fig. S1b). Such a polar residue at a buried position of the coiled coil can influence the oligomerization state and helix orientation (Woolfson, 2005 ▸). As shown experimentally, a single asparagine residue at position a of the GCN4 leucine zipper and peptide ‘velcro’ (ACID-P1/BASE-p1 heterodimer) determines a two-stranded parallel arrangement for these two coiled coils (O’Shea et al., 1991 ▸; Harbury et al., 1993 ▸; Lumb & Kim, 1995 ▸; Oakley & Kim, 1998 ▸). In a two-stranded parallel coiled coil, this asparagine makes a stabilizing side chain–side chain hydrogen bond to the corresponding asparagine in the partner helix, while an antiparallel arrangement would result in two buried and noncomplemented asparagine side chains (Woolfson, 2005 ▸). Thus, the removal of this conserved buried asparagine in the N-terminally truncated fragment of JIP3-LZII_3-10 would result in a loss of structural uniqueness, leading to a mixture of parallel and antiparallel LZII. We propose that this conserved buried asparagine residue might be a structural determinant specifying the parallel orientation of LZII of JIP3/JIP4.
Our study highlights that N-terminal truncation of LZII of JIP3/JIP4 can change its coiled-coil orientation without affecting its overall stability. Modelling and structural analysis suggest that ARF6 will not recognize the antiparallel arrangement observed for JIP3-LZII_3-10. We guess that other JIP3/JIP4-LZII partners, such as kinesin 1 and the dynein–dynactin complex, would also be impacted by such a structural rearrangement. Here, we draw attention to the fact that LZII of JIP3/JIP4 is a versatile structural motif, modifications of which can impact partner recognition and thus biological function.
Supplementary Material
PDB reference: LZII fragment from JIP3, 4pxj
Supporting Information.. DOI: 10.1107/S2053230X16001576/tt5076sup1.pdf
Acknowledgments
X-ray data collection was performed with the help of the staff of the PROXIMA 1 beamline at the SOLEIL synchrotron, Saint-Aubin, France. This work has benefited from the facilities of the LEBS/IMAGIF platform, providing access to the TECAN crystallization robot. This work was supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01. This work was supported by grant SFI20121205592 from ARC to JM and grants SFRH/BD/84016/2012 and EXCL/QEQ-COM/0394/2012 from FCT to PAF, CM and MJR.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. (1984). J. Chem. Phys. 81, 3684–3690.
- Bowman, A. B., Kamal, A., Ritchings, B. W., Philp, A. V., McGrail, M., Gindhart, J. G. & Goldstein, L. S. (2000). Cell, 103, 583–594. [DOI] [PubMed]
- Brown, H. M., Van Epps, H. A., Goncharov, A., Grant, B. D. & Jin, Y. (2009). Dev. Neurobiol. 69, 174–190. [DOI] [PMC free article] [PubMed]
- Byrd, D. T., Kawasaki, M., Walcoff, M., Hisamoto, N., Matsumoto, K. & Jin, Y. (2001). Neuron, 32, 787–800. [DOI] [PubMed]
- Case, D. A. et al. (2012). Amber 12. University of California, San Francisco, USA.
- Cavalli, V., Kujala, P., Klumperman, J. & Goldstein, L. S. B. (2005). J. Cell Biol. 168, 775–787. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Combet, C., Blanchet, C., Geourjon, C. & Deléage, G. (2000). Trends Biochem. Sci. 25, 147–150. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL. http://www.pymol.org.
- D’Souza-Schorey, C. & Chavrier, P. (2006). Nature Rev. Mol. Cell Biol. 7, 347–358. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W. & Gascuel, O. (2010). Syst. Biol. 59, 307–321. [DOI] [PubMed]
- Harbury, P., Zhang, T., Kim, P. & Alber, T. (1993). Science, 262, 1401–1407. [DOI] [PubMed]
- Isabet, T., Montagnac, G., Regazzoni, K., Raynal, B., El Khadali, F., England, P., Franco, M., Chavrier, P., Houdusse, A. & Ménétrey, J. (2009). EMBO J. 28, 2835–2845. [DOI] [PMC free article] [PubMed]
- Ito, M., Yoshioka, K., Akechi, M., Yamashita, S., Takamatsu, N., Sugiyama, K., Hibi, M., Nakabeppu, Y., Shiba, T. & Yamamoto, K. I. (1999). Mol. Cell. Biol. 19, 7539–7548. [DOI] [PMC free article] [PubMed]
- Jagadish, N., Rana, R., Selvi, R., Mishra, D., Garg, M., Yadav, S., Herr, J. C., Okumura, K., Hasegawa, A., Koyama, K. & Suri, A. (2005). Biochem. J. 389, 73–82. [DOI] [PMC free article] [PubMed]
- Jeppesen, G. M. & Hoerber, J. K. H. (2012). Biochem. Soc. Trans. 40, 438–443. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Katoh, K. & Standley, D. M. (2013). Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed]
- Kelkar, N., Gupta, S., Dickens, M. & Davis, R. J. (2000). Mol. Cell. Biol. 20, 1030–1043. [DOI] [PMC free article] [PubMed]
- Kelkar, N., Standen, C. L. & Davis, R. J. (2005). Mol. Cell. Biol. 25, 2733–2743. [DOI] [PMC free article] [PubMed]
- Lee, C. M., Onésime, D., Reddy, C. D., Dhanasekaran, N. & Reddy, E. P. (2002). Proc. Natl Acad. Sci. USA, 99, 14189–14194. [DOI] [PMC free article] [PubMed]
- Lumb, K. J. & Kim, P. S. (1995). Biochemistry, 34, 8642–8648. [DOI] [PubMed]
- Marchesin, V., Castro-Castro, A., Lodillinsky, C., Castagnino, A., Cyrta, J., Bonsang-Kitzis, H., Fuhrmann, L., Irondelle, M., Infante, E., Montagnac, G., Reyal, F., Vincent-Salomon, A. & Chavrier, P. (2015). J. Cell Biol. 211, 339–358. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McDonnell, A. V., Jiang, T., Keating, A. E. & Berger, B. (2006). Bioinformatics, 22, 356–358. [DOI] [PubMed]
- Montagnac, G., Sibarita, J.-B., Loubéry, S., Daviet, L., Romao, M., Raposo, G. & Chavrier, P. (2009). Curr. Biol. 19, 184–195. [DOI] [PubMed]
- Myers, K. R. & Casanova, J. E. (2008). Trends Cell Biol. 18, 184–192. [DOI] [PMC free article] [PubMed]
- Nguyen, Q., Lee, C. M., Le, A. & Reddy, E. P. (2005). J. Biol. Chem. 280, 30185–30191. [DOI] [PubMed]
- Oakley, M. G. & Kim, P. S. (1998). Biochemistry, 37, 12603–12610. [DOI] [PubMed]
- O’Shea, E. K., Klemm, J. D., Kim, P. S. & Alber, T. (1991). Science, 254, 539–544. [DOI] [PubMed]
- Roberts, A. J., Kon, T., Knight, P. J., Sutoh, K. & Burgess, S. A. (2013). Nature Rev. Mol. Cell Biol. 14, 713–726. [DOI] [PMC free article] [PubMed]
- Roe, D. R. & Cheatham, T. E. III (2013). J. Chem. Theory Comput. 9, 3084–3095. [DOI] [PubMed]
- Sakamoto, R., Byrd, D. T., Brown, H. M., Hisamoto, N., Matsumoto, K. & Jin, Y. (2005). Mol. Biol. Cell, 16, 483–496. [DOI] [PMC free article] [PubMed]
- Shankar, S., Mohapatra, B. & Suri, A. (1998). Biochem. Biophys. Res. Commun. 243, 561–565. [DOI] [PubMed]
- Woolfson, D. N. (2005). Adv. Protein Chem. 70, 79–112. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: LZII fragment from JIP3, 4pxj
Supporting Information.. DOI: 10.1107/S2053230X16001576/tt5076sup1.pdf