

The purification, crystallization and initial X-ray crystallographic studies of the N-terminal and M domains of the α-catenin homologue HMP-1 from C. elegans are described.
Keywords: α-catenin, HMP-1, adherens junction, Caenorhabditis elegans
Abstract
Adherens junctions transmit mechanical force between cells. In these junctions, β-catenin binds to cadherins and to the N-terminal domain of α-catenin, which in turn binds to actin filaments via its C-terminal domain. The middle (M) domain of α-catenin plays an important role in responding to mechanical tension. The nematode Caenorhabditis elegans contains α- and β-catenin homologues called HMP-1 and HMP-2, respectively, but HMP-1 behaves differently from its mammalian homologue. Thus, structural and biochemical studies of HMP-1 have been initiated to understand the mechanism of HMP-1 and the evolution of α-catenin. The N-terminal domain of HMP-1 in complex with the minimal HMP-1-binding region of HMP-2 was purified and crystallized. These crystals diffracted to 1.6 Å resolution and belonged to space group P3121, with unit-cell parameters a = b = 57.1, c = 155.4 Å. The M domain of HMP-1 was also purified and crystallized. The M-domain crystals diffracted to 2.4 Å resolution and belonged to space group P212121, with unit-cell parameters a = 72.8, b = 81.5, c = 151.4 Å. Diffraction data were collected and processed from each crystal, and the structures were solved by molecular replacement.
1. Introduction
Cell–cell adhesion is a defining characteristic of multicellular organisms that dictates tissue integrity, strength and morphology. In epithelia, cell–cell contacts known as adherens junctions (AJs) contain classical cadherin cell adhesion molecules that are linked to the actin cytoskeleton. The extracellular regions of cadherins from opposing cells bind to each other, while their cytoplasmic tails bind to β-catenin. β-Catenin in turn binds to α-catenin, a filamentous actin (F-actin) binding protein (Pokutta et al., 2008 ▸). α-Catenin also interacts with a number of other F-actin-binding proteins such as vinculin. The cadherin/catenin/F-actin assembly transmits mechanical forces between cells, enabling coordinated cytoskeletal rearrangements and cellular movements such as those that occur during tissue morphogenesis. However, the molecular mechanism by which the cadherin–catenin complex mediates mechanical and chemical signals to effect cytoskeletal architecture is poorly understood.
The cadherin/β-catenin/α-catenin module is conserved throughout metazoans. However, biochemical and biophysical studies of α-catenins from mammals, fish, insects and nematodes have revealed critical differences such as in oligomerization properties and in the allosteric regulation of actin-binding activity by β-catenin (Miller, Pokutta et al., 2013 ▸; Pokutta et al., 2014 ▸; Desai et al., 2013 ▸; Miller, Clarke et al., 2013 ▸). The AJ complex of the nematode Caenorhabditis elegans comprises the cadherin homologue HMR-1, the β-catenin homologue HMP-2 and the α-catenin homologue HMP-1. Structural studies of HMP-1 have been initiated in order to understand how α-catenin has changed during evolution.
Vertebrate and insect α-catenins comprise three domains, designated N (N-terminal), M (middle) and ABD (C-terminal actin-binding domain), each comprised of a series of α-helical bundles. The mammalian N domain binds to β-catenin and also serves as a dimerization domain (Pokutta & Weis, 2000 ▸). It consists of two four-helix bundles that share a continuous long α-helix. β-Catenin binding and homodimerization are mutually exclusive. Importantly, the mammalian αE-catenin dimer binds robustly to F-actin, whereas β-catenin binding weakens the affinity of α-catenin for F-actin in solution (Drees et al., 2005 ▸; Yamada et al., 2005 ▸). However, the E-cadherin/β-catenin/αE-catenin module interacts strongly with F-actin when tension is applied (Buckley et al., 2014 ▸). In contrast, C. elegans HMP-1 is a stable monomer, yet retains the ability to bind to HMP-2 (Kwiatkowski et al., 2010 ▸). It is not yet known whether the C. elegans cadherin–catenin complex exhibits the force-dependent regulation of actin binding found in the vertebrate system.
The mammalian α-catenin M domain contains three four-helix bundles linked by short loops (Rangarajan & Izard, 2013 ▸; Ishiyama et al., 2013 ▸) and contains binding sites for several other F-actin-binding proteins including vinculin, afadin and ZO-1 (Choi et al., 2012 ▸; Itoh et al., 1997 ▸; Pokutta et al., 2002 ▸; Yonemura et al., 2010 ▸; Yao et al., 2015 ▸; Weiss et al., 1998 ▸). A variety of cellular, biochemical and single-molecule experiments have shown that the M domain undergoes force-dependent conformational changes that are needed to expose its vinculin-binding site (Choi et al., 2012 ▸; Yao et al., 2014 ▸; Yonemura et al., 2010 ▸). Although it is clear that vertebrate α-catenin serves as a force transducer, it is not clear whether HMP-1 plays a similar role.
Here, we report the overexpression and crystallization of the HMP-1 N domain (residues 2–275) complexed with the minimal HMP-1-binding region of HMP-2 (residues 36–79), and of the HMP-1 M domain (residues 270–646). The HMP-1 N and M domains have 26 and 42% sequence identity to the corresponding domains of αE-catenin, respectively. By comparing the domain structures of HMP-1 and αE-catenin, we hope to provide molecular insight into the mechanism of HMP-1.
2. Materials and methods
2.1. Macromolecule production
The clones for the genes encoding full-length HMP-1 and HMP-2 were kindly provided by Dr Jeff Hardin (University of Wisconsin). To express the N-terminal domain of HMP-1 (HMP-1N; residues 2–275), a PCR product was amplified using 5′-CACGAATTCAAATGCCAACTCAACAGCTGAAGC-3′ forward and 5′-CACCTCGAGTCACAAATCGGTATCGTACCAATTGTGATTAGG-3′ reverse primers (restriction sites are shown in bold) and subcloned into the pGEX-TEV expression vector, which contains a Tobacco etch virus (TEV) protease-cleavable glutathione S-transferase (GST) affinity tag. The minimal HMP-1-binding region of HMP-2 was designed based on a βα-catenin chimeric protein (Pokutta & Weis, 2000 ▸). An expression construct spanning residues 36–79 of HMP-2 (HMP-236–79), which correspond to residues 109–152 of mouse β-catenin, was obtained by subcloning the PCR product from the PCR reaction with 5′-CCACGAATTCAGACTTCTGCTGCAGAAGCCACAAAT-3′ forward and 5′-CGACTCGAGTCACGAGAGAGTCCTGACATATCG-3′ reverse primers into pGEX-TEV vector. The M domain of HMP-1 (HMP-1M; residues 275–646) was PCR-amplified using 5′-CCACTCTAGACGGCGATCTTATCAATGAAATCGATACTTT-3′ forward and 5′-CACCCTCGAGTCAATCATTCATACTTCGGTTCATTAAAAGTG-3′ reverse primers and was cloned into pGEX-TEV vector. All three recombinant constructs were verified by DNA sequencing.
Each recombinant plasmid was transformed into competent Escherichia coli Rosetta (DE3) cells and a single colony was picked and grown overnight in Luria–Bertani (LB) medium containing ampicillin and chloramphenicol at 310 K. A one-hundredth volume of the overnight culture was inoculated into new LB medium containing antibiotics and the cells were grown until the OD600 reached about 0.7. Protein expression was induced by the addition of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and was followed by an additional 4 h incubation at 303 K. Cells were harvested by centrifugation at 3500g for 15 min and resuspended in phosphate-buffered saline (PBS) containing protease-inhibitor cocktail (Roche). The resuspended cells of HMP-1M, or a mixture of the resuspended cells of HMP-1N and HMP-236–79 (1:2 ratio culture volume), were lysed using an EmulsiFlex-C3 homogenizer (Avestin Inc., Ottawa, Canada). Each lysate was centrifuged at 26 500g for 30 min at 277 K and the resulting supernatant was incubated for 1 h with glutathione agarose (G-agarose) beads (Pierce) which had been pre-equilibrated with PBS buffer. The column was washed with ten column volumes of PBSTR buffer (PBS, 1 M NaCl, 5 mM DTT, 0.05% Tween 20) followed by two column volumes of cleavage buffer (30 mM Tris–HCl pH 8.0, 100 mM NaCl, 3 mM DTT). TEV protease was added into the column [20:1(w:w) substrate:TEV] to remove the GST tag, and tag-free protein was collected from the column after overnight incubation at 277 K.
After TEV protease treatment, HMP-1N complexed with HMP-236–79 and excess HMP-236–79 were obtained in a flowthrough fraction. This mixture (∼20 ml) was concentrated to ∼5 ml using Centricon membranes (10 000 molecular-weight cutoff) and loaded onto a Superdex 200 HiLoad 26/600 column equilibrated with a buffer consisting of 20 mM HEPES pH 8.0, 150 mM NaCl, 1 mM DTT. The protein complex eluted as a single peak and the fractions were pooled and concentrated to 15 mg ml−1 for crystallization (Fig. 1 ▸ a).
Figure 1.

Purification of the HMP-1N–HMP-236–79 complex and of HMP-1M. Superdex S200 gel-filtration profiles and peak fractions on an SDS–PAGE gel are shown for the HMP-1N–HMP-236–79 complex (a) and HMP-1M (b).
To further purify tag-free HMP-1M, the sample collected from the G-agarose column was loaded onto a HiTrap Q anion-exchange column (GE Healthcare) which had been pre-equilibrated with a buffer consisting of 20 mM Tris–HCl pH 8.0, 1 mM DTT, 20 mM NaCl, and HMP-1M was eluted using a linear gradient of 50–350 mM NaCl and pooled. The pooled fractions were loaded onto a HiLoad 26/60 Superdex 200 size-exclusion chromatography column equilibrated with a buffer consisting of 25 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. The protein eluted as a monomer in a sharp symmetric peak (Fig. 1 ▸ b). The fractions were collected and concentrated to 25 mg ml−1 for crystallization.
2.2. Crystallization
Initial crystallization screening of the HMP-1N–HMP-236–79 complex and HMP-1M was carried out with Phoenix (Art Robbins Instruments) or Mosquito (TTP Labtech) crystallization robots using commercial crystallization screens. An initial hit for the HMP-1N–HMP-236–79 complex was found in 0.1 M bis-tris pH 5.5, 0.2 M lithium sulfate, 25% PEG 3350 from a 96-well sitting-drop plate at 293 K. Larger but multi-layered crystals were obtained in a 24-well hanging-drop Linbro plate by equilibrating 2 µl drops consisting of 1 µl protein complex solution and 1 µl reservoir solution against 500 µl reservoir solution consisting of 0.1 M citrate pH 5.6, 0.2 M lithium sulfate, 20% PEG 3350. Freshly grown multiple crystal clusters were used for seeding experiments. To make the seed stock, the plate clusters were transferred to an Eppendorf tube containing 0.1 ml reservoir solution and seed bead (Hampton Research) and crushed by vortexing. The seed stock and its serial dilutions (1:5, 1:10, 1:20 and 1:50 ratios) were used for streak-seeding into pre-equilibrated drops consisting of 1 µl protein solution (11 mg ml−1) and 1 µl reservoir solution with 80% of the precipitant concentration for a normal crystallization condition. Single crystals appeared after 48 h incubation at 298 K (Fig. 2 ▸ a).
Figure 2.

Crystals of the HMP-1N–HMP-236–79 complex and HMP-1M. (a) HMP-1N–HMP-236–79 complex crystals grown from 0.1 M citrate pH 5.6, 0.2 M lithium sulfate, 25% PEG 3350 at 298 K (left) and after improvement by streak-seeding (right). (b) HMP-1M crystals obtained from 0.1 M bis-tris pH 6.5, 0.2 M sodium chloride, 25% PEG 3350 at 295 K (left) and single plate-like crystals obtained after streak-seeding (right).
HMP-1M crystals were first observed in 0.1 M bis-tris pH 6.5, 0.2 M NaCl, 25% PEG 3350 from a 96-well sitting-drop vapour-diffusion plate at 295 K. Crystals were reproduced in a 24-well hanging-drop Linbro plate. Lowering the concentration of PEG 3350 to 22% and changing the incubation temperature from 295 to 298 K improved the size of the crystals. To reduce the nucleation and multiplicity of crystals, a streak-seeding experiment was performed as described above. Larger diffraction-quality crystals appeared from the seeded drops after 24 h incubation at 298 K (Fig. 2 ▸ b).
2.3. Data collection and processing
Crystals of the HMP-1N–HMP-236–79 complex were mounted on cryoloops and flash-cooled in liquid nitrogen using perfluoropolyether oil (PFO) as a cryoprotectant. A 1.6 Å resolution diffraction data set was collected on beamline 12-2 at Stanford Synchrotron Radiation Lightsource (SSRL). The diffraction images, each of which was obtained by a 0.5 s exposure with 0.2° oscillation at a wavelength of 1.0332 Å on a Pilatus 6M detector, were indexed and integrated using XDS (Kabsch, 2010 ▸) and scaled using SCALA (Evans, 2006 ▸).
The HMP-1M crystals were cryoprotected with 20% glycerol in reservoir solution. A single flash-cooled crystal was used to obtain a 2.4 Å resolution diffraction data set on beamline 7A at Pohang Accelerator Laboratory (PAL). Each diffraction image was collected as a 3 s exposure with 1° oscillation at a wavelength of 0.97934 Å on an ADSC Q270 detector. A total of 140 diffraction images were integrated and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▸) and SCALEPACK (Otwinowski & Minor, 1997 ▸), respectively. The data-collection statistics are summarized in Table 1 ▸.
Table 1. Data collection and processing.
Values in parentheses are for the outer shell.
| HMP-1N–HMP-236–79 | HMP-1M | |
|---|---|---|
| Wavelength (Å) | 1.0332 | 0.97934 |
| Detector | Pilatus 6M | ADSC Q270 |
| Crystal-to-detector distance (mm) | 300 | 300 |
| Total rotation range (°) | 100 | 140 |
| Space group | P3121 | P212121 |
| a, b, c (Å) | 57.1, 57.1, 155.4 | 72.8, 81.5, 151.4 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| Resolution range (Å) | 40–1.6 (1.67–1.60) | 40–2.4 (2.49–2.40) |
| No. of unique reflections | 39608 (3775) | 35766 (3683) |
| Completeness (%) | 99.7 (99.0) | 99.4 (98.9) |
| Multiplicity | 5.3 (5.0) | 3.9 (3.8) |
| Solvent content (%) | 40 | 54 |
| 〈I/σ(I)〉 | 24.3 (3.7) | 13.4 (5.3) |
| R merge † | 0.027 (0.36) | 0.085 (0.26) |
| CC1/2 ‡ | 0.999 (0.886) | 0.997 (0.911) |
| Overall B factor from Wilson plot (Å2) | 29.4 | 26.8 |
R
merge = 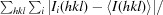
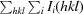 , where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
, where I
i(hkl) is the ith measurement of reflection hkl and 〈I(hkl)〉 is the weighted mean of all measurements of I(hkl).
CC1/2 is the Pearson correlation coefficient between random half data sets (Diederichs & Karplus, 2013 ▸).
3. Results and discussion
Full-length HMP-1 was initially expressed and purified for structural studies, but the expression level was very low and only small amounts of soluble protein could be obtained. Moreover, the full-length protein degraded readily during purification, suggesting flexibility that might interfere with crystallization. Indeed, by sequence homology to the mouse protein, the C-terminal actin-binding domain is connected to the M domain by a ∼30-amino-acid linker. To obtain crystallizable protein, the N-terminal and the M domains were designed based on secondary-structure prediction and sequence conservation with mouse αE-catenin. The N-terminal domain of HMP-1 (HMP-1N; residues 2–275) and the HMP-1 M domain (HMP-1M; residues 275–646) were each expressed in E. coli as a well behaved, soluble protein.
To characterize the interaction between HMP-1 and HMP-2, attempts were made to form a stable complex of HMP-1 and HMP-2. A minimal HMP-1-binding construct of HMP-2, residues 36–79 (HMP-236–79), was designed, expressed in E. coli and co-purified with HMP-1N after co-lysis of a 2 l culture of GST-HMP-236–79 and a 1 l culture of GST-HMP-1N. In this way, HMP-1N is fully saturated with HMP-236–79 and a homogeneous complex can be purified. After removal of the GST affinity tag, excess HMP-236–79, of ∼5 kDa, was partially removed during the concentration process using a 10 000 molecular-weight cutoff Centricon and the complex was further purified by size-exclusion chromatography. HMP-236–79 co-eluted with the 31 kDa HMP-1N and complex formation was confirmed by SDS–PAGE (Fig. 1 ▸ a). The HMP-1N–HMP-236–79 complex was initially crystallized as multiple layers of plates in 0.1 M bis-tris pH 5.5, 0.2 M lithium sulfate, 25% PEG 3350 at 293 K. Optimization of the crystallization conditions, including changing the buffer to citrate buffer and increasing the temperature to 298 K, improved the size of the crystals but still produced multilayered crystals. Streak-seeding was employed to produce single crystals with overall dimensions of 0.1 × 0.2 × 0.3 mm (Fig. 2 ▸ a).
HMP-1M was purified by G-agarose affinity, anion-exchange and size-exclusion chromatography. It eluted from the size-exclusion column as a monomer and the final yield of purified HMP-1M was about 8 mg per litre of cell culture (Fig. 1 ▸ a). An initial crystallization hit was found in 0.1 M bis-tris pH 6.5, 0.2 M sodium chloride, 25% PEG 3350 at 295 K and larger crystals were obtained by lowering the PEG concentration to 20–22%. However, the crystals grew as clusters of thin plates even after additive screening and optimization of the conditions. Streak-seeding was then carried out and produced single plate-like crystals (Fig. 2 ▸ b). Thus, for both cases reported here multi-layered crystal clusters were improved to diffraction-quality single crystals by streak-seeding.
Diffraction data were measured at 100 K for the HMP-1N–HMP-236–79 complex and HMP-1M to 1.6 and 2.4 Å resolution, respectively (Fig. 3 ▸, Table 1 ▸). The space group of the HMP-1N complex crystal was determined to be P3121 or P3221. The expected molecular mass of a 1:1 heterodimeric complex deduced from the amino-acid sequences is 36 kDa. The Matthews coefficient (Matthews, 1968 ▸) calculated from the unit-cell parameters suggest that the HMP-1N–HMP-236–79 complex crystals contain one complex in the asymmetric unit with 40% solvent content (V M = 2.03 Å3 Da−1). The HMP-1M crystals belonged to the orthorhombic space group P212121. The Matthews coefficient (VM = 2.66 Å3 Da−1) implies that there are two 42 kDa HMP-1M molecules in the asymmetric unit with 54% solvent content. The data-collection statistics are presented in Table 1 ▸.
Figure 3.

Diffraction images of the HMP-1N–HMP-236–79 complex (a) and HMP-1M (b).
Each structure was solved by molecular replacement (MR) using Phaser (McCoy et al., 2007 ▸). The mouse βα-catenin chimeric protein structure (PDB entry 1dow; Pokutta & Weis, 2000 ▸) was used as the search model for the HMP-1N–HMP-236–79 complex. A solution was only obtained in space group P3121. Initial refinement of the MR solution using PHENIX (Adams et al., 2010 ▸) resulted in an R work and R free of 49 and 54%, respectively. Extra electron density for the N-terminal amino acids 13–45 of HMP-1, which were absent in the search model, is visible and manual model building followed by structure refinement is under way. To solve the HMP-1M domain structure by MR, the two crystallographically independent copies of the M domain (residues 276–631) from the dimeric αE-catenin structure (PDB entry 4igg; Rangarajan & Izard, 2013 ▸) were superimposed and used as an ensemble search model. As suggested by the Matthews coefficient, two MR solutions were obtained and initial refinement by PHENIX gave an R work and R free of 47 and 53%, respectively. Further refinement and model building for each structure are in progress. Currently, the R work and R free values are below 30%.
Acknowledgments
This work was supported by the Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2013R1A1A2061541; HJC), a grant from the Basic Research Laboratory (NRF-2014R1A4A10052590) through the National Research Foundation of Korea (HJC) and grant GM09463 from the US National Institutes of Health (WIW). Portions of this work were performed at the Stanford Synchrotron Radiation Lightsource (SSRL). The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Experiments at PLS-II were supported in part by MSIP and POSTECH.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Buckley, C. D., Tan, J., Anderson, K. L., Hanein, D., Volkmann, N., Weis, W. I., Nelson, W. J. & Dunn, A. R. (2014). Science, 346, 1254211. [DOI] [PMC free article] [PubMed]
- Choi, H.-J., Pokutta, S., Cadwell, G. W., Bobkov, A. A., Bankston, L. A., Liddington, R. C. & Weis, W. I. (2012). Proc. Natl Acad. Sci. USA, 109, 8576–8581. [DOI] [PMC free article] [PubMed]
- Desai, R., Sarpal, R., Ishiyama, N., Pellikka, M., Ikura, M. & Tepass, U. (2013). Nature Cell Biol. 15, 261–273. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (2013). Acta Cryst. D69, 1215–1222. [DOI] [PMC free article] [PubMed]
- Drees, F., Pokutta, S., Yamada, S., Nelson, W. J. & Weis, W. I. (2005). Cell, 123, 903–915. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ishiyama, N., Tanaka, N., Abe, K., Yang, Y. J., Abbas, Y. M., Umitsu, M., Nagar, B., Bueler, S. A., Rubinstein, J. L., Takeichi, M. & Ikura, M. (2013). J. Biol. Chem. 288, 15913–15925. [DOI] [PMC free article] [PubMed]
- Itoh, M., Nagafuchi, A., Moroi, S. & Tsukita, S. (1997). J. Cell Biol. 138, 181–192. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kwiatkowski, A. V., Maiden, S. L., Pokutta, S., Choi, H.-J., Benjamin, J. M., Lynch, A. M., Nelson, W. J., Weis, W. I. & Hardin, J. (2010). Proc. Natl Acad. Sci. USA, 107, 14591–14596. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miller, P. W., Clarke, D. N., Weis, W. I., Lowe, C. J. & Nelson, W. J. (2013). Curr. Top. Membr. 72, 267–311. [DOI] [PMC free article] [PubMed]
- Miller, P. W., Pokutta, S., Ghosh, A., Almo, S. C., Weis, W. I., Nelson, W. J. & Kwiatkowski, A. V. (2013). J. Biol. Chem. 288, 22324–22332. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pokutta, S., Choi, H.-J., Ahlsen, G., Hansen, S. D. & Weis, W. I. (2014). J. Biol. Chem. 289, 13589–13601. [DOI] [PMC free article] [PubMed]
- Pokutta, S., Drees, F., Takai, Y., Nelson, W. J. & Weis, W. I. (2002). J. Biol. Chem. 277, 18868–18874. [DOI] [PMC free article] [PubMed]
- Pokutta, S., Drees, F., Yamada, S., Nelson, W. J. & Weis, W. I. (2008). Biochem. Soc. Trans. 36, 141–147. [DOI] [PMC free article] [PubMed]
- Pokutta, S. & Weis, W. I. (2000). Mol. Cell, 5, 533–543. [DOI] [PubMed]
- Rangarajan, E. S. & Izard, T. (2013). Nature Struct. Mol. Biol. 20, 188–193. [DOI] [PMC free article] [PubMed]
- Weiss, E. E., Kroemker, M., Rudiger, A. H., Jockusch, B. M. & Rüdiger, M. (1998). J. Cell Biol. 141, 755–764. [DOI] [PMC free article] [PubMed]
- Yamada, S., Pokutta, S., Drees, F., Weis, W. I. & Nelson, W. J. (2005). Cell, 123, 889–901. [DOI] [PMC free article] [PubMed]
- Yao, M., Qiu, W., Liu, R., Efremov, A. K., Cong, P., Seddiki, R., Payre, M., Lim, C. T., Ladoux, B., Mège, R.-M. & Yan, J. (2014). Nature. Commun. 5, 4525. [DOI] [PubMed]
- Yonemura, S., Wada, Y., Watanabe, T., Nagafuchi, A. & Shibata, M. (2010). Nature Cell Biol. 12, 533–542. [DOI] [PubMed]