

Abstract
In 2010 the identities of thousands of anti-Plasmodium compounds were released publicly to facilitate malaria drug development. Understanding these compounds’ mechanisms of action—i.e., the specific molecular targets by which they kill the parasite—would further facilitate the drug development process. Given that kinases are promising anti-malaria targets, we screened ~14,000 cell-active compounds for activity against five different protein kinases. Collections of cell-active compounds from GlaxoSmithKline (the ~13,000-compound Tres Cantos Antimalarial Set, or TCAMS), St. Jude Children’s Research Hospital (260 compounds), and the Medicines for Malaria Venture (the 400-compound Malaria Box) were screened in biochemical assays of Plasmodium falciparum calcium-dependent protein kinases 1 and 4 (CDPK1 and CDPK4), mitogen-associated protein kinase 2 (MAPK2/MAP2), protein kinase 6 (PK6), and protein kinase 7 (PK7). Novel potent inhibitors (IC50 < 1 μM) were discovered for three of the kinases: CDPK1, CDPK4, and PK6. The PK6 inhibitors are the most potent yet discovered for this enzyme and deserve further scrutiny. Additionally, kinome-wide competition assays revealed a compound that inhibits CDPK4 with few effects on ~150 human kinases, and several related compounds that inhibit CDPK1 and CDPK4 yet have limited cytotoxicity to human (HepG2) cells. Our data suggest that inhibiting multiple Plasmodium kinase targets without harming human cells is challenging but feasible.
Introduction
While screens of compound libraries for anti-Plasmodium activity are nothing new [1], there has been a recent trend toward public disclosure of all hit compounds arising from these screens [2–4]. These disclosures facilitate follow-up studies of these “cell-active” compounds and accelerate progress toward new antimalarial drugs. Nevertheless, many challenges remain in developing compounds with activity against culture-grown Plasmodium cells into clinically effective drugs [5]. Among these is identifying the compounds’ mechanism of action, i.e., the specific molecular targets by which they kill the parasite. While knowledge of compounds’ targets is not absolutely necessary for drug development, it can enable detailed protein structure studies, inform work on toxicology and acquisition of resistance, and hasten identification of suitable “backup compounds” [5].
Plasmodium kinases have great potential as drug targets. Despite the ubiquity of ATP binding sites, selective and potent inhibition of individual kinases has been achievable for both infectious and non-infectious diseases [6,7]; thus, kinases as a class are considered “druggable.” Furthermore, the Plasmodium kinome includes many potentially exploitable differences with respect to the human kinome [8], and kinome-wide essentiality data [9,10] further enable prioritization of possible Plasmodium kinase targets.
Based on these considerations and precedents for successful soluble expression [11–14], we selected five P. falciparum kinases (Table 1) with which to screen cell-active compound collections from GlaxoSmithKline (GSK), St. Jude Children’s Research Hospital, and the Medicines for Malaria Venture (MMV). Two of the kinases, CDPK4 and PK7, are not essential in the erythrocyte stages of the life cycle and thus are unlikely to be any compound’s primary target in these stages. However, an ideal malaria drug is active against multiple life-cycle stages—for example, inhibiting CDPK4 or PK7 in the sexual (gametocyte-to-oocyst) stages and acting at some other target(s) in the erythrocyte stages. Such dual- activity compounds could further the malaria eradication agenda [15] by both treating clinical malaria and blocking transmission.
Table 1. P. falciparum protein kinases selected for biochemical high-throughput screening.
| Target name (abbreviation) | PlasmoDB ID (old ID) | Key evidence for importance |
|---|---|---|
| Calcium-Dependent Protein Kinase 1 (CDPK1) | PF3D7_0217500 (PFB0815w) | Essentiality in erythrocyte stages of the life cycle has been validated genetically and chemically [16,9]. |
| Calcium-Dependent Protein Kinase 4 (CDPK4) | PF3D7_0717500 (PF07_0072) | CDPK4 is essential in gametocytes, though not in erythrocyte stages [17,9,10]. |
| Mitogen-Associated Protein Kinase 2 (MAPK2) | PF3D7_1113900 (PF11_0147) | MAPK2 is essential in erythrocyte stages (in P. falciparum, though not P. berghei) and in male gametogenesis [18–20]. |
| Protein Kinase 6 (PK6) | PF3D7_1337100 (MAL13P1.185) | PK6 is essential in erythrocyte stages [9,10]. |
| Protein Kinase 7 (PK7) | PF3D7_0213400 (PFB0605w) | PK7 knockouts fail to complete oocyst development [21]. They also grow slowly in erythrocyte stages, though PK7 is not essential in these stages [9,10]. |
Methods
Compounds
Collections of anti-Plasmodium compounds described in previous reports [3,4,22] were generously provided by GlaxoSmithKline (the 13,000-compound Tres Cantos Antimalarial Set, or TCAMS), St. Jude Children’s Research Hospital (260 compounds), and the Medicines for Malaria Venture (the 400-compound Malaria Box). Compounds for primary screens were provided as 1 mM stocks in dimethyl sulfoxide (DMSO), with 25 nL/well lyophilized in assay-ready plates (GSK/TCAMS); as 50 μM stocks in assay buffer (St. Jude); and as 10 mM stocks in DMSO (MMV). Compounds for dose-response studies were provided as 1000X stocks in DMSO, with 25 nL/well lyophilized in assay-ready plates (GSK/TCAMS).
Possible kinase substrates and control inhibitors
Bovine proteins β-casein, histone H2A, histone III-S, and myelin basic protein (MBP), as well as Poly(Glu,Tyr), staurosporine, and GW8510 were purchased from Sigma-Aldrich (St. Louis, MO, USA). MEK-1 peptide substrate ADPDHDHTGFLTEYVATRWRR as well as kinase inhibitors 1NA-PP1 and adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP) were purchased from Santa Cruz Biotech (Dallas, TX, USA). Peptide RRRKKSPRKRA was from Genscript (Piscataway, NJ, USA); Syntide-2 (PLARTLSVAGLPGKK) was from American Peptide Company (Sunnyvale, CA, USA). Inhibitors BRB-796, hymenialdisine, and olomoucine were from Calbiochem (now part of EMD Millipore).
Expression and purification of proteins
Genes for each kinase listed in Table 1 were cloned, expressed, and purified essentially as previously described [12,23,14]. An expression plasmid for PK6 was provided by Christian Doerig. Full-length proteins with N-terminal 6-Histidine tags were purified via nickel affinity column chromatography.
General biochemical screening assay format
Biochemical assays were performed in white 384-well plates (Nunc). Liquid transfers were done manually with 8-channel pipets. Compounds were initially tested in a single-point screen (final concentration 1 μM). Compounds exhibiting >50% inhibition at 1 μM were then tested at concentrations of 10, 3.3, 1.1, 0.37, 0.12, 0.041, 0.014, 0.0046, 0.0015, 0.0005, and 0.00017 μM to determine the concentration at which 50% inhibition occurs (IC50). (In the PK6 and PK7 screens, Z’ factors were suboptimal, so apparent hits were retested at 1 μM before proceeding with dose-response tests.) All incubations were at room temperature (~20°C). The catalytic activity of each kinase was considered proportional to ATP consumed, as determined from measurements of residual [ATP] with the luciferase-based reagent Kinase-Glo (Promega) following incubation. Luminescence (proportional to residual [ATP]) was measured on the plate readers FLx800 (BioTek Instruments, Winooski, VT, USA) and MicroBeta2 (PerkinElmer, Waltham, MA, USA). Incubation times were chosen to ensure near-complete ATP depletion in the absence of inhibition, thus maximizing the difference in signal between inhibited and uninhibited wells.
CDPK1 and CDPK4 screens
As described previously [14], final concentrations were 10 μM ATP, 0.1% bovine serum albumen (BSA) (w/v), 10 mM MgCl2, 2 mM CaCl2, and 6.6 nM or 208 nM of CDPK1 and CDPK4, respectively, in a buffer of 20 mM HEPES (pH 7.5). 40 μM Syntide-2 was used as a substrate; BKI-1 [14] was used as a control inhibitor, while reaction mixtures lacking Syntide-2 were included as an additional negative control. Incubation time was 90 minutes. Reactions were terminated by adding excess EGTA (5 mM), which sequesters Ca2+ and halts calcium-dependent kinase activity.
MAPK2 screen
Final concentrations were 1 μM ATP, 0.5 mM DTT, 1 mM MgCl2, 0.5 mg/mL BSA, and 10 μg/ml MAP2 in a buffer of 50 mM HEPES (pH 7.0). 0.5 mg/mL histone III-S served as the substrate [11]; 100 μM AMP-PNP, an ATP analog, was used as a control inhibitor. Incubation time was 4 hours.
PK6 screen
Final concentrations were 1.5 μM ATP, 5 mM MnCl2, and 15 μg/ml PK6 in a buffer of 100 mM Tris-HCl (pH 7.5). 50 μg/mL MBP was provided as the substrate [12]; 10 μM staurosporine was used as a control inhibitor. Incubation time was 3 hours and 40 minutes.
PK7 screen
Final concentrations were 1 μM ATP, 2 mM DTT, 20 mM MgCl2, 2 mM MnCl2, 0.01% BSA, and 6 μg/ml PK7 in a buffer of 20 mM Tris-HCl (pH 7.5). The enzyme itself was the only substrate present (since autophosphorylation occurs [13]); 100 μM 1NA-PP1 [24] was used as a control inhibitor. Incubation time was 3 hours.
Determination of PK7’s Km
To estimate PK7’s Km for ATP, we coupled its ADP production to ADP use by pyruvate kinase (PK) and subsequent oxidation of NADH by lactate dehydrogenase (LDH). The rate at which absorbance at 340 nm decreased was considered indicative of PK7 activity. Aside from the reagents listed above, these assays also included 0.15 NADH, 0.244 mM phosphoenolpyruvate (PEP), 5 U/mL PK, 7.5 U/mL LDH, and ATP concentrations ranging from 3 mM to 1.37 μM.
32P labeling assays
To confirm that inhibition of ATP consumption reflected inhibition of substrate phosphorylation, selected PK6 inhibitors were studied in 32P labeling assays conducted in parallel to the luminescence-based assays. Concentrations were as noted above, with compounds present at 1 μM, except that 8 μCi/mL γ-32P ATP was used in place of the usual 1.5 μM unlabeled ATP. Incubation time was 1 hour. 4.6 μL from each well was spotted onto a negatively charged phosphocellulose membrane, attracting the positively charged MBP substrate. The membrane was washed three times for 10 minutes with 0.5% phosphoric acid, dried, and exposed to a phosphoscreen that was scanned with a Typhoon FLA 9000 (GE). Radioactivity on the membrane was considered proportional to phosphorylation of MBP with 32P ATP.
Chemoproteomics
Kinobead competition assays were employed to determine the selectivity of selected hit compounds against the human and P. falciparum kinomes. Kinobeads were prepared as described [25,26]. The chemoproteomic inhibition binding experiments were performed as previously described [27]. Briefly, Kinobeads were washed and equilibrated in lysis buffer (50 mM Tris-Cl, pH 7.4, 0.4% Igepal-CA630, 1.5 mM MgCl2, 5% Glycerol, 150 mM NaCl, 25 mM NaF, 1 mM Na3VO4, 1 mM DTT, and 1 complete EDTA-free protease inhibitor tablet (Roche) per 25 ml). They were incubated at 4°C for 1 hour with 1 ml (5 mg) K562 extract, which was pre-incubated with compound or DMSO (vehicle control). Beads were transferred to disposable columns (MoBiTec), washed extensively with lysis buffer and eluted with SDS sample buffer. Proteins were alkylated, separated on 4–12% Bis-Tris NuPAGE (Life Technologies), stained with colloidal Coomassie, and quantified by isobaric tagging and LC-MS/MS. Digestion, labeling with TMT isobaric mass tags, peptide fractionation, and mass spectrometric analyses were performed essentially as described [27]. Criteria for protein quantification were: a minimum of 2 sequence assignments matching to unique peptides was required (FDR for quantified proteins <<0.1%), Mascot ion score > 15, signal to background ratio of the precursor ion > 4, signal to interference > 0.5 [28]. Reporter ion intensities were multiplied by ion accumulation time, yielding an area value proportional to the number of reporter ions present in the mass analyzer. Peptide fold changes were corrected for isotope purity as described and adjusted for interference caused by co-eluting nearly isobaric peaks as estimated by the signal-to-interference measure [29]. Protein quantification was achieved using a sum-based bootstrap algorithm [30]. Apparent dissociation constants were determined by taking into account the protein depletion by the beads as described [27].
Data analysis, statistics, and archiving
Km was calculated from Lineweaver-Burk plots. IC50 values were generally calculated from dose-response curves of three independent experiments with Prism 3 software (GraphPad). Z’ factors were calculated as described by Zhang et al. [31]. The parts of Additional File 1 representing our newly reported data have been [will be] deposited in ChEMBL.
Results
Optimization of CDPK1, CDPK4, and PK6 assays was conducted previously ([14] and C. Bodenreider, unpublished data); here we note highlights of MAPK2 and PK7 assay development.
MAPK2 characterization and assay development
Our stock of recombinant MAPK2 showed little activity at 37°C in comparison with room temperature (~20°C). 1% glycerol enhanced MAPK2 activity but was left out of the screening assay for simplicity. Since the P. falciparum MAPK2 may share some features with cyclin-dependent kinases (CDKs), various CDK inhibitors (hymenialdisine, GW8510, olomoucine) as well as the MAP kinase inhibitor BIRB796 were tested for possible use as a control inhibitor. None gave consistent inhibition at concentrations of 10–30 μM, so the ATP analog AMP-PNP was used instead.
PK7 characterization and assay development
Determining a kinase’s affinity for ATP helps one assess the likelihood of finding compounds that can compete directly with ATP in its active site. The Km of PK7 for ATP has not been reported previously; our assays yielded a value of 46 ± 3 μM. We also looked for protein or peptide substrates that would enhance PK7’s baseline rate of ATP consumption (the baseline rate presumably being due to PK7 autophosphorylation). MBP, β-casein, and histone H2A have been reported to be phosphorylated by PK7 in vitro [13] but did not noticeably augment PK7’s intrinsic ATPase activity. The same was true of peptide RRRKKSPRKRA, which conforms to a previously reported consensus sequence (though the consensus sequence was not itself tested as a substrate) [24]; the peptide ADPDHDHTGFLTEYVATRWRR, a substrate of MEKs (a.k.a. MAP kinase kinases), to which PK7 is related; and the generic tyrosine kinase substrate Poly(Glu,Tyr), which was tried for completeness, despite uncertainties about tyrosine phosphorylation in Plasmodium [32]. In the absence of an ATPase-enhancing substrate, we screened compounds for their ability to inhibit PK7’s intrinsic ATPase activity, presumably reflecting autophosphorylation.
High-throughput screening
The ~14,000 anti-Plasmodium compounds noted above were tested for possible inhibition of the five P. falciparum protein kinases noted above. Table 2 provides an overview of the screens. Z’ factors (based on 16 positive and 8–16 negative control wells per plate) were generally good, especially considering that all pipetting was done manually. While no hits were obtained for MAPK2 and only 2 hits were found for PK7, numerous hits were identified for CDPK1, CDPK4, and PK6 (Table 2).
Table 2. Summary of biochemical screens of P. falciparum protein kinases.
| Target | median Z’ | # of initial hits (hit rate) | # of confirmed sub-μM hits |
|---|---|---|---|
| CDPK1 | 0.71 | 220 (1.6%) | 181 |
| CDPK4 | 0.84 | 77 (0.6%) | 56 |
| MAPK2 | 0.66 | 0 (0%) | 0 |
| PK6 | 0.34 | 83 (0.6%) | 65 |
| PK7 | 0.48 | 2 (0.01%) | 2 |
As might have been expected from the similarity of the ATP binding sites in CDPK1 and CDPK4 [14], there was considerable overlap among the compounds that inhibited CDPK1 and those that inhibited CDPK4 (Fig 1). More surprisingly, many inhibitors acted both against CDPK1 and PK6. A complete listing of all sub-micromolar hits is given in S1 File.
Fig 1. Venn diagrams showing overlapping and non-overlapping targets of hit compounds.

225 compounds had IC50’s below 1 μM against at least one kinase (left); a subset of 72 compounds had IC50’s below 100 nM against at least one kinase (right).
Independent confirmation of inhibition
Since our screening assays measured ATP consumption rather than phosphorylation per se, we sought to confirm that inhibition of ATP use would reflect inhibition of phosphorylation. Low-throughput tests of compounds that did and did not inhibit PK6 revealed a strong correlation (R2 = 0.91) between inhibition of ATP consumption and inhibition of MBP phosphorylation as measured via 32P labeling (Fig 2).
Fig 2. Correlation of ATP depletion (measured with Kinase-Glo) with MBP phosphorylation (measured with 32P-ATP).

Eight compounds were studied for possible inhibition of PK6. Compounds that impaired ATP use also impaired MBP phosphorylation. Each data point represents an average of two independent experiments conducted on separate days.
Analysis of scaffolds
Structurally related hits were clustered manually into scaffolds. 196 hits were grouped into scaffolds A through L (Fig 3), while the other 29 hits were either singletons or members of very small clusters (<6 members). Our clusters were broadly consistent with those generated previously [3], though our tendency was toward fewer distinct scaffolds with more members. For instance, scaffold A includes members of 7 clusters previously generated with molecular frameworks and members of 9 clusters previously generated with Daylight fingerprinting (Additional File 1).
Fig 3. Clustering of P. falciparum protein kinase hits into chemical scaffolds.

Inhibition of HepG2 cell growth at compound concentrations of 10 μM were previously reported by Gamo et al. [3]. For some scaffolds, target counts exceed the number of hits because some compounds hit more than one target.
Many of the compounds covered by Fig 3 were previously recognized as likely P. falciparum kinase inhibitors based on their effects on human kinases [3]. For example, scaffold D inhibits Epidermal Growth Factor Receptor (EGFR), scaffold G inhibits Akt (a.k.a. Protein Kinase B), and scaffold K inhibits Pyruvate Dehydrogenase Kinase (PDK); it is likely that these scaffolds also target one or more P. falciparum kinases structurally similar to the human kinases. On the other hand, none of scaffold I’s 16 compounds and only 1 of scaffold B’s 10 compounds were previously annotated as probable kinase inhibitors based on historical screening data [3]. The finding that these scaffolds inhibit at least one P. falciparum kinase (primarily CDPK1, so far) thus constitutes additional progress toward identifying their specific target(s).
Integration of new data with previous data
The results reported here add to what is already known about these publicly disclosed antimalarial compounds, permitting further insights. For example, regarding the possibility of a compound that inhibits multiple Plasmodium kinases and thus acts at multiple life-cycle stages, we can ask whether multi-kinase inhibitors are inevitably cytotoxic. Fig 4 shows that inhibitors of multiple kinases do tend to be more cytotoxic to human HepG2 cells; however, many of the dual-activity compounds have low to moderate cytotoxicity at a concentration of 10 μM.
Fig 4. Human cytotoxicity of inhibitors of 1, 2, or 3 of the P. falciparum kinases studied.

Inhibition of HepG2 cell growth at compound concentrations of 10 μM were previously reported by Gamo et al. [3].
In terms of cytotoxicity, some scaffolds look more appealing than others. A comparison of scaffolds D and G revealed that, while compounds in both scaffolds cover a wide range of potencies against CDPK4, most scaffold-D compounds are fairly cytotoxic to human HepG2 cells, whereas most scaffold-G compounds are more benign (Fig 5). A similar trend can be seen with scaffold-F and scaffold-H inhibitors of CDPK1; members of scaffold F generally appear less cytotoxic to HepG2 cells (Fig 5). These data may be useful in prioritizing scaffolds during drug development.
Fig 5. A comparison of different CDPK inhibitors’ cytotoxicity to human cells.

Inhibition of HepG2 cell growth at compound concentrations of 10 μM is shown for CDPK4 inhibitors in scaffolds D and G (top) and for CDPK1 inhibitors in scaffolds F and H (bottom).
Correlations between compounds’ IC50s against a given molecular target and EC50s against P. falciparum cell growth, if found, would suggest that the target may indeed represent the compounds’ primary mechanism of action. However, IC50s against CDPK1, CDPK4 and PK6 did not strongly correlate with previously reported EC50s against P. falciparum 3D7 cells, either overall (S1 Fig; R2≤0.1 for each enzyme) or within individual scaffolds (data not shown). These poor correlations could be due to one or more of several factors, such as (A) the limited ranges of IC50s and EC50s studied; (B) polypharmacology, i.e., compounds’ actions at additional (unknown) targets, perhaps (unlike CDPK4 and PK7) expressed strongly in the erythrocyte life-cycle stages tested in the EC50 assays; and (C) compounds’ varied bioavailability in the EC50 assays, e.g., due to variations in membrane permeance or binding to components of blood used in the EC50 assays.
Assessment of compound promiscuity
The possibility that an inhibitor might target multiple Plasmodium kinases but leave the human kinome untouched, however appealing, is difficult to evaluate with biochemical assays alone. Moreover, while strong inhibition of HepG2 cell growth might indicate that a compound inhibits human kinases as well, such growth inhibition might also involve mechanisms unrelated to kinases. To obtain direct profiles of selected compounds’ activity against the P. falciparum and human kinomes, we tested these compounds in kinobead competition assays.
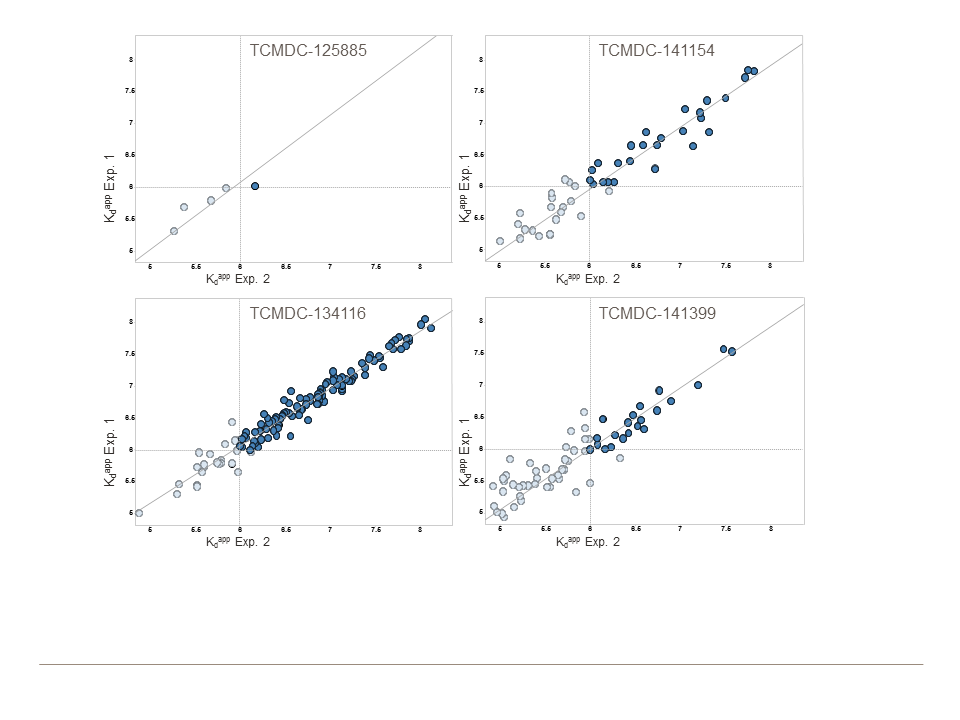
The kinobead results are illustrated in Fig 6 and summarized numerically in Table 3. Interestingly, although the four tested compounds had similar effects on HepG2 cell growth, inhibiting it by 33–48% at 10 μM, their effects on human kinases varied greatly. At the extremes, TCMDC-12885 had a “clean” profile, while TCMDC-134116 bound to two thirds of the human kinases tested with high affinity (pKd ≥ 6).
Fig 6. Assessment of compound promiscuity with human kinases.

Kinobeads were incubated with K562 cell extract either in the presence of vehicle (DMSO) or TCAMS compound, respectively (20 μM-0.03 μM). Protein kinases captured by the beads (140–150 kinases per experiment) were quantified following tryptic digestion, isobaric peptide tagging, and LC-MS/MS analysis. Kinases were identified as potential targets by virtue of their reduced capture in the presence of excess TCAMS compounds. Apparent dissociation constants (Kd’s) were calculated from the extent to which capture of each kinase was reduced at each compound concentration. Kd values from duplicate experiments generally agreed with each other quite well (S2 Fig). Colored bands indicate kinase-ligand complexes with apparent pKd’s of ≥6, with darker shades denoting higher pKd’s. Kinases that did not have an apparent pKd of ≥6 for any of the compounds are not represented; only names of every other targeted kinase are shown due to space limitations. These results are summarized numerically in Table 3.
Table 3. Summary of kinobead competition assays (results reflect two independent experiments).
| Compound | Inhibition of HepG2 cell growth at 10 μM [3] | Human kinases quantified on kinobeads (N = 2) | Human kinases with apparent pKd ≥ 4.7 (N = 2) | Human kinases with apparent pKd ≥ 6 (N = 2) |
|---|---|---|---|---|
| TCMDC-125885 | 33% | 153 | 5 | 1 |
| TCMDC-134116 | 43% | 136 | 126 | 96 |
| TCMDC-141154 | 48% | 146 | 47 | 26 |
| TCMDC-141399 | 47% | 147 | 63 | 24 |
Discussion
We attained mixed results in our effort to link anti-Plasmodium compounds with possible P. falciparum protein kinase targets. At one extreme, none of the 14,000 compounds gave significant inhibition of MAPK2 or PK7, with the exception of two members of an imidazopyridazine scaffold previously reported to inhibit PK7 and many other kinases [33]. These findings could mean that (A) our stocks of MAPK2 and/or PK7 were not well-folded; (B) sampling of chemical space was inadequate for finding inhibitors; (C) these enzymes are not very druggable; and/or (D) these enzymes adopt different, more druggable conformations in a cellular context with interacting with other proteins. We believe that explanation (A) is unlikely to be true, in part because thermal melt assays of MAPK2 and PK7 (data not shown) revealed characteristics of stable proteins (low fluorescence at baseline, high melting temperature, large change in fluorescence during melting) [34]. Explanation (B) also seems somewhat unlikely, at least for MAPK2, because a thermal melt screen of this enzyme against 2,000 diverse kinase inhibitors gave no hit compounds that raised the melting temperature by >2°C (data not shown), whereas an unstable yet druggable protein should have been hit in this screen. Thus, the simplest explanation of our data may be that MAPK2 and PK7 have low druggability in vitro (D) or perhaps in general (C).
On the other hand, our data include the most numerous, diverse, and potent PK6 inhibitors reported to date. Previous studies of PK6 [12,35] have not included extensive searches for chemical inhibitors. Our study thus complements previous reports in revealing many potential “tool compounds” for further study of PK6.
CDPK1 and CDPK4 have been the focus of much recent research, and potent inhibitors of each are already known [36,37,14,38,39]. Specificity of inhibition has been achieved with compounds that are accommodated by these enzymes’ unusually small “gatekeeper residues” in the ATP binding pocket (threonine for CDPK1, serine for CDPK4). The present study provides further biochemical characterization of the CDPK1 and CDPK4 ATP binding sites. Given the similarity of these binding sites in CDPK1 and CDPK4 [14], it surprised us that only 25% of the CDPK1 hits (46 of 181) had sub-micromolar IC50s against CDPK4. Since the “gatekeeper residue” is slightly larger in CDPK1 (threonine) than in CDPK4 (serine), it was also surprising that CDPK1 had many more hits than CDPK4 (181 vs. 56). It seems that, in the vicinity of the CDPK1 and CDPK4 gatekeeper residues, compound binding efficacy may be determined at least as much by the strength of interactions with amino acids as by steric limitations of the binding pocket [40].
The data presented here complement previous efforts to link hits from cell-based P. falciparum screens to specific molecular targets. Of the 76 compounds associated with 16 specific protein targets in previous reports [2,4,41,42], only one compound is a member of any scaffold shown in Fig 3. Scaffold A fits the structure of compound “CK-8” (also called SJ000111331 and GNF-Pf-4995), a modest inhibitor of the P. falciparum choline kinase (IC50 = 12 μM) [41]. Our connection of scaffolds A through L to specific protein kinases establishes novel hypotheses about these scaffolds’ exact sites of action.
As with previous biochemical and biophysical screens of anti-Plasmodium compounds [4,41,42], our results do not prove that the kinases studied here are the only or primary targets through which the associated compounds kill P. falciparum cells. The hypothesis that a specific kinase is a compound’s primary target could be tested by evolving resistance to the compound and determining whether the resistance-enabling mutants occur in the gene of the hypothesized target [43,44].
Finally, our data offer some indication as to the likelihood of developing a multi-kinase-targeting compound that is efficacious against malaria but safe in humans. Most individual compounds that inhibited three kinases were highly cytotoxic to human liver cells, but some dual-acting compounds, especially in scaffold G, were not (Figs 4 and 5). Ease of achieving joint inhibition of CDPK1 and CDPK4 was mixed, as 82% of CDPK4 hits also inhibited CDPK1 but only 25% of CDPK1 hits inhibited CDPK4 (Fig 1). While many of the multi-acting compounds were members of scaffold D, which generally looked cytotoxic (Fig 5), EGFR inhibitors based on this scaffold have been patented as possible lung cancer drugs, so clinically safe compounds based on scaffolds like this one may be possible. On the whole, our data provide a basis for both optimism and cautiousness regarding the concept of a multi-kinase-targeting malaria drug.
Supporting Information
(PNG)
{kind=link}
X and Y axes are both labeled with the negative logarithm of Kd values (pKd).
(PNG)
{kind=link}
This Microsoft Excel file includes IC50s, scaffold information, and previously collected data downloaded from ChEMBL (https://www.ebi.ac.uk/chembl/).
(XLS)
This Microsoft Excel file includes a table of the experimental setup and apparent dissociation constants determined by taking into account protein depletion by the beads.
(XLSX)
Acknowledgments
This project benefited greatly from the contributions of many collaborators from around the world. Anti-Plasmodium compounds were generously provided by GlaxoSmithKline (Francisco-Javier Gamo, Maria- Esperanza Herreros-Aviles, and David Wilson), St. Jude Children’s Research Hospital (W. Armand Guiguemde and R. Kip Guy), and the Medicines for Malaria Venture (Jeremy Burrows, Simon Macdonald, Thomas Spangenberg, Tim Wells, and Paul Willis) / Scynexis (Paul Kowalczyk). Ariadna Santander (University of Washington) coordinated Materials Transfer Agreements with these institutions. Christian Doerig (Monash University) provided pre-publication release of kinase essentiality data as well as a plasmid for expressing PK6. Ben Turk (Yale University) contributed sage advice and pilot testing of peptide substrates of PK7. Christophe Bodenreider (Novartis Institute for Tropical Diseases, Singapore) helped optimize the PK6 assay. Lynn Barrett, Cassie Bryan, Steve Nakazawa Hewitt, David Leibly, and Trang Nhu Nguyen (University of Washington) contributed to expression and purification of the protein kinases. Finally, this paper benefited from the helpful suggestions of anonymous reviewers.
Abbreviations
- AMP-PNP
adenosine 5′-(β,γ-imido)triphosphate
- BSA
bovine serum albumen
- CDPK1
calcium-dependent protein kinase 1
- CDPK4
calcium-dependent protein kinase 4
- EGFR
epidermal growth factor receptor
- IC50
50% inhibitory concentration
- LDH
lactate dehydrogenase
- MAPK2
mitogen-associated protein kinase 2
- MBP
myelin basic protein
- MMV
Medicines for Malaria Venture
- P. falciparum
Plasmodium falciparum
- PDK
pyruvate dehydrogenase kinase
- PEP
phosphoenolpyruvate
- PK
pyruvate kinase
- PK6
protein kinase 6
- PK7
protein kinase 7
- SGC
Structural Genomics Consortium
Data Availability
All relevant data are within the paper and its Supporting Information files. Data have also been submitted to ChEMBL, CDD Public, and PubMed at the following URLs: ChEMBL: (https://www.ebi.ac.uk/chemblntd/), CDD Public: (https://www.collaborativedrug.com/pages/public_access), PubChem: (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159585) (CDPK1), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159588) (CDPK4), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159589) (MAPK2), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159586) (PK6), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159587) (PK7).
Funding Statement
This study was funded in part by the following sources: the Medicines for Malaria Venture (mmv.org; 11/2500 to WCVV); the National Institute for Allergy and Infectious Diseases (niaid.nih.gov; R01AI1089441 to WCVV); the National Institute for General Medical Science (nigms.nih.gov; R01GM086858 to DJM); the University of Washington Plein Endowment for Geriatric Pharmacy Research (to KRK); and the National Cancer Institute (cancer.gov; F32CA174346 to SEL). The funders had no role in study design, data collection and analysis, or decision to publish. The Medicines for Malaria Venture (MMV), which partially funded this study, employs author DL. Commercial companies Cellzome GmbH and GlaxoSmithKline provided support in the form of salary for authors MJLM, SGD, MB, and GD, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the Author Contributions section.
References
- 1.Kinnamon KE, Rothe WE. Biological screening in the U.S. Army antimalarial drug development program. Am J Trop Med Hyg 1975;24: 174–178. [DOI] [PubMed] [Google Scholar]
- 2.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A 2008;105: 9059–9064. 10.1073/pnas.0802982105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010;465: 305–310. 10.1038/nature09107 [DOI] [PubMed] [Google Scholar]
- 4.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, et al. Chemical genetics of Plasmodium falciparum. Nature 2010;465: 311–315. 10.1038/nature09099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guiguemde WA, Shelat AA, Garcia-Bustos JF, Diagana TT, Gamo FJ, Guy RK. Global phenotypic screening for antimalarials. Chem Biol 2012;19: 116–129. 10.1016/j.chembiol.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr Opin Cell Biol 2009;21: 317–324. 10.1016/j.ceb.2009.01.015 [DOI] [PubMed] [Google Scholar]
- 7.Schreiber M, Res I, Matter A. Protein kinases as antibacterial targets. Curr Opin Cell Biol 2009;21: 325–330. 10.1016/j.ceb.2009.01.026 [DOI] [PubMed] [Google Scholar]
- 8.Doerig C, Abdi A, Bland N, Eschenlauer S, Dorin-Semblat D, Fennell C, et al. Malaria: targeting parasite and host cell kinomes. Biochim Biophys Acta 2010;1804: 604–612. 10.1016/j.bbapap.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 9.Tewari R, Straschil U, Bateman A, Bohme U, Cherevach I, Gong P, et al. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe 2010;8: 377–387. 10.1016/j.chom.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solyakov L, Halbert J, Alam MM, Semblat JP, Dorin-Semblat D, Reininger L, et al. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat Commun 2011;2: 565 10.1038/ncomms1558 [DOI] [PubMed] [Google Scholar]
- 11.Dorin D, Alano P, Boccaccio I, Ciceron L, Doerig C, Sulpice R, et al. An atypical mitogen-activated protein kinase (MAPK) homologue expressed in gametocytes of the human malaria parasite Plasmodium falciparum. Identification of a MAPK signature. J Biol Chem 1999;274: 29912–29920. [DOI] [PubMed] [Google Scholar]
- 12.Bracchi-Ricard V, Barik S, Delvecchio C, Doerig C, Chakrabarti R, Chakrabarti D. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem J 2000;347 Pt 1: 255–263. [PMC free article] [PubMed] [Google Scholar]
- 13.Dorin D, Semblat JP, Poullet P, Alano P, Goldring JP, Whittle C, et al. PfPK7, an atypical MEK-related protein kinase, reflects the absence of classical three-component MAPK pathways in the human malaria parasite Plasmodium falciparum. Mol Microbiol 2005;55: 184–196. [DOI] [PubMed] [Google Scholar]
- 14.Ojo KK, Pfander C, Mueller NR, Burstroem C, Larson ET, Bryan CM, et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest 2012;122: 2301–2305. 10.1172/JCI61822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso PL B Q, Bell D, Berman P, Breman JG, de Savigny D, Drakeley C, Evans T, Block MA, Hanson K, Kachur SP, Kress D, Macdonald M, Newman RD, Nyonator FK, Pagnoni F, Perkins M, Schapira A, Schellenberg D, Slutsker L, Sullivan D, Tanner M, Teklehaimanot A, Teuscher T, Mendis K, Brown G. A research agenda for malaria eradication: health systems and operational research. PLoS Med 2011;8: e1000397 10.1371/journal.pmed.1000397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato N, Sakata T, Breton G, Le Roch KG, Nagle A, Andersen C, et al. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol 2008;4: 347–356. 10.1038/nchembio.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 2004;117: 503–514. [DOI] [PubMed] [Google Scholar]
- 18.Rangarajan R, Bei AK, Jethwaney D, Maldonado P, Dorin D, Sultan AA, et al. A mitogen-activated protein kinase regulates male gametogenesis and transmission of the malaria parasite Plasmodium berghei. EMBO Rep 2005;6: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tewari R, Dorin D, Moon R, Doerig C, Billker O. An atypical mitogen-activated protein kinase controls cytokinesis and flagellar motility during male gamete formation in a malaria parasite. Mol Microbiol 2005;58: 1253–1263. [DOI] [PubMed] [Google Scholar]
- 20.Dorin-Semblat D, Quashie N, Halbert J, Sicard A, Doerig C, Peat E, et al. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Mol Microbiol 2007;65: 1170–1180. [DOI] [PubMed] [Google Scholar]
- 21.Dorin-Semblat D, Sicard A, Doerig C, Ranford-Cartwright L. Disruption of the PfPK7 gene impairs schizogony and sporogony in the human malaria parasite Plasmodium falciparum. Eukaryot Cell 2008;7: 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TN, Willis P. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS ONE 2013;8: e62906 10.1371/journal.pone.0062906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong A, et al. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol 2007;151: 100–110. [DOI] [PubMed] [Google Scholar]
- 24.Merckx A, Echalier A, Langford K, Sicard A, Langsley G, Joore J, et al. Structures of P. falciparum protein kinase 7 identify an activation motif and leads for inhibitor design. Structure 2008;16: 228–238. 10.1016/j.str.2007.11.014 [DOI] [PubMed] [Google Scholar]
- 25.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature biotechnology 2007;25: 1035–1044. [DOI] [PubMed] [Google Scholar]
- 26.Bergamini G, Bell K, Shimamura S, Werner T, Cansfield A, Muller K, et al. A selective inhibitor reveals PI3Kgamma dependence of T(H)17 cell differentiation. Nature chemical biology 2012;8: 576–582. 10.1038/nchembio.957 [DOI] [PubMed] [Google Scholar]
- 27.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nature biotechnology 2011;29: 255–265. 10.1038/nbt.1759 [DOI] [PubMed] [Google Scholar]
- 28.Savitski MM, Fischer F, Mathieson T, Sweetman G, Lang M, Bantscheff M. Targeted data acquisition for improved reproducibility and robustness of proteomic mass spectrometry assays. Journal of the American Society for Mass Spectrometry 2010;21: 1668–1679. 10.1016/j.jasms.2010.01.012 [DOI] [PubMed] [Google Scholar]
- 29.Savitski MM, Mathieson T, Zinn N, Sweetman G, Doce C, Becher I, et al. Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. Journal of proteome research 2013;12: 3586–3598. 10.1021/pr400098r [DOI] [PubMed] [Google Scholar]
- 30.Savitski MM, Sweetman G, Askenazi M, Marto JA, Lang M, Zinn N, et al. Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Analytical chemistry 2011;83: 8959–8967. 10.1021/ac201760x [DOI] [PubMed] [Google Scholar]
- 31.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4: 67–73. [DOI] [PubMed] [Google Scholar]
- 32.Talevich E, Tobin AB, Kannan N, Doerig C. An evolutionary perspective on the kinome of malaria parasites. Philos Trans R Soc Lond B Biol Sci 2012;367: 2607–2618. 10.1098/rstb.2012.0014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bouloc N, Large JM, Smiljanic E, Whalley D, Ansell KH, Edlin CD, et al. Synthesis and in vitro evaluation of imidazopyridazines as novel inhibitors of the malarial kinase PfPK7. Bioorg Med Chem Lett 2008;18: 5294–5298. 10.1016/j.bmcl.2008.08.043 [DOI] [PubMed] [Google Scholar]
- 34.Crowther GJ, He P, Rodenbough PP, Thomas AP, Kovzun KV, Leibly DJ, et al. Use of thermal melt curves to assess the quality of enzyme preparations. Anal Biochem 2010;399: 268–275. 10.1016/j.ab.2009.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manhani KK, Arcuri HA, da Silveira NJ, Uchoa HB, de Azevedo WF Jr, Canduri F. Molecular models of protein kinase 6 from Plasmodium falciparum. J Mol Model 2005;12: 42–48. [DOI] [PubMed] [Google Scholar]
- 36.Lemercier G, Fernandez-Montalvan A, Shaw JP, Kugelstadt D, Bomke J, Domostoj M, et al. Identification and characterization of novel small molecules as potent inhibitors of the plasmodial calcium-dependent protein kinase 1. Biochemistry 2009;48: 6379–6389. 10.1021/bi9005122 [DOI] [PubMed] [Google Scholar]
- 37.Larson ET, Ojo KK, Murphy RC, Johnson SM, Zhang Z, Kim JE, et al. Multiple determinants for selective inhibition of apicomplexan calcium-dependent protein kinase CDPK1. J Med Chem 2012;55: 2803–2810. 10.1021/jm201725v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z, Ojo KK, Johnson SM, Larson ET, He P, Geiger JA, et al. Benzoylbenzimidazole-based selective inhibitors targeting Cryptosporidium parvum and Toxoplasma gondii calcium-dependent protein kinase-1. Bioorg Med Chem Lett 2012;22: 5264–5267. 10.1016/j.bmcl.2012.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lourido S, Zhang C, Lopez MS, Tang K, Barks J, Wang Q, et al. Optimizing small molecule inhibitors of calcium-dependent protein kinase 1 to prevent infection by Toxoplasma gondii. J Med Chem 2013;56: 3068–3077. 10.1021/jm4001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keyloun KR, Reid MC, Choi R, Song Y, Fox AM, Hillesland HK, et al. The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014;141: 1499–1509. 10.1017/S0031182014000857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowther GJ, Napuli AJ, Gilligan JH, Gagaring K, Borboa R, Francek C, et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol Biochem Parasitol 2011;175: 21–29. 10.1016/j.molbiopara.2010.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theobald AJ, Caballero I, Coma I, Colmenarejo G, Cid C, Gamo FJ, et al. Discovery and biochemical characterization of Plasmodium thioredoxin reductase inhibitors from an antimalarial set. Biochemistry 2012;51: 4764–4771. 10.1021/bi3005076 [DOI] [PubMed] [Google Scholar]
- 43.Spillman NJ, Allen RJ, McNamara CW, Yeung BK, Winzeler EA, Diagana TT, et al. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 2013;13: 227–237. 10.1016/j.chom.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015;522: 315–320. 10.1038/nature14451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG)
X and Y axes are both labeled with the negative logarithm of Kd values (pKd).
(PNG)
This Microsoft Excel file includes IC50s, scaffold information, and previously collected data downloaded from ChEMBL (https://www.ebi.ac.uk/chembl/).
(XLS)
This Microsoft Excel file includes a table of the experimental setup and apparent dissociation constants determined by taking into account protein depletion by the beads.
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. Data have also been submitted to ChEMBL, CDD Public, and PubMed at the following URLs: ChEMBL: (https://www.ebi.ac.uk/chemblntd/), CDD Public: (https://www.collaborativedrug.com/pages/public_access), PubChem: (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159585) (CDPK1), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159588) (CDPK4), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159589) (MAPK2), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159586) (PK6), (https://pubchem.ncbi.nlm.nih.gov/bioassay/1159587) (PK7).