

Abstract
The polymyxin lipodecapeptides colistin and polymyxin B have become last resort therapies for infections caused by highly drug-resistant Gram-negative bacteria. Unfortunately, their utility is compromised by significant nephrotoxicity and polymyxin-resistant bacterial strains. We have conducted a systematic activity–toxicity investigation by varying eight of the nine polymyxin amino acid free side chains, preparing over 30 analogues using a novel solid-phase synthetic route. Compounds were tested against a panel of Gram-negative bacteria and counter-screened for in vitro cell toxicity. Promising compounds underwent additional testing against primary kidney cells isolated from human kidneys to better predict their nephrotoxic potential. Many of the new compounds possessed equal or better antimicrobial potency compared to polymyxin B, and some were less toxic than polymyxin B and colistin against mammalian HepG2 cells and human primary kidney cells. These initial structure–activity and structure–toxicity studies set the stage for further improvements to the polymyxin class of antibiotics.
Introduction
Bacterial sepsis kills millions of people every year. In the United States, patients hospitalized for septicemia or sepsis are more than eight times as likely to die during their hospitalization compared to those hospitalized for other diagnoses,1 with over 200,000 deaths in 2008.2 Over one-third of patients with sepsis treated in an intensive care unit die in the hospital.3 Sepsis is the most expensive condition treated in U.S. hospitals, costing more than $20 billion in 2011.4 Sepsis is most commonly caused by Gram-positive (G+ve) Staphylococcus aureus and Streptococcus pyogenes and Gram-negative (G-ve) Klebsiella spp., Escherichia coli, and Pseudomonas aeruginosa.5 Unfortunately, there has been an ominous rise in highly drug-resistant G-ve bacteria that are able to overcome almost every known antibiotic, threatening a return to a “preantibiotic” era if new therapies are not discovered. The current clinical pipeline is mainly populated by G+ve drug candidates.6 One possible approach to antibiotic development is to revisit antibiotics discovered in the “golden age” of antibiotic discovery (the 1950s–1970s) to see if they can be optimized or improved.
In 1947 two American groups independently discovered antibiotic substances produced by Bacillus polymyxa.7−9 The same year a group in England described a new antibiotic they named aerosporin isolated from a strain at first thought to be B. aerosporus, but later shown to be identical to the strain B. polymyxa.10 Many examples of polymyxin derivatives have since been isolated from nature, all of them with various degrees of activity against bacteria.11−15 A review by Stansly published in 1949 on early human clinical use of four distinct polymyxin antibiotic complexes A, B, C, and D concluded that there is “little doubt that polymyxins have exhibited effective therapeutic activity in certain infections of man produced by Gram-negative bacteria. It is apparent, however, that their application in this field is likely to be limited because of the occurrence of untoward reactions elicited by material used in the clinical trials.”16 Nonetheless, two of the polymyxins, polymyxin B 1 and polymyxin E (colistin) 2, entered clinical use and in recent years have become last-resort antibiotics for multidrug resistant G-ve infections, fueling a surge of interest in their use.17−22 The main limitation of the polymyxins is nephrotoxicity, with adverse renal toxicity observed in over 50% of cases in some studies.23,24 This has led to a systematic effort to improve dosing regimens.25,26
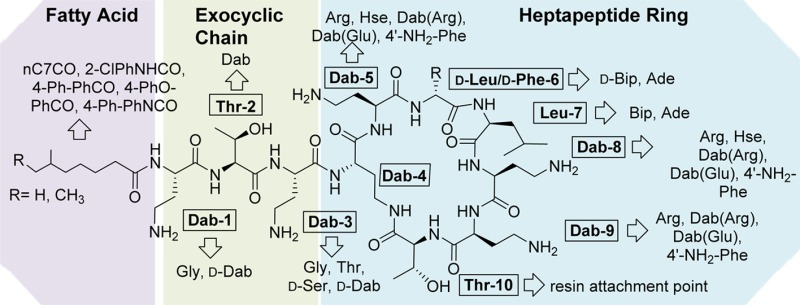
Polymyxins 1 and 2 are both positively charged macrocyclic peptide antibiotics composed of a heptapeptide ring and three amino acid exocyclic tail capped by a mixture of two fatty acids (see Figure 1), differing only in a d-Phe/d-Leu alteration at position-6. They are highly basic, containing six 2,4-diaminobutyric acid (Dab) residues, including one residue whose side chain is used for lactam formation with the C-terminal Thr residue. While polymyxin B is administered as the parent compound sulfate salt, colistin is almost always used as a polymethanesulfonylated prodrug, colistin methanesulfonate sodium, which spontaneously hydrolyses to release polymyxin E 2. Given that the polymyxins are natural products produced by fermentation, their composition does vary considerably between manufacturers.27
Figure 1.

Structures of polymyxin B 1 and polymyxin E (colistin) 2.
The antimicrobial activity of the polymyxins is initiated by binding to Lipid A, the membrane-anchoring component of lipopolysaccharide (LPS) found in the outer membrane of G-ve bacteria.17,28,29 Displacement of membrane-stabilizing divalent cations and insertion of the fatty acyl chain expands and weakens the bacterial outer membrane, causing cell leakage and allowing polymyxin entry.17,30,31 Once inside the cell, further modes of action have been postulated, including the generation of hydroxyl radical production32 and inhibition of the respiratory enzyme NADH-quinone oxidoreductase.33,34
Unfortunately, resistance to the polymyxins is appearing and has been identified in most major pathogens, including A. baumannii, K. pneumoniae, and P. aeruginosa.17 The main mechanisms of resistance involve alterations to LPS, in particular via addition of phosphoethanolamine and/or l-4-amino-arabinose to Lipid A, which lowers the binding affinity with the Dab residues of the polymyxins due to reduction of negative charge.17,35,36 This increase in resistance, coupled with the growing importance of polymyxins in treating highly drug-resistant G-ve infections, has led to a number of efforts in recent years to prepare polymyxin analogues to understand structure–activity relationships, improve potency, reduce nephrotoxicity, and overcome resistance. Despite their length of time in use, there have been relatively few structure–activity relationship (SAR) studies conducted to date compared to other classes of antibiotics, with most publications focusing on the readily modified exocyclic tail (Pfizer,37−39 Novacta Biosystems Ltd./Cab-Tab Anti-Infectives Ltd.,40−42 Northern Antibiotics,43−45 Sakura et al.46) or selective/nonselective modifications of the multiple Dab side chains (Northern Antibiotics,43−45 Pfizer,37−39,47 and Schering-Plough48). Biosource Pharm Inc./Cubist developed a method for fatty acid deacylation and linear tail amino acid hydrolysis, followed by reconstruction of the linear chain.49−54 From this study, CB182,80455 entered Phase I clinical trials, but development was discontinued in 2009. In one of the few studies to investigate changes within the polymyxin ring system, Li et al. at Monash University patented a set of polymyxin derivatives that retain activity against polymyxin-resistant strains; this group centered their efforts on the variation of hydrophobicity at positions 6 and 7 (numbering as in Figure 1).56−58 Early efforts at polymyxin SAR were summarized by Storm et al. in 1977,59 while more recent developments were reviewed by Velkov et al. in 201030 and Vaara in 2013.60
A set of novel polymyxin derivatives developed by Northern Antibiotics, with the exocyclic Dab-1 residue removed/substituted and the fatty acid tail altered, were found to be less toxic than polymyxin B61 based on cytotoxicity testing in the porcine renal proximal tubular cell line LLC-PK1. It was proposed that this was due to a reduction in the number of positive charges coupled with alteration of hydrophobicity of the fatty acid. Based on these results and the improved activity of the Monash compounds, we decided to investigate the effects of alterations at other positions within the polymyxins, with a particular focus on modifying the basic Dab residues contained within the ring to alter their basicity. Given that the Dab residues are proposed to be integral to binding to Lipid A and that mutation of Lipid A to introduce phosphoethanolamine is a key polymyxin resistance mechanism, we also wanted to examine whether introducing zwitterionic residues or substituents capable of hydrogen-bonding at these positions might help overcome resistance.
Twenty-six new analogues were prepared and assayed. In addition, six key compounds from the literature were also prepared to provide a systematic SAR comparison assessed under common conditions. Some of the compounds retained better antimicrobial activity against polymyxin-resistant strains compared to polymyxin B and colistin. Most importantly, some of the most promising compounds showed less cytotoxicity than polymyxin B and colistin when tested in mammalian cell lines, and less toxicity than polymyxin B, colistin, and gentamicin in a cytotoxicity assay potentially predictive of nephrotoxicity as measured by LDH and GGT release from human primary kidney cells.
Results and Discussion
Chemistry
A total of 32 compounds was synthesized (six of these have been reported previously46−48,53,54,57,58) with the structures presented in Tables 1 and S1 (Supporting Information). All compounds possessed >95% purity, as determined by LCMS analysis using both ELSD and UV (210 nm) detection. The synthetic routes toward the final compounds in this library are summarized in Schemes 1, 2, and S2 (Supporting Information). The first solid-phase total synthesis of polymyxin B1 was developed in 1999 by Sharma62 using Fmoc-Thr(tBu)-SASRIN resin, with the linear peptide cleaved from the resin and cyclized in solution. In the current study we present a new synthetic route to the polymyxins employing an on-resin cyclization that allows for alteration of any substituent position other than Thr-10 (see Scheme 1 for a representative synthesis of 10). The first step toward this library of compounds was the O-allyl ester protection of the C-terminal amino acid Fmoc-l-Thr-OH 3 (Thr10 in the final product). The side-chain alcohol of the protected amino acid 4 was then loaded onto a dihydropyran DHP HM resin (3,4-dihydro-2H-pyran-2-yl-methoxymethyl polystyrene). The peptide sequence was constructed by standard solid-phase peptide synthesis (SPPS) using sequential Fmoc deprotection followed by amino acid attachment using a systematic double coupling protocol, including a capping step with acetic anhydride and pyridine (50:50 v/v) to avoid the formation of deletion products due to unreacted free amino groups. On-resin cyclization was conducted following incorporation of the Fmoc-Dab(Alloc) residue by in situ deprotection of both Thr10 allyl ester and the Dab-4 side chain amine Nγ-Alloc protecting groups (Scheme 1, step vii), followed by overnight cyclization using DPPA and DIPEA in DMF at room temperature. Fmoc deprotection of the cyclized product then allowed completion of the exocyclic linear tail by further amino acid couplings. Following each step, an analytical sample of resin was cleaved with TFA/Et3SiH/H2O (95:1:4) followed by LCMS analysis to confirm the coupling with the corresponding amino acid. The final coupling capped the peptides with the appropriate fatty acid. Cleavage of the cyclic decapeptides from the resin with concomitant side chain deprotection was achieved with the addition of a solution of TFA/Et3SiH/H2O (95:1:4) for 30 min at room temperature, providing crude compounds that were then HPLC-purified to afford the final polymyxin analogue TFA salts as white powders; total yields were generally between 5 and 30% based on the initial resin loading.
Table 1. Compound Structures.


All amino acids are l-isomers unless otherwise indicated. Pmx-B: polymyxin B.
11 = polymyxin B3.
12 = compound 5 in O’Dowd et al.47
13 = compound 5 in Leese et al.54
15 = compound 18 in Leese et al.53
18 is similar to compound 24 (retains PmxB mixture of fatty acids) in Weinstein et al.48
28 = dl-OctGly version of l-Ade compound 2 in Li et al.57
30 = dl-OctGly version of d-Ade compound 1 in Li et al.58
Scheme 1. General On-Resin Cyclization Synthetic Route Exemplified by Synthesis of 10.

Reagent and conditions: (i) Cs2CO3, MeOH, H2O, pH 7-8; (ii) allyl bromide, dry DMF; (iii) DHP polystyrene resin, PPTS, dry DCE; (iv) 30% piperidine, DMF; (v) solid-phase peptide synthesis (SPPS) with corresponding amino acid (Table S3), HCTU, DIPEA; (vi) acetic anhydride, pyridine; (vii) Pd(PPh3), PhSiH3; (viii) DPPA, DIPEA, DMF; (ix) 4-Ph-PhCO2H, HCTU, DIPEA; (x) TFA/Et3SiH/H2O (95:1:4).
Scheme 2. General On-Resin Cyclization Synthetic Route with Selective Dab Modification Used for Synthesis of Compounds 18, 19, 22, 23, 31, and 32.

Reagent and conditions: (iv) 30% piperidine, DMF; (v) solid-phase peptide synthesis (SPPS) with corresponding amino acid (Table S3), HCTU, DIPEA; (vi) acetic anhydride, pyridine; (vii) Pd(PPh3), PhSiH3; (viii) DPPA, DIPEA, DMF; (ix) n-C7CO, HCTU, DIPEA; (x) 2% H2NNH2 in DMF; (xi) peptide coupling with corresponding amino acid: Boc-l-Arg(Pbf)-OH or Boc-l-Glu(OtBu)-OH, HCTU, DIPEA; (xii) TFA/Et3SiH/H2O (95:1:4).
For some syntheses, an orthogonal protecting group was employed for one of the multiple Dab residues, allowing for selective modification of individual Dab side chains. Thus, side-chain ivDde-protected Dab was used at positions P5, P8, or P9 (see Scheme 2 for generic route; Scheme S1 for explicit synthesis of 18 and 19). The P4 Alloc-protected Dab was deprotected and used for cyclization as before, with the fully protected resin-bound peptide treated with 2% hydrazine to selectively unmask the desired Dab residue. This was then derivatized by coupling with Boc-Arg(Pbf)-OH or Boc-Glu(OtBu)-OH, with global deprotection and purification as previously described.
Biological Activity
The 32 compounds, along with vancomycin, gentamicin, colistin, and polymyxin B, were assessed against a panel of predominantly G-ve bacteria, with S. aureus used as a representative G+ve strain. Five antibiotic-sensitive and resistant ATCC reference strains covering the G-ve ESKAPE pathogens (E. coli, K. pneumoniae, A. baumannii, and P. aeruginosa) were used, along with polymyxin-resistant clinical isolates of K. pneumoniae, A. baumannii, and P. aeruginosa (Table S2). Notably, compounds that were resyntheses of those previously reported demonstrated similar activity to the earlier literature values (footnotes in Tables 2 and 3). Counter-screening for general cytotoxicity was conducted using human liver hepatocellular carcinoma cells (HepG2). Our previous studies have demonstrated that cytotoxicity measurements against these immortalized cells were not predictive of nephrotoxicity,63,64 but they do provide an indication of potential general cell toxicity.
Table 2. Summary of MIC against a Panel of Sensitive and Resistant Bacteria and Cytotoxicity (CC50) against HepG2 Cells for a Set of Compounds with Diversity in the Exocyclic Chain and Fatty Acid Tail.
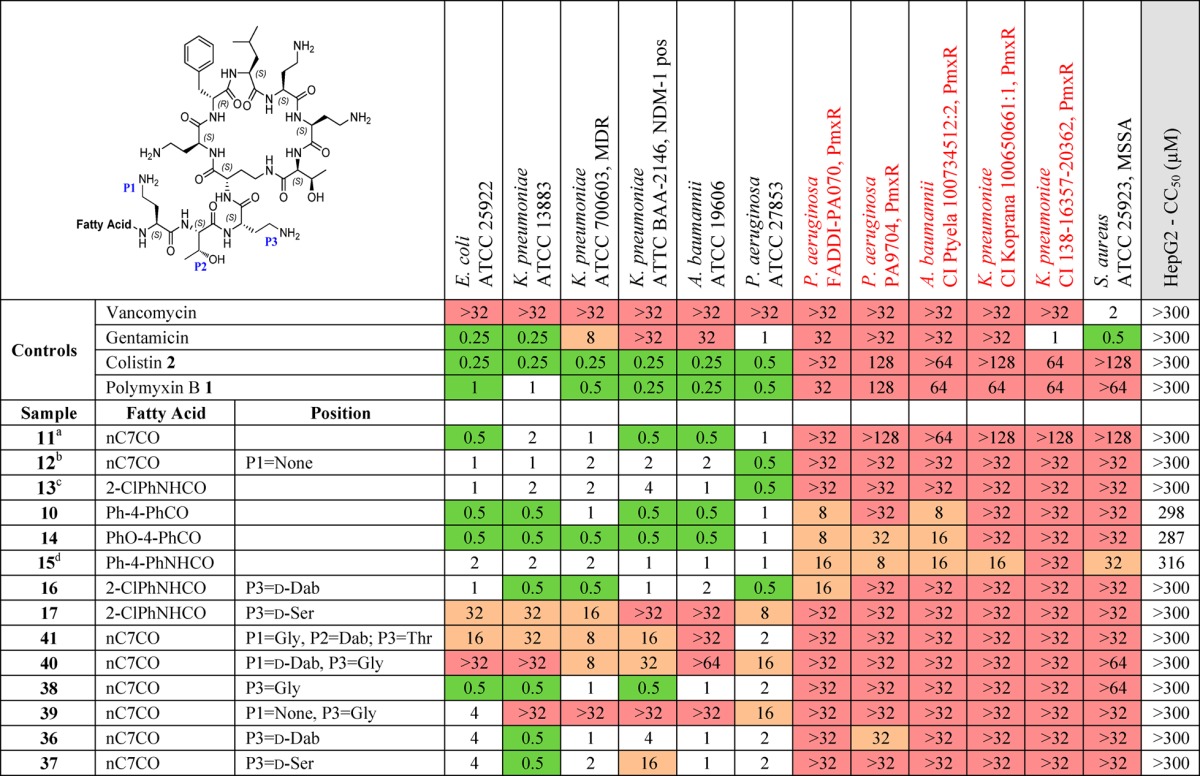
All amino acids are l-isomers unless otherwise indicated. ND: not determined. PmxR: polymyxin resistant. CI: clinical isolate. MSSA: methicillin sensitive S. aureus. MICs were determined visually after 18 h of incubation at 37 °C, with the MIC defined as the lowest compound concentration at which no bacterial growth was visible. MIC in μg/mL. 2-Chlorophenyl carbamoyl (2-ClPhNHCO), 4-biphenyl-benzoyl (Ph-4-PhCO), 4-phenoxybenzoyl (PhO4-PhCO), 4-biphenyl carbamoyl (Ph-4-PhNHCO), octanoyl (nC7CO).
11 is polymyxin B3. Literature MIC E. coli = 0.5 and P. aeruginosa = 1.0 nmol/mL. E. coli IFO12734, and P. aeruginosa IFO3080 from the Institute for Fermentation, Osaka, Japan.46
12 is compound 5 in O’Dowd et al.; MIC E. coli = 0.03, P. aeruginosa = 4, S. aureus = >16 μg/mL.47
13 is compound 5 in Leese et al.; MIC90 P. aeruginosa (100 strains) = 2, A. baumannii (81 strains) = 4, E. coli (80 strains) = 2, K. pneumoniae (81 strains) = 4 μg/mL.54
15 is compound 18 in Leese et al.; MIC E. coli = 2.5, P. aeruginosa ATCC 27853 = 2.5 μg/mL; MIC values were determined by serial 2-fold broth dilution method.53
Table 3. Summary of MIC against a Panel of Sensitive and Resistant Bacteria and Cytotoxicity (CC50) against HepG2 Cells for a Set of Compounds with Diversity in the Heptapeptide Ring.
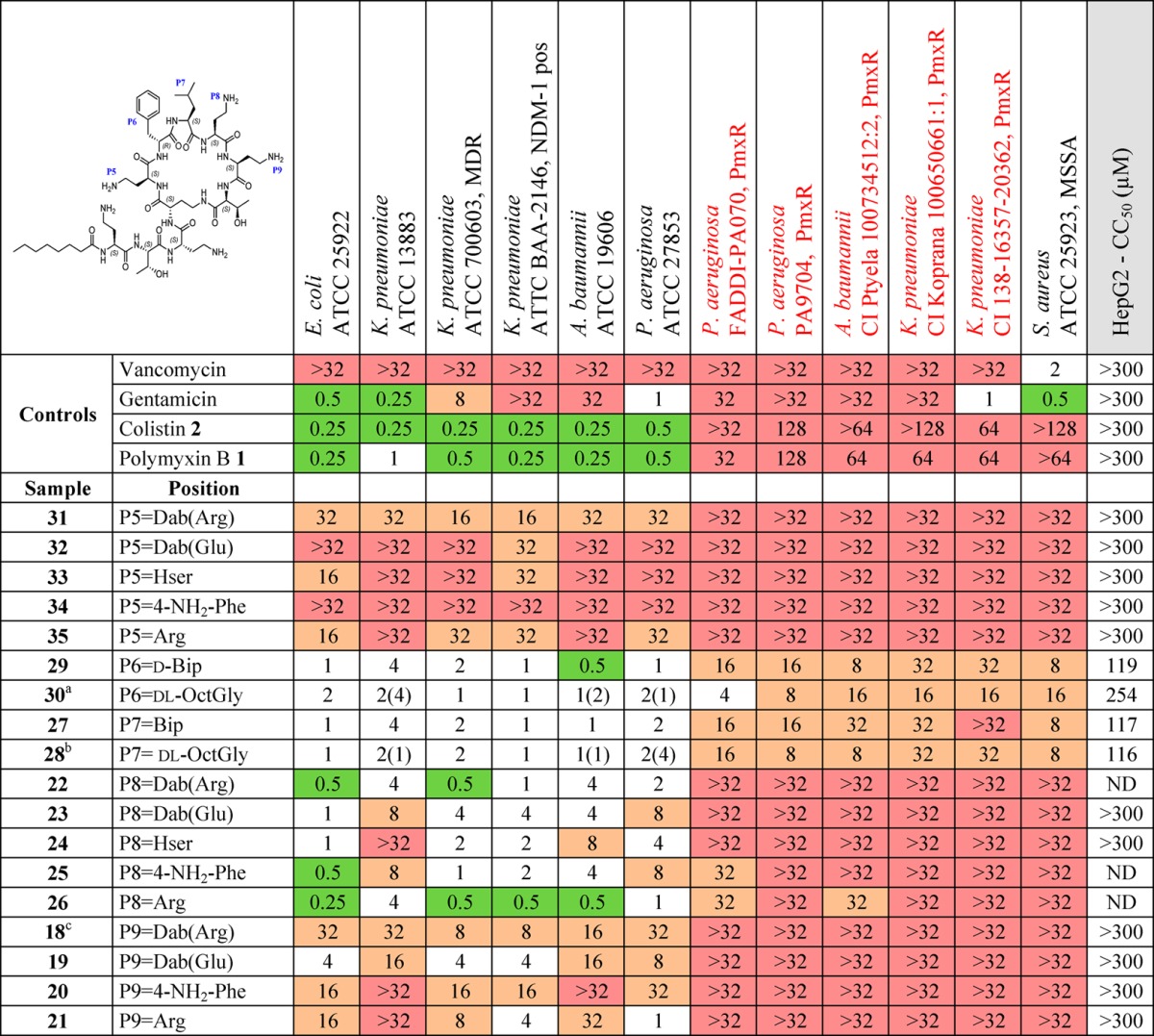
All amino acids are l-isomers unless otherwise indicated. ND: not determined. PmxR: polymyxin resistant. CI: clinical isolate. MSSA: methicillin resistant S. aureus. MICs were determined visually after 18 h of incubation at 37 °C, with the MIC defined as the lowest compound concentration at which no bacterial growth was visible. MIC in μg/mL. 2-Chlorophenyl carbamoyl (2-ClPhNHCO), 4-biphenyl-benzoyl (Ph-4-PhCO), 4-phenoxybenzoyl (PhO4-PhCO), 4-biphenyl carbamoyl (Ph-4-PhNHCO), octanoyl (nC7CO).
Values in parentheses are reported MIC for compound 2 = l-Ade version of 28 in Li et al.57
Values in parentheses are reported MIC for compound 1 = d-Ade version of 30 in Li et al.58
18 similar to compound 24 (retains polymyxin B fatty acid) in Weinstein et al.; MIC E. coli = 0.16, P. aeruginosa = 0.03, S. aureus = 2 μg/mL.48
Alterations to the fatty acid tail (Table 2) had minimal effect on activity against the reference strains, but biphenyl or biphenyl ether substituents (10, 14, and 15) did promote improved activity against polymyxin-resistant strains, albeit accompanied by greater cytotoxicity. In the linear exocyclic peptide tail (Table 2), replacement of Dab-3 with d-Dab (36, 16) or Gly (38) had minimal effect. Analogue 38 is particularly interesting as it retained MIC activity among the reference strains (0.5–2 μg/mL for most strains) with low cytotoxicity seen in HepG2 cells (CC50 > 300 μM). This is the only polymyxin B example where Dab-3 has been replaced by a nonbasic amino acid, where the amino acid replacement also potentially impacts on the final structural geometry. The effects of substitution of Dab-3 with d-Ser depended on the fatty acid; with the heptanoic acid 37 activity was slightly decreased (vs 11), but with o-chlorophenylurea 17 most activity was lost. The inversion of l-Dab-1 to d-Dab-1 in 40 abrogated most activity, a much larger effect than seen with the inversion of l-Dab-3 to d-Dab-3 in 36/16. Finally, replacing the linear chain Dab-Thr-Dab sequence with Gly-Dab-Thr in 41 was also detrimental to antibacterial activity.
For alterations within the heptapeptide ring system (Table 3), replacement of d-Phe/d-Leu-6 or Leu-7 with the more lipophilic residues dl-Ade or Bip/d-Bip (27–30) replicated the results of Velkov et al.,56 where these substitutions restored activity against polymyxin-resistant strains. However, these changes also caused increased cytotoxicity against HepG2 cell lines. The effects of altering the ring Dab residues varied significantly depending on which Dab was altered. Replacement of Dab-5 with homoserine (Hse), Arg, or 4′-amino-Phe (33–35) or acylation with Arg or Glu (31, 32), almost completely abolished activity (16 to >32 μg/mL against all tested strains). The same modifications at Dab-9 (18–21) gave more variable results, retaining moderate levels of activity (4–16 μg/mL for most strains). The Dab-8 position was most tolerant of alterations, with even neutral Hse 24 and zwitterionic Dab(Glu) 23 retaining good activity (1–8 μg/mL for most strains). Unfortunately, the hypothesis that a zwitterionic substituent might overcome Lipid A resistance modifications was not correct, with all of the Dab(Glu) substitutions inactive against the polymyxin-resistant strains. The Dab-8 replacement with Arg-8 26 (Table 3) had the best activity of all the analogues made and showed some activity against the polymyxin-resistant strains. While not as potent as the Arg-8 26 analogue, the 4′-amino-Phe-8 analogue 25 also showed good activity; this is an interesting result due to the aromatic nature of the replacement for the otherwise aliphatic amino acid Dab. Further investigation to elaborate on the SAR trends observed with these type of compounds is in progress.
Nephrotoxicity
Traditional cytotoxicity assays have been confirmed as improper indicators for nephrotoxicity screening.63,64 Therefore, in addition to general cytotoxicity screening assessed using HepG2 cells, we selected four compounds with promising antimicrobial potency and reasonable cytotoxicity against the HepG2 cell lines for further assessment for toxicity using primary kidney cells freshly isolated from human kidneys (see Table 4). This assay, measuring LDH and GGT release, has been reported to be a better in vitro model for predicting in vivo nephrotoxicity.63−65 We also assessed cytotoxicity against immortalized HEK293 cells. Colistin, polymyxin B, and gentamicin were employed as control nephrotoxic compounds and clearly showed the greatest toxicity in both primary renal cell assay readouts (Figure 2A,B), in contrast to their apparent lack of cytotoxicity in the HepG2/HEK293 assays. Compounds 10 and 14, with nonalkyl fatty acids, had shown greater cytotoxicity than polymyxin B and colistin, but induced almost no release of LDH or GGT at 100 μg/mL in the primary kidney cells. Compound 38 was the least cytotoxic (HepG2/HEK293 assays) of the four new analogues tested, but possessed intermediate levels of “nephrotoxicity” against primary kidney cells, as did 15 which had similar cytotoxicity against HepG2 cells as 10 and 14. The results support our previous assessment of in vitro predictors of nephrotoxicity, in that there is little correlation between general cytotoxicity measurements and the human kidney results.63,64
Table 4. Summary of MIC, Cytotoxicity CC50 Data (HepG2 and HEK293 Cell Lines) and Nephrotoxicity CC50 Data Measuring LDH (A) and GGT (B) Release from Primary Human Kidney Cells Using Lead Compounds (10, 14, 15, and 38) Compared to Control Compounds (Colistin, Polymyxin B, and Gentamicin).
| sample | K. pneumoniae ATCC 13883 | K. pneumoniae ATCC 700603, MDR | K. pneumoniae ATTC BAA-2146, NDM-1 pos | HepG2 CC50 (μM) | HEK293 CC50 (μM) | LDH release CC50 (μg/mL) | GGT release CC50 (μg/mL) |
|---|---|---|---|---|---|---|---|
| gentamicin | 0.25 | 8 | >32 | >300 | >300 | 33 | 33 |
| 1 polymyxin B | 1 | 0.5 | 0.25 | >300 | >300 | 23 | 177 |
| 2 colistin | 0.25 | 0.25 | 0.25 | >300 | >300 | 25 | 48 |
| 10 | 0.5 | 1 | 0.5 | 298 | 297 | >128 | >128 |
| 14 | 0.5 | 0.5 | 0.5 | 287 | 296 | >128 | >128 |
| 15 | 2 | 2 | 1 | 316 | >300 | >128 | >128 |
| 38 | 0.5 | 1 | 0.5 | >300 | >300 | 122 | >128 |
Figure 2.

In vitro nephrotoxicity studies measuring LDH (A) and GGT (B) release from primary human kidney cells using lead compounds (10, 14, 15, and 38) compared to control compounds (colistin, polymyxin B, and gentamicin).
Conclusions
A new solid-phase synthesis of the polymyxin class of antibiotics has been developed based on attachment to the resin through the side chain of Thr-10, allowing for on-resin cyclization, variation at any other position, and selective derivatization of the side chains of individual Dab residues. Thirty-two analogues were prepared and tested against a panel of reference and clinical isolates, including polymyxin-resistant MDR strains. Cytotoxicity screening was performed against the HepG2 cell line. The results confirmed that more lipophilic groups at P6/P7 improved activity against polymyxin-resistant strains but increased cytotoxicity against mammalian cells. Similar effects were observed by replacement of the alkyl fatty acid with biaryl substituents. Further testing of these compounds by assessing cytotoxicity in primary human renal cells as a potential surrogate for nephrotoxicity conversely found reduced toxicity compared to the increased general cytotoxicity seen in an immortalized cell line. Large differences were found when varying the five free side-chain Dab residues: Dab-5 could not be replaced or altered without loss of activity, while alterations at Dab-9 also severely affected potency, though to a slightly less extent. In contrast Dab-8 tolerated a range of basic, neutral, or zwitterionic replacements. Similarly, Dab-3 could be replaced by nonbasic Gly or Ser with minimal effects, while inversion of the configuration of Dab-1 abolished activity. Further studies of this class of compounds will investigate combinations of promising substituent alterations, particularly those that reduce the overall basic character, improve activity against polymyxin-resistant bacteria and demonstrate reduced potential for nephrotoxicity in human kidney cells.
Experimental Section
Synthetic Materials and Methods
Experimental procedures are described in the Supporting Information. All chemicals were obtained from commercial suppliers and used without further purification. LC–MS analyses were conducted using Agilent Technologies 1200 Series Instrument with a G1316A variable wavelength detector set at λ = 210 nm, 1200 Series ELSD, 6110 quadrupole ESI–MS, using an Agilent Eclipse XDB-Phenyl column (3 × 100 mm, 3.5 μm particle size, flow rate 1 mL/min, the mobile phases 0.05% formic acid in water and 0.05% formic acid in acetonitrile). Compound purification was done using a Agilent 1260 Infinity Preparative HPLC with a G1365D multiple wavelength detector set at λ = 210 nm and an Agilent Eclipse XDB-Phenyl column 21.2 × 100 mm, 5 μm particle size. Ultraviolet/visible spectra were recorded with a Varian Cary 50 Bio spectrophotometer. Identities of final products were confirmed by MS/MS spectra, obtained using an API QSTAR Pulsar Hybrid LC–MS/MS System, high resolution mass spectrometry (HRMS), performed on a Bruker Micro TOF mass spectrometer using (+)-ESI calibrated to sodium formate, and by 1H (600 MHz) and 2D NMR spectra, obtained using a Bruker Avance-600 spectrometer equipped with a TXI cryoprobe in D2O, referenced externally with NaOAc (δH 1.90 and 8.44; 10 mg in 500 μL D2O) and then internally with the HDO resonance at δ 4.77.66 Final purity of more than 95% for all compounds was confirmed by LC–MS analysis using both ELSD and UV (210 nm) detection.
Minimum Inhibitory Concentration (MIC) Determination
Bacteria were either obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) or independent academic clinical isolate collections, as listed in Table S2. Bacteria were cultured in nutrient broth (NB; Bacto Laboratories, catalog no. 234000) or Muller Hinton broth (MHB; Bacto Laboratories, catalog no. 211443) at 37 °C overnight with shaking (∼180 rpm). A sample of each culture was then diluted 50-fold in fresh MHB and incubated at 37 °C for 1.5–3 h with shaking (∼180 rpm). Compound stock solutions were prepared as 0.64 or 2.56 mg/mL in water. The compounds, at twice the final desired concentration, were serially diluted 2-fold across the wells of 96-well plates (Polystyrene, Corning, catalogue No. 3370). Mid log phase bacterial cultures (after 1.5–3 h incubation) were diluted to a final concentration of 5 × 105 colony forming units (CFU)/mL, and 50 μL was added to each well giving a final compound concentration range of 32 μg/mL to 0.015 μg/mL (128–0.06 μg/mL for colistin and polymyxin B against the polymyxin resistant clinical isolates). MICs were determined visually after 18 h of incubation at 37 °C, with the MIC defined as the lowest compound concentration at which no bacterial growth was visible.
Cytotoxicity Assay (Resazurin Assay)
Cytotoxicity to HEK293 and HepG2 cells was determined using the resazurin assay.67,68 In brief, HEK293 and HepG2 cells were seeded at 4000 cells/well in black-wall, clear-bottom 384-well plates (Corning, Australia) and incubated for 24 h at 37 °C, 5% CO2. Compounds were then added into each well. After 24 h incubation, 5 μM resazurin was added per well and incubated at 37 °C for 2 h. Fluorescence intensity was then read using a Polarstar Omega with excitation/emission 560/590. The data were then analyzed using GraphPad Prism 6 software. CC50 values were determined using GraphPad Prism 6 software (Inc. La Jolla, CA).
Nephrotoxicity Assay (Lactate Dehydrogenase (LDH) and Gamma Glutamyl Transferase (GGT) Release in Primary Cells Freshly Isolated from Human Kidneys)
Primary human renal proximal tubule epithelial (hPT) cells were isolated and used for in vitro prediction of drug-induced nephrotoxicity63,64,69 by measuring the release of metabolism enzymes, LDH and GGT, as previously described.70
Acknowledgments
M.A.C. is an NHMRC Principal Research Fellow (APP1059354). Research conducted by A.G.G., M.S.B., A.G.E., A.M.K., S.R., J.X.H., M.A.T.B., and M.A.C. was supported by NHMRC grants APP1005350 and APP1045326 and NIH grant R21AI098731/R33AI098731. M.S.B., J.X.H., and M.A.T.B. were supported by a Wellcome Trust Seeding Drug Discovery Award (094977/Z/10/Z). L.L. is supported by NIH grant R21AI098731/R33AI098731. We thank Wang Hengzhuang (University Hospital of Copenhagen, Denmark), Ilias Karaiskos, and Helen Giamarellou (sixth Dept. of Internal Medicine, Hygeia General Hospital, Athens, Greece), Cely S. Abboud (Instituto Dante Pazzanese de Cardiologia, São Paulo, Brazil), and Roger L. Nation (Institute of Pharmaceutical Sciences, Monash University, Australia) for providing the Gram-negative clinical isolates for antimicrobial testing. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Glossary
Abbreviations Used
- ATCC
American Type Culture Collection
- Dab
2,3-diaminobutyric acid
- DIPEA
diisopropylethylamine
- DMF
dimethylformamide
- E. coli
Escherichia coli
- GGT
γ-glutamyltransferase
- G-ve
Gram-negative
- G+ve
Gram-positive
- HCTU
(2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate)
- HepG2
human liver hepatocellular carcinoma cells
- HEK293
human embryonic kidney 293
- HK2
human kidney proximal tubule epithelial cell line
- ivDde
1-(4,4-dimethyl-2,6-dioxocyclohexylidene)-3-methylbutyl
- LDH
lactate dehydrogenase
- P. aeruginosa
Pseudomonas aeruginosa
- S. aureus
Staphylococcus aureus
- S. pyogenes
Streptococcus pyogenes
- SPPS
solid-phase peptide synthesis
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01593.
The authors declare no competing financial interest.
Supplementary Material
References
- Hall M. J.; Williams S. N.; DeFrances C. J.; Golosinskiy A. Inpatient care for septicemia or sepsis: a challenge for patients and hospitals. NCHS Data Brief 2011, 1–8. [PubMed] [Google Scholar]
- Lagu T.; Rothberg M. B.; Shieh M. S.; Pekow P. S.; Steingrub J. S.; Lindenauer P. K. Hospitalizations, costs, and outcomes of severe sepsis in the United States 2003 to 2007. Crit. Care Med. 2012, 40, 754–761. 10.1097/CCM.0b013e318232db65. [DOI] [PubMed] [Google Scholar]
- Vincent J. L.; Marshall J. C.; Namendys-Silva S. A.; Francois B.; Martin-Loeches I.; Lipman J.; Reinhart K.; Antonelli M.; Pickkers P.; Njimi H.; Jimenez E.; Sakr Y.; investigators I. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir. Med. 2014, 2, 380–6. 10.1016/S2213-2600(14)70061-X. [DOI] [PubMed] [Google Scholar]
- Torio C. M.; Andrews R. M.. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2011: Statistical Brief #160. In Healthcare Cost and Utilization Project (HCUP) Statistical Briefs, Rockville, MD, 2011. [Google Scholar]
- Opal S. M.; Garber G. E.; LaRosa S. P.; Maki D. G.; Freebairn R. C.; Kinasewitz G. T.; Dhainaut J. F.; Yan S. B.; Williams M. D.; Graham D. E.; Nelson D. R.; Levy H.; Bernard G. R. Systemic host responses in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated). Clin. Infect. Dis. 2003, 37, 50–58. 10.1086/375593. [DOI] [PubMed] [Google Scholar]
- Butler M. S.; Blaskovich M. A.; Cooper M. A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 2013, 66, 571–591. 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- Benedict R. G.; Langlykke A. F. Antibiotic activity of Bacillus polymyxa. J. Bacteriol. 1947, 54, 24. [PubMed] [Google Scholar]
- Stansly P. G.; Schlosser M. E. Studies on polymyxin: Isolation and identification of Bacillus polymyxa and differentiation of polymyxin from certain known antibiotics. J. Bacteriol. 1947, 54, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansly P. G.; Shepherd R. G.; White H. J. Polymyxin: a new chemotherapeutic agent. Bull. Johns Hopkins Hosp. 1947, 81, 43–54. [PubMed] [Google Scholar]
- Ainsworth G. C.; Brown A. M.; Brownlee G. Aerosporin, an antibiotic produced by Bacillus aerosporus Greer. Nature 1947, 159, 263. 10.1038/160263a0. [DOI] [PubMed] [Google Scholar]
- Shoji J.; Hinoo H.; Wakisaka Y.; Koizumi K.; Mayama M.; Matsuura S. Isolation of two new polymyxin group antibiotics. Studies on antibiotics from the genus Bacillus. XX. J. Antibiot. 1977, 30, 1029–1034. 10.7164/antibiotics.30.1029. [DOI] [PubMed] [Google Scholar]
- Shoji J.; Kato T.; Hinoo H. The structure of polymyxin S. Studies on antibiotics from the genus Bacillus. XXI. J. Antibiot. 1977, 30, 1035–1041. 10.7164/antibiotics.30.1035. [DOI] [PubMed] [Google Scholar]
- Wilkinson S.; Lowe L. A. Structures of the polymyxins A and the question of identity with the polymyxins M. Nature 1966, 212, 311. 10.1038/212311a0. [DOI] [PubMed] [Google Scholar]
- Parker W. L.; Rathnum M. L.; Dean L. D.; Nimeck M. W.; Brown W. E.; Meyers E. Polymyxin F, a new peptide antibiotic. J. Antibiot. 1977, 30, 767–769. 10.7164/antibiotics.30.767. [DOI] [PubMed] [Google Scholar]
- Shoji J.; Kato T.; Hinoo H. The structure of polymyxin T1. Studies on antibiotics from the genus Bacillus. XXII. J. Antibiot. 1977, 30, 1042–1048. 10.7164/antibiotics.30.1042. [DOI] [PubMed] [Google Scholar]
- Stansly P. G. The polymyxins; a review and assessment. Am. J. Med. 1949, 7, 807–818. 10.1016/0002-9343(49)90419-2. [DOI] [PubMed] [Google Scholar]
- Velkov T.; Roberts K. D.; Nation R. L.; Thompson P. E.; Li J. Pharmacology of polymyxins: new insights into an ’old’ class of antibiotics. Future Microbiol. 2013, 8, 711–724. 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadar B.; Kocsis B.; Nagy K.; Szabo D. The renaissance of polymyxins. Curr. Med. Chem. 2013, 20, 3759–3773. 10.2174/09298673113209990185. [DOI] [PubMed] [Google Scholar]
- Nation R. L.; Velkov T.; Li J. Colistin and polymyxin B: peas in a pod, or chalk and cheese?. Clin. Infect. Dis. 2014, 59, 88–94. 10.1093/cid/ciu213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassamali Z.; Rotschafer J. C.; Jones R. N.; Prince R. A.; Danziger L. H. Polymyxins: wisdom does not always come with age. Clin. Infect. Dis. 2013, 57, 877–883. 10.1093/cid/cit367. [DOI] [PubMed] [Google Scholar]
- Nation R. L.; Li J. Colistin in the 21st century. Curr. Opin. Infect. Dis. 2009, 22, 535–543. 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas S.; Brunel J. M.; Dubus J. C.; Reynaud-Gaubert M.; Rolain J. M. Colistin: an update on the antibiotic of the 21st century. Expert Rev. Anti-Infect. Ther. 2012, 10, 917–934. 10.1586/eri.12.78. [DOI] [PubMed] [Google Scholar]
- Spapen H.; Jacobs R.; Van Gorp V.; Troubleyn J.; Honore P. M. Renal and neurological side effects of colistin in critically ill patients. Ann. Intensive Care 2011, 1, 14. 10.1186/2110-5820-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlos C. P.; Mendes G. E.; Miquelin A. R.; Luz M. A.; da Silva C. G.; van Rooijen N.; Coimbra T. M.; Burdmann E. A. Macrophage depletion attenuates chronic cyclosporine A nephrotoxicity. Transplantation 2010, 89, 1362–1370. 10.1097/TP.0b013e3181da0587. [DOI] [PubMed] [Google Scholar]
- Sorli L.; Luque S.; Grau S.; Berenguer N.; Segura C.; Montero M. M.; Alvarez-Lerma F.; Knobel H.; Benito N.; Horcajada J. P. Trough colistin plasma level is an independent risk factor for nephrotoxicity: a prospective observational cohort study. BMC Infect. Dis. 2013, 13, 380. 10.1186/1471-2334-13-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation R. L.; Li J.; Cars O.; Couet W.; Dudley M. N.; Kaye K. S.; Mouton J. W.; Paterson D. L.; Tam V. H.; Theuretzbacher U.; Tsuji B. T.; Turnidge J. D. Framework for optimization of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect. Dis. 2015, 15, 225–234. 10.1016/S1473-3099(14)70850-3. [DOI] [PubMed] [Google Scholar]
- Brink A. J.; Richards G. A.; Colombo G.; Bortolotti F.; Colombo P.; Jehl F. Multicomponent antibiotic substances produced by fermentation: implications for regulatory authorities, critically ill patients and generics. Int. J. Antimicrob. Agents 2014, 43, 1–6. 10.1016/j.ijantimicag.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Pristovsek P.; Kidric J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: an NMR and molecular modeling study. J. Med. Chem. 1999, 42, 4604–4613. 10.1021/jm991031b. [DOI] [PubMed] [Google Scholar]
- Newton B. A. The properties and mode of action of the polymyxins. Bacteriol. Rev. 1956, 20, 14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T.; Thompson P. E.; Nation R. L.; Li J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R. E.; Chapple D. S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson T. R.; Liu X.; Schroeder M. R.; Kraft C. S.; Burd E. M.; Weiss D. S. Rapid killing of Acinetobacter baumannii by polymyxins is mediated by a hydroxyl radical death pathway. Antimicrob. Agents Chemother. 2012, 56, 5642–5649. 10.1128/AAC.00756-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deris Z. Z.; Akter J.; Sivanesan S.; Roberts K. D.; Thompson P. E.; Nation R. L.; Li J.; Velkov T. A secondary mode of action of polymyxins against Gram-negative bacteria involves the inhibition of NADH-quinone oxidoreductase activity. J. Antibiot. 2014, 67, 147–151. 10.1038/ja.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi T.; Murase Y.; Mori M.; Shiomi K.; Omura S.; Paranagama M. P.; Kita K. Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: quinone oxidoreductase from the Gram-positive bacterium Mycobacterium smegmatis. J. Biochem. 2009, 146, 491–499. 10.1093/jb/mvp096. [DOI] [PubMed] [Google Scholar]
- Beceiro A.; Llobet E.; Aranda J.; Bengoechea J. A.; Doumith M.; Hornsey M.; Dhanji H.; Chart H.; Bou G.; Livermore D. M.; Woodford N. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob. Agents Chemother. 2011, 55, 3370–3379. 10.1128/AAC.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. S.; Suzuki Y.; Jones M. B.; Marshall S. H.; Rudin S. D.; van Duin D.; Kaye K.; Jacobs M. R.; Bonomo R. A.; Adams M. D. Genomic and transcriptomic analyses of colistin-resistant clinical isolates of Klebsiella pneumoniae reveal multiple pathways of resistance. Antimicrob. Agents Chemother. 2015, 59, 536–543. 10.1128/AAC.04037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee T. V.; Brown M. F.; Starr J. T.; Ackley D. C.; Abramite J. A.; Aubrecht J.; Butler A.; Crandon J. L.; Dib-Hajj F.; Flanagan M. E.; Granskog K.; Hardink J. R.; Huband M. D.; Irvine R.; Kuhn M.; Leach K. L.; Li B.; Lin J.; Luke D. R.; MacVane S. H.; Miller A. A.; McCurdy S.; McKim J. M. Jr.; Nicolau D. P.; Nguyen T. T.; Noe M. C.; O’Donnell J. P.; Seibel S. B.; Shen Y.; Stepan A. F.; Tomaras A. P.; Wilga P. C.; Zhang L.; Xu J.; Chen J. M. Discovery of Dap-3 polymyxin analogues for the treatment of multidrug-resistant Gram-negative nosocomial infections. J. Med. Chem. 2013, 56, 5079–5093. 10.1021/jm400416u. [DOI] [PubMed] [Google Scholar]
- Magee T. V.; Chen J. M.; Martinez C. A.; Li Z. B. Polymyxin Derivates Useful As Antibacterial Agents. US20120316105, 2012.
- Chen J. M.; Magee T. V.; Martinez C. A. Polymyxin derivatives useful as antibacterial agents. WO/2012/168820, 2012.
- Saadi M.; Duperchy E.; Brown P.; Dawson M. J.; Wadman S. N. Polymyxin derivatives. WO/2013/072695, 2013.
- Saadi M.; Duperchy E.; Brown P.; Dawson M. J.; Wadman S. N. Compounds. US20150031602, 2015.
- Brown P.; Dawson M.; Simonovic M.; Boakes S.; Duperchy E. Polymyxin derivatives and their use in combination therapy together with different antibiotics. WO/2014/188178, 2014.
- Vaara M.; Fox J.; Loidl G.; Siikanen O.; Apajalahti J.; Hansen F.; Frimodt-Moller N.; Nagai J.; Takano M.; Vaara T. Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob. Agents Chemother. 2008, 52, 3229–3236. 10.1128/AAC.00405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaara M. S.; Vaara T. I. Polymyxin derivatives and uses thereof. US8329645, 2012.
- Vaara M. S.; Vaara T. I. Polymyxin derivatives and uses thereof. US20140134669, 2014.
- Sakura N.; Itoh T.; Uchida Y.; Ohki K.; Okimura K.; Chiba K.; Sato Y.; Sawanishi H. The contribution of the N-terminal structure of polymyxin B peptides to antimicrobial and lipopolysaccharide binding activity. Bull. Chem. Soc. Jpn. 2004, 77, 1915–1924. 10.1246/bcsj.77.1915. [DOI] [Google Scholar]
- O’Dowd H.; Kim B.; Margolis P.; Wang W.; Wu C.; Lopez S. L.; Blais J. Preparation of tetra-Boc-protected polymyxin B nonapeptide. Tetrahedron Lett. 2007, 48, 2003–2005. 10.1016/j.tetlet.2007.01.071. [DOI] [Google Scholar]
- Weinstein J.; Afonso A.; Moss E. Jr.; Miller G. H. Selective chemical modifications of polymyxin B. Bioorg. Med. Chem. Lett. 1998, 8, 3391–3396. 10.1016/S0960-894X(98)00612-X. [DOI] [PubMed] [Google Scholar]
- Leese R. A.; Francis N.; Curran W.; Borders D. B.; Jarolmen H. Peptide Antibiotics amd Methods for Making Same. WO/2006/083317, 2006.
- Curran W. V.; Liu C. M.; Bombardier A. C. D.; Leese R. A.; Hwang Y. S.; Lippa B. S.; Zhang Y.. Antibiotic compositions for the treatment of gram negative infections. US8415307, 2013.
- Curran W. V.; Liu C. M.; Bombardier A. C. D.; Leese R. A.; Hwang Y. S.; Lippa B. S.; Zhang Y. Antibiotic compositions for the treatment of gram negative infections. US20140073559, 2014.
- Curran W. V.; Liu C. M.; Bombardier A. C. D.; Leese R. A.; Hwang Y. S.; Lippa B. S.; Zhang Y. Antibiotic compositions for the treatment of gram negative infections. US8906866, 2014.
- Leese R. A.; Francis N.; Curran W. V.; Borders D. B.; Jarolmen H. Peptide antibiotics and methods for making same. US8889826B2, 2014.
- Leese R. A. Antibiotic compositions for the treatment of gram negative infections. US 8343912B2, 2013.
- Quale J.; Shah N.; Kelly P.; Babu E.; Backer M.; Rosas-Garcia G.; Salamera J.; George A.; Bratu S.; Landman D. Activity of polymyxin B and the novel polymyxin analogue CB-182,804 against cntemporary gram-negative pathogens in New York City. Microb. Drug Resist. 2012, 18, 132–136. 10.1089/mdr.2011.0163. [DOI] [PubMed] [Google Scholar]
- Velkov T.; Roberts K. D.; Nation R. L.; Wang J.; Thompson P. E.; Li J. Teaching ’old’ polymyxins new tricks: new-generation lipopeptides targeting gram-negative ’superbugs’. ACS Chem. Biol. 2014, 9, 1172–1177. 10.1021/cb500080r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Velkov T.; Nation R. L; Thompson P. E. Antimicrobial compounds. WO/2010/130007, 2010.
- Li J.; Velkov T.; Nation R. L.; Thompson P. E.; Roberts K. D. Antimicrobial compounds. WO/2012/051663, 2012.
- Storm D. R.; Rosenthal K. S.; Swanson P. E. Polymyxin and related peptide antibiotics. Annu. Rev. Biochem. 1977, 46, 723–763. 10.1146/annurev.bi.46.070177.003451. [DOI] [PubMed] [Google Scholar]
- Vaara M. Novel derivatives of polymyxins. J. Antimicrob. Chemother. 2013, 68, 1213–1219. 10.1093/jac/dkt039. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Tulkens P. M.; Denamur S.; Vaara T.; Vaara M. Novel polymyxin derivatives are less cytotoxic than polymyxin B to renal proximal tubular cells. Peptides 2012, 35, 248–252. 10.1016/j.peptides.2012.03.033. [DOI] [PubMed] [Google Scholar]
- Sharma S. K.; Wu A. D.; Chandramouli N.; Fotsch C.; Kardash G.; Bair K. W. Solid-phase total synthesis of polymyxin B1. J. Pept. Res. 1999, 53, 501–506. 10.1034/j.1399-3011.1999.00045.x. [DOI] [PubMed] [Google Scholar]
- Huang J. X.; Blaskovich M. A.; Cooper M. A. Cell- and biomarker-based assays for predicting nephrotoxicity. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1621–1635. 10.1517/17425255.2014.967681. [DOI] [PubMed] [Google Scholar]
- Huang J. X.; Kaeslin G.; Ranall M. V.; Blaskovich M. A.; Becker B.; Butler M. S.; Little M. H.; Lash L. H.; Cooper M. A. Evaluation of biomarkers for in vitro prediction of drug-induced nephrotoxicity: comparison of HK-2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol. Res. Perspect. 2015, 3, e00148. 10.1002/prp2.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lash L. H.; Putt D. A.; Zalups R. K. Biochemical and functional characteristics of cultured renal epithelial cells from uninephrectomized rats: factors influencing nephrotoxicity. J. Pharmacol. Exp. Ther. 2001, 296, 243–251. [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- McMillian M. K.; Li L.; Parker J. B.; Patel L.; Zhong Z.; Gunnett J. W.; Powers W. J.; Johnson M. D. An improved resazurin-based cytotoxicity assay for hepatic cells. Cell Biol. Toxicol. 2002, 18, 157–173. 10.1023/A:1015559603643. [DOI] [PubMed] [Google Scholar]
- O’Brien J.; Wilson I.; Orton T.; Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- Cummings B. S.; Lash L. H. Metabolism and toxicity of trichloroethylene and S-(1,2-dichlorovinyl)-l-cysteine in freshly isolated human proximal tubular cells. Toxicol. Sci. 2000, 53, 458–66. 10.1093/toxsci/53.2.458. [DOI] [PubMed] [Google Scholar]
- Lash L. H.; Putt D. A.; Cai H. Drug metabolism enzyme expression and activity in primary cultures of human proximal tubular cells. Toxicology 2008, 244, 56–65. 10.1016/j.tox.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.