

Abstract
A signaling pathway connects ovarian hormones to preparation of the mammalian uterus for pregnancy.
Reproduction in mammals is coordinated by an intricate sequence of events and signals that culminate in implantation of the embryo in the uterus (1–3). To succeed, these signals must be produced at the correct time, in the proper place, and in the amounts needed, and must be received by the appropriate targets. Perturbation of any of these factors can prevent establishment of pregnancy, resulting in infertility. On page 912 of this issue, Li et al. (4) show that the transcription factor Hand2 regulates signals that establish conditions in the mammalian uterus to support pregnancy.
Uterine tissue (see the figure) consists of an outer muscle layer (myometrium) which contracts during labor to facilitate delivery. The inner lumen is lined by an epithelial cell layer from which emanate glandular structures, also lined by epithelial cells. The embryo must attach to, and then invade through, the epithelial layer to establish a pregnancy, and the glands secrete factors essential to the process. Stromal cells between the outer and inner layers expand and differentiate into decidual cells to support fetal and placental development.
Figure. Implantation signals.
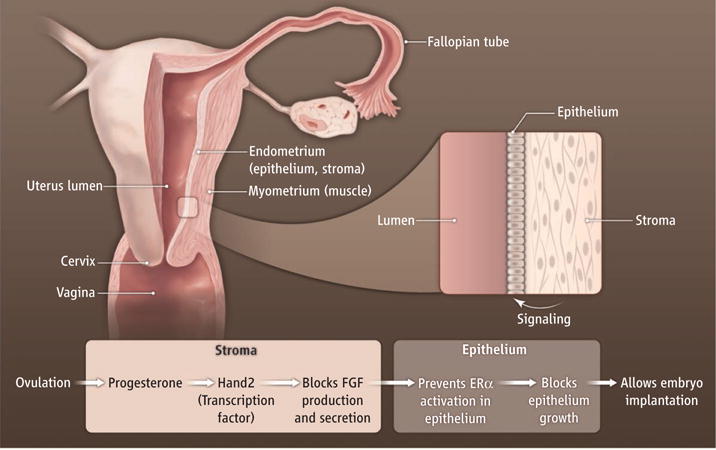
Stromal cells respond to progesterone during pregnancy by activating the transcription factor Hand2. This triggers a signaling cascade that blocks estrogen receptor (ERα) signaling in uterine epithelial cells, thereby creating an epithelium that is receptive for embryo implantation.
The ovarian hormones estrogen and progesterone have relatively complementary roles, reflecting their temporal and cyclical fluctuations. Estrogen production surges just before ovulation, and induces growth of the uterine epithelial cells that will come into contact with a developing embryo. Progesterone increases after ovulation and further prepares the endometrial tissue (the stromal and epithelial cells) for pregnancy by both dampening the proliferative effect of estrogen on the epithelial cells, and acting with estrogen to stimulate proliferation of stromal decidual tissues. This convergence of hormonal and other signals with the arrival of the embryo at the proper time is called the “implantation window.”
The responses of uterine cells to estrogen and progesterone are mediated in part by receptors that are transcription factors. The estrogen receptor (ER) is found in all uterine cell types, whereas progesterone receptor (PR) expression varies depending on the cell type and phase of the ovarian cycle.
As in earlier studies with PR-deficient mice (1, 3), Li et al. show that when progesterone is unable to decrease estrogen’s proliferative effect on the endometrial epithelium, pregnancy cannot be established. Disrupting this effect of progesterone is the basis for the estrogen-containing “morning after pill.” In certain clinical conditions, estrogen in the absence of progesterone (“unopposed estrogen”) may damage the endometrium. Hormone-replacement therapies that lack progesterone, or anovulatory conditions (such as polycystic ovarian syndrome) that lengthen the estrogenic portion of the menstrual cycle, are associated with increased incidence of endometrial cancers (5). However, the signaling pathways by which estrogen and progesterone successfully establish uterine receptivity have not been fully elucidated.
Estrogen, acting through ERα in the stromal cells, increases the production of paracrine factors, including insulin-like growth factor 1 (IGF-1), which then trigger the proliferation of epithelial cells (6, 7). Estrogen also maintains the epithelia by preventing programmed cell death of epithelial cells (8). These mechanisms have been established through numerous approaches including ablation of certain progesterone target genes (Ihh, Couptf2, Fkbp4, and Errfi1), leading to loss of uterine receptivity (3, 9, 10). Overall, these findings emphasize that proper progesterone-mediated regulation of multiple uterine stromal signals is critical for controlling the epithelial estrogenic responses that allow embryo implantation.
To identify the progesterone-regulated signaling pathways that underlie implantation, Li et al. used microarray gene profiling analysis of progesterone-responsive transcription at the implantation window in the mouse. Comparison of mRNA in receptive uterine tissue to that in tissue treated with the PR antagonist RU486 revealed altered expression of Hand2. Selective ablation of the Hand2 gene in uterine cells showed that the transcription factor plays a key role in establishing a receptive endometrium. Microarray analysis also revealed misregulation of signaling by fibroblast growth factor (FGF) in the uterine tissue of Hand2-deficient mice compared to that in normal mice. Previous studies had indicated a role for FGF signaling in ovine implantation (2), indicating that the mechanism may have broader application to other mammals. Additionally, expression of Fgf2 transcript in the stroma surrounding an implanting blastocyst was previously reported in mice (11). However, Li et al. link regulation of the FGF signals to progesterone regulation of Hand2, which helps to explain how progesterone establishes uterine receptivity.
Whether there is a general role for Hand2 and FGF signaling in establishing receptive endometria of other mammals remains to be determined. It will be important to evaluate whether perturbing this pathway might lead to adverse clinical outcomes, such as infertility, endometriosis, or malignancy. Indeed, mutations in the genes encoding FGF9 and an FGF receptor (FGFR2) are associated with endometrial cancer (12, 13). Whether mutations in the Hand2 gene can explain clinical dysfunction also will require further research.
Reproduction and endometrial function involve intricate interplay between different cell types that are too complex to adequately study with cell culture approaches. The findings of Li et al. relied on the development of elaborate in vivo models. Such approaches promise to open important new avenues of investigation into this critical window of receptivity.
References
- 1.Wang H, Dey SK. Nat Rev Genet. 2006;7:185. doi: 10.1038/nrg1808. [DOI] [PubMed] [Google Scholar]
- 2.Bazer FW, Spencer TE, Johnson GA, Burghardt RC, Wu G. Reproduction. 2009;138:195. doi: 10.1530/REP-09-0158. [DOI] [PubMed] [Google Scholar]
- 3.Lim HJ, Wang H. J Clin Invest. 2010;120:1004. doi: 10.1172/JCI41210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, et al. Science. 2011;331:912. doi: 10.1126/science.1197454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schindler AE. Maturitas. 2009;62:334. doi: 10.1016/j.maturitas.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 6.Cunha GR, Cooke PS, Kurita T. Arch Histol Cytol. 2004;67:417. doi: 10.1679/aohc.67.417. [DOI] [PubMed] [Google Scholar]
- 7.Murphy LJ, Ghahary A. Endocr Rev. 1990;11:443. doi: 10.1210/edrv-11-3-443. [DOI] [PubMed] [Google Scholar]
- 8.Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Proc Natl Acad Sci USA. 2010;107:19272. doi: 10.1073/pnas.1013226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee DK, et al. Mol Endocrinol. 2010;24:930. doi: 10.1210/me.2009-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim TH, Lee DK, Franco HL, Lydon JP, Jeong JW. Biol Reprod. 2010;82:706. doi: 10.1095/biolreprod.109.081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paria BC, et al. Proc Natl Acad Sci USA. 2001;98:1047. doi: 10.1073/pnas.98.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollock PM, et al. Oncogene. 2007;26:7158. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krejci P, Prochazkova J, Bryja V, Kozubik A, Wilcox WR. Hum Mutat. 2009;30:1245. doi: 10.1002/humu.21067. [DOI] [PMC free article] [PubMed] [Google Scholar]