

Abstract
Background
Papillary renal cell carcinoma, accounting for 15% of renal cell carcinoma, is a heterogeneous disease consisting of different types of renal cancer, including tumors with indolent, multifocal presentation and solitary tumors with an aggressive, highly lethal phenotype. Little is known about the genetic basis of sporadic papillary renal cell carcinoma; no effective forms of therapy for advanced disease exist.
Methods
We performed comprehensive molecular characterization utilizing whole-exome sequencing, copy number, mRNA, microRNA, methylation and proteomic analyses of 161 primary papillary renal cell carcinomas.
Results
Type 1 and Type 2 papillary renal cell carcinomas were found to be different types of renal cancer characterized by specific genetic alterations, with Type 2 further classified into three individual subgroups based on molecular differences that influenced patient survival. MET alterations were associated with Type 1 tumors, whereas Type 2 tumors were characterized by CDKN2A silencing, SETD2 mutations, TFE3 fusions, and increased expression of the NRF2-ARE pathway. A CpG island methylator phenotype (CIMP) was found in a distinct subset of Type 2 papillary renal cell carcinoma characterized by poor survival and mutation of the fumarate hydratase (FH) gene.
Conclusions
Type 1 and Type 2 papillary renal cell carcinomas are clinically and biologically distinct. Alterations in the MET pathway are associated with Type 1 and activation of the NRF2-ARE pathway with Type 2; CDKN2A loss and CIMP in Type 2 convey a poor prognosis. Furthermore, Type 2 papillary renal cell carcinoma consists of at least 3 subtypes based upon molecular and phenotypic features.
Kidney cancer, or renal cell carcinoma, is not a single disease, but is made up of a number of different types of cancer characterized by different genetic drivers, and each with a different histology, clinical course, and response to therapy.1,2 Papillary renal cell carcinoma, which accounts for 15-20% of kidney cancers, is a heterogeneous disease with differing histological subtypes and variations in both disease progression as well as patient outcomes. Papillary renal cell carcinoma has two main sub-types; type 1, which is often multifocal, characterized by papillae and tubular structures covered with small cells containing basophilic cytoplasm and small, uniform oval nuclei3 whereas type 2 is more heterogeneous, contains papillae covered by large cells with eosinophilic cytoplasm and large spherical nuclei with prominent nucleoli.3,4 While papillary renal cell carcinoma in some patients is indolent, bilateral, and multifocal, other patients present with solitary lesions that have an aggressive clinical course. Little is known about the genetic basis of the sporadic forms of papillary renal cell carcinoma and there are currently no effective forms of therapy for patients with advanced disease.
Much of our prior knowledge of the genetic basis of papillary renal cell carcinoma is based on the study of inherited papillary renal cell carcinoma. Hereditary papillary renal cell carcinoma, a rare disorder presenting with an increased risk of Type 1 disease,4 is characterized by activating germline mutations of the MET gene.5 Somatic MET mutations are found in 13%-15% of non-hereditary papillary renal cell carcinomas.6,7 Hereditary leiomyomatosis and renal cell carcinoma, a hereditary cancer syndrome in which affected individuals are at risk of developing an aggressive form of Type 2 papillary renal cell carcinoma,8,9 is caused by germline mutation of the tricarboxylic acid (TCA) cycle enzyme gene, fumarate hydratase (FH).10 These aggressive tumors are characterized by increased oxidative stress11 and activation of the NRF2/antioxidant response element (ARE) pathway.12 Mutations in the genes that regulate the NRF2/ARE pathway, such as CUL3 and NFE2L2 (NRF2), have also been found in sporadic papillary renal cell carcinoma.13
We present an integrative genomic analysis of 161 papillary renal cell carcinoma tumors that provides molecular insights into tumor classification, will affect clinical recommendations, and may suggest paths to the development of mechanistically-based therapies.
Methods
Patients
Tumors were selected from 161 patients. Pathology review was performed to classify the tumors as Type 1, Type 2 or uncharacterized papillary renal cell carcinoma (see the Methods section of the Supplementary Appendix). The clinical and genetic characteristics of these patients are described in Table S1 in the Supplementary Appendix.
Analytic Platforms
Whole exome sequence, copy number, miRNA and mRNA expression, and CpG methylation data were generated (Table S2 in the Supplementary Appendix). Details for all analyses are available in the Methods section of the Supplementary Appendix. All data sets are available at the Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga).
Results
Histological Sub-typing
Pathological review of the161 tumors identified 75 Type 1, 60 Type 2, and 26 cases that could not be classified as Type 1 or Type 2. Consistent with previous studies3,14, the Type 1 tumors were predominately Stage I, whereas the Type 2 tumors were frequently Stage III/IV (Fig. S1 in the Supplementary Appendix).
Somatic Alterations Underscore Molecular Differences between Type 1 and Type 2 Papillary Renal Cell Carcinoma
Copy Number Alterations
Single nucleotide polymorphism (SNP) array-based profiling of somatic copy number alterations revealed distinctive patterns across three main tumor subgroups. One subgroup, predominantly composed of Type 1 and lower grade tumors, was defined by multiple chromosomal gains, including nearly universal gain of chromosomes 7 and 17, and lower frequency gain of chromosomes 2, 3, 12, 16, and 20 (Fig. 1a, and Fig. S2 in the Supplementary Appendix). The other two subgroups were predominately Type 2 tumors. While one subgroup had few copy number alterations, the other was characterized by a high degree of aneuploidy with multiple chromosomal losses, including frequent chromosome 9p loss, and associated with poorer survival (p<0.0001) (Fig. 1a, and Fig. S2 in the Supplementary Appendix).
Figure 1. Somatic alterations in papillary renal cell carcinoma underlie molecular differences between Type 1 and Type 2 cancers.
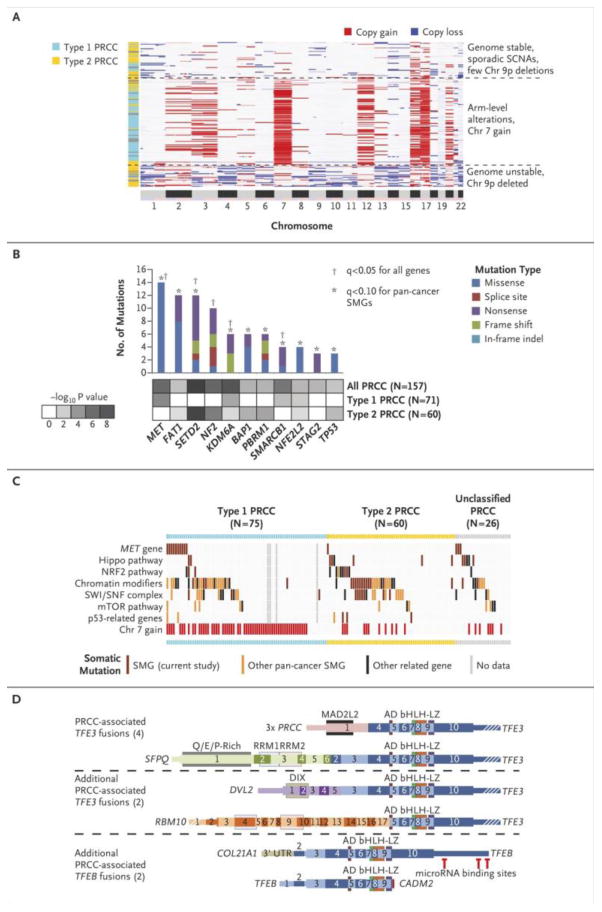
(a) Unsupervised clustering of DNA copy profiles of papillary renal cell carcinoma, revealing three molecular subtypes, one of which is highly enriched for Type 1 tumors (light blue) and the other two, for Type 2 tumors (yellow). (b) Significantly mutated genes (SMGs) in papillary renal cell carcinoma, determined by considering all genes (q<0.1, range 0.0-1.0) or focusing on the set of 260 genes previously implicated in cancer by large-scale, pan-cancer exome analyses26 (q<0.1). p-values by MutSig 2CV algorithm. (c) Pathway-centric view of gene mutations in papillary renal cell carcinoma, involving key pathways and genes implicated in cancer, either in this present study or elsewhere as indicated.26 The tumors were classified by histological type from left to right: Type 1 (light blue), Type 2 (yellow) or unclassified papillary renal cell carcinoma (gray), and from top to bottom by gene or pathway altered. Pathways and genes represented include: MET, HIPPO pathway (NF2, SAV1, WWC1), NRF2 pathway (NFE2L2, KEAP1, CUL3, SIRT1, FH), chromatin modification (CREBBP, DOTL1, EHMT1/2, EP300, EZH1/2, KAT2A/B, KDM1A/B, KDM4A/B, KDM5A/B/C, KDM6A/B, MLL1/2/3/4/5, NSD1, SETD2, SMYD4, SRCAP), SWI/SNF complex (ACTB, ACTL6A/B, ARID1A/B, ARID2, BCL6A/B/C, BCL11A/B, BRD7/9, DPF1/2/3, PHF10, PBRM1, SMARCA2/4, SMARCB1, SMARCC1/2, SMARCD1/2/3, SMARCE1), mTOR pathway (MTOR, PIK3CA, PTEN, STK11, TSC1, TSC2), and p53 pathway (ATM, CDKN1A, CDKN2A, FBXW7, RB1, TP53). (d) Fusion gene analysis identified TFE3 or TFEB fusions in 8 PRCC tumors, including two novel gene fusion partners for TFE3, DVL2 and RBM10, and two novel gene fusion partners for TFEB, COL21A1 and CADM2. Schematic versions of these fusions demonstrate the exons and functional domains that are present within the different gene fusions and the position of potential miR binding sites in TFEB. The retained exons of TFE3 or TFEB are colored in shades of blue. Thin regions represent non-coding sequence, while thick regions represent the translated reading frame and white strips indicate the region is no longer to scale. AD = Strong Transcription Activation Domain, bHLH = Basic Helix-Loop-Helix Domain, LZ = Leucine Zipper Domain, MAD2L2 = Mitotic Arrest Deficient-Like 2 Interaction Domain, DIX = Dishevelled and Axin Domain, RMM = RNA-Recognition Motif.
Whole Exome-Sequencing
Whole-exome sequencing identified 10,380 putative somatic mutations in 157 tumors with an average of 1.45 non-silent mutations per megabase (https://tcga-data.nci.nih.gov/tcgafiles/ftp_auth/distro_ftpusers/anonymous 8/tumor/kirp/gsc/hgsc.bcm.edu/illuminaga_dnaseq_curated/mutations/hgsc.bcm.edu_KIRP.IlluminaGA_DNASeq_curated.Level_2.1.0.0/). An initial screen for significantly mutated genes using MutSig 2.0CV with q-values <0.1 (q-values range from 0 to 1) identified 5 significantly mutated genes (MET, SETD2, NF2, KDM6A and SMARCB1) recurrently mutated in papillary renal cell carcinoma representing 24% of cases (Fig. 1b). Further analysis, performed restricting the multiple hypothesis testing to genes previously associated with cancer by the PanCan2115, identified 6 additional significantly mutated genes (FAT1, BAP1, PBRM1, STAG2, NFE2L2 and TP53) with 36% of cases demonstrating mutation of at least one (Fig. 1b). Mutation of these significantly mutated genes was demonstrated not to be sub-clonal (Table S3 in the Supplementary Appendix).
Hippo and Chromatin Modifier Pathways
Several papillary renal cell carcinoma significantly mutated genes are components of well-known cancer-associated pathways or complexes, including NF2 in the Hippo signaling pathway, SMARCB1 and PBRM1 in the SWI/SNF complex, and SETD2, KDM6A and BAP1 in several chromatin modifier pathways. Assessment of genes within these pathways (Table S4 in the Supplementary Appendix) demonstrated a high number of mutations in both Type 1 and Type 2 tumors involving SWI/SNF complex (19.7% and 26.7% respectively), chromatin modifier pathways (35.2% and 38.3% respectively) and Hippo signaling pathway (2.8% and 10.0% respectively) (Fig. 1c).
TFE3 and TFEB Gene Fusions
Gene fusions involving TFE3/TFEB have previously been associated with papillary renal cell carcinoma (reviewed in Kaufman, et al.16). We identified gene fusions in 17 (10.6%) tumors, including 8 with TFE3/TFEB gene fusions (Table S5 in the Supplementary Appendix). Four of the TFE3 fusions involved known fusion partners, PRCC and SFPQ, and 2 involved novel fusion partners, RBM10 and DVL2 (Fig. 1d). These TFE3 fusion tumors demonstrated varying degrees of increased mRNA expression for known TFE3 transcriptional targets, including CTSK, BIRC7, DIAPH1 and HIF1A (Fig. S3 in the Supplementary Appendix). The two TFEB fusions both involved novel fusion partners, COL21A1 and CADM2, with the COL21A1-TFEB fusion resulting in a similar construct to the known MALAT1-TFEB fusions16 and the TFEB-CADM2 fusion resulting in a novel truncated version of TFEB that had lost several microRNA binding sites (Fig. 1d). The TFEB fusion tumors demonstrated high mRNA expression of the TFEB transcription factor and a known target gene, CTSK (Fig. S4 in the Supplementary Appendix). Seven of the TFE3/TFEB fusions were identified in the Type 2 tumors (7/60 - 11.7%).
Papillary Renal Cell Carcinoma Type-Specific Alterations
Type 1:MET
We found mutation of MET in seventeen tumors, three of which were germline. Most mutations were in the tyrosine kinase domain (14/17) and were found primarily in Type 1 tumors (13/75, 18.6%) (Fig. 2a-b). In addition, an alternate MET RNA transcript that replaces canonical exons 1 and 2 with a novel first exon spliced to canonical exon 3 (Fig. 2a) was identified in 8 tumors (4 Type 1, 3 Type 2 and 1 unclassified). This isoform represented the majority of transcripts in 2 tumors and a fraction in the remaining 6 tumors and was recently observed to produce a stable, shortened protein in gastric cancer cell lines (Fig. S5a in the Supplementary Appendix).17 Exons 1 and 2 of MET encode the hepatocyte growth factor ligand binding domain and, analogous to the epidermal growth factor receptor, EGFRvIII, isoform18, this isoform may result in ligand-independent MET activation. In addition, gene fusions involving MET were found in three cases (Table S5 in the Supplementary Appendix). Potentially driven in part by trisomy of chromosome 7, MET mRNA expression and protein phosphorylation (pY1235) were significantly higher in Type 1 vs. Type 2 PRCCs (p<1E-9 and p<0.01, respectively, t-test) (Fig. S5b in the Supplementary Appendix). Altered MET status (defined as mutation, splice variant, or gene fusion) or increased chromosome 7 copy number (encoding MET but which may also involve other genes) was identified in 81.3% of Type 1 PRCC. GISTIC2.0 analysis identified loss of 1p36 in 18 (11.1%) papillary renal cell carcinomas involved candidate tumor suppressor ERRFI1, a negative regulator of EGFR (Fig. S6 in the Supplementary Appendix), which deletions significantly co-occurred (p=0.021, Fisher's exact) with chromosome 7 gain and EGFR amplification.
Figure 2. Alterations in papillary renal cell carcinoma involving MET oncogene.
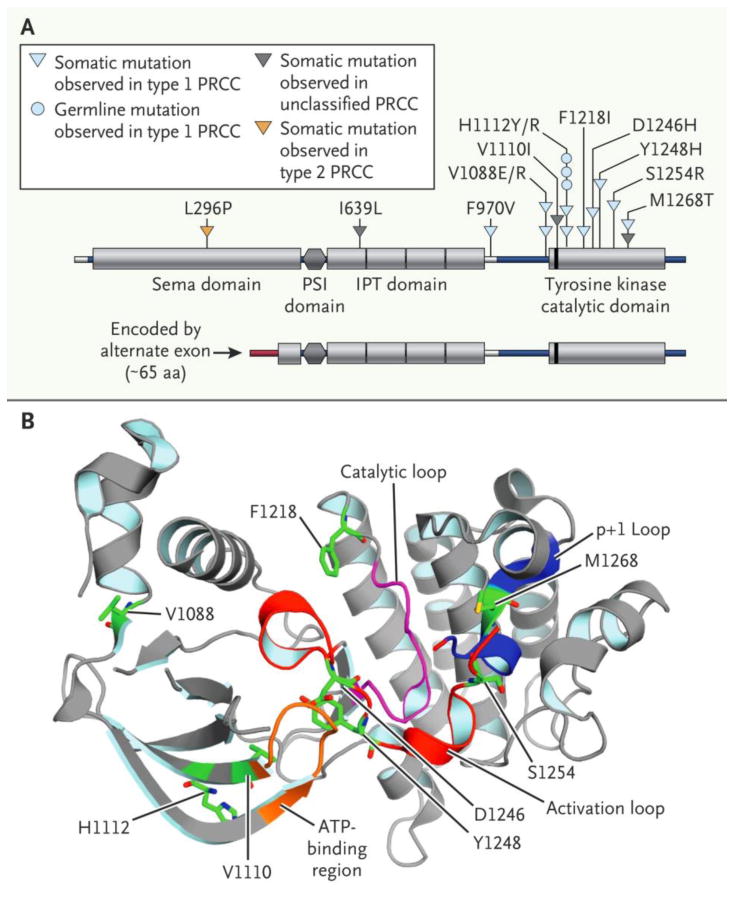
(a) Schematic of somatic mutations in MET, along with germline variant H1112R previously implicated in hereditary papillary renal cell carcinoma 37, and the novel RNA transcript variant of MET lacking the canonical exons 1 and 2, but containing a novel exon 1 that splices to the canonical exon 3. (b) Crystal structure for the MET tyrosine kinase catalytic domain (RCSB-PDB 3I5N38), on which are mapped the residues altered in papillary renal cell carcinoma. All amino acid numbering is based on the MET protein sequences.
Type 2: CDKN2A
GISTIC2.0 analysis identified focal loss of 9p21 in 13 (8.1%) papillary renal cell carcinomas resulting in loss of CDKN2A (Fig. S7a in the Supplementary Appendix). We found mutation or promoter hypermethylation of CDKN2A in 11 tumors (Fig. S7b in the Supplementary Appendix), including 3 of the tumors with focal loss of 9p21, resulting in 21 tumors (13.0%) defined as having CDKN2A alteration (Fig. S7c in the Supplementary Appendix). CDKN2A alteration was strongly associated with Type 2 histology with 25.0% (15/60) of Type 2 tumors demonstrating alterations. CDKN2A- altered tumors demonstrated both increased levels of phosphorylated retinoblastoma protein (Rb) and increased expression of cell cycle-related genes consistent with the predicted consequences of CDKN2A loss (Fig. S7d in the Supplementary Appendix). Univariate analysis of patients with CDKN2A altered tumors demonstrated significantly decreased overall survival when compared with all papillary renal cell carcinomas (p<1E-10, Fig. S7e in the Supplementary Appendix) as well as when compared with only Type 2 tumors (p<0.0001, Fig. S7f in the Supplementary Appendix). In addition, increased expression of microRNA miR-10b-5p correlated with lower expression of its target CDKN2A (Fig. S8 in the Supplementary Appendix).
Type 2: SETD2, BAP1 and PBRM1
Type 2 tumors were associated with mutations in the chromatin modifying genes, SETD2, BAP1 and PBRM1, which are frequently mutated in clear cell kidney tumors in combination with chromosome 3p loss.19 Mutations of BAP1 and PBRM1 were mutually exclusive but PBRM1 mutations were frequently concurrent with SETD2 mutations (Fig. S9 in the Supplementary Appendix). Although loss of chromosome 3p was also associated with Type 2 papillary renal cell carcinoma, only 3 of 13 Type 2 papillary renal cell carcinomas with SETD2, BAP1 or PBRM1 mutation demonstrated loss and no promoter hypermethylation was observed (Fig. S9 in the Supplementary Appendix).
CpG Island Methylator Phenotype (CIMP) in Type 2 Tumors
Nine tumors (5.6%) had increased DNA methylation at loci that were unmethylated in matched normal tissue. This represents a novel kidney-associated CpG island methylator phenotype (CIMP20) that included universal hypermethylation of the CDKN2A promoter (Fig. 3a). Eight of nine tumors were Type 2 papillary renal cell carcinomas. In five tumors we found germline and/or somatic mutation of FH (55.6%). We found decreased expression of FH mRNA and increased expression of genes associated with cell cycle progression and response to hypoxia in all nine (Fig. 3a). Notably, CIMP tumors had at an earlier age of presentation and had decreased survival compared with other papillary renal cell carcinoma patients (Fig. 3b). Fumarate hydratase-deficient Type 2 tumors in hereditary leiomyomatosis and renal cell carcinoma patients are characterized by a Warburg-like metabolic shift to glycolysis-dependent metabolism and an increased expression of hypoxia related genes.21,22 Similarly, CIMP papillary renal cell carcinoma tumors demonstrated increased expression of key genes involved in glycolysis (HK1, LDHA and PDK1), the pentose phosphate pathway (G6PD) and the fatty acid synthesis (FASN) (Fig. S10 in the Supplementary Appendix). In addition, there was decreased expression of the majority of genes for the Krebs cycle and the adenosine monophosphate-activated kinase (AMPK) complex, a suppressor of fatty acid synthesis (Fig. 3d-e). Protein expression data for G6PD, FASN and AMPK correlated with the mRNA expression (Fig. 3e).
Figure 3. DNA methylation profiling uncovers a subset of papillary renal cell carcinoma manifesting a CpG Methylator Phenotype (CIMP).
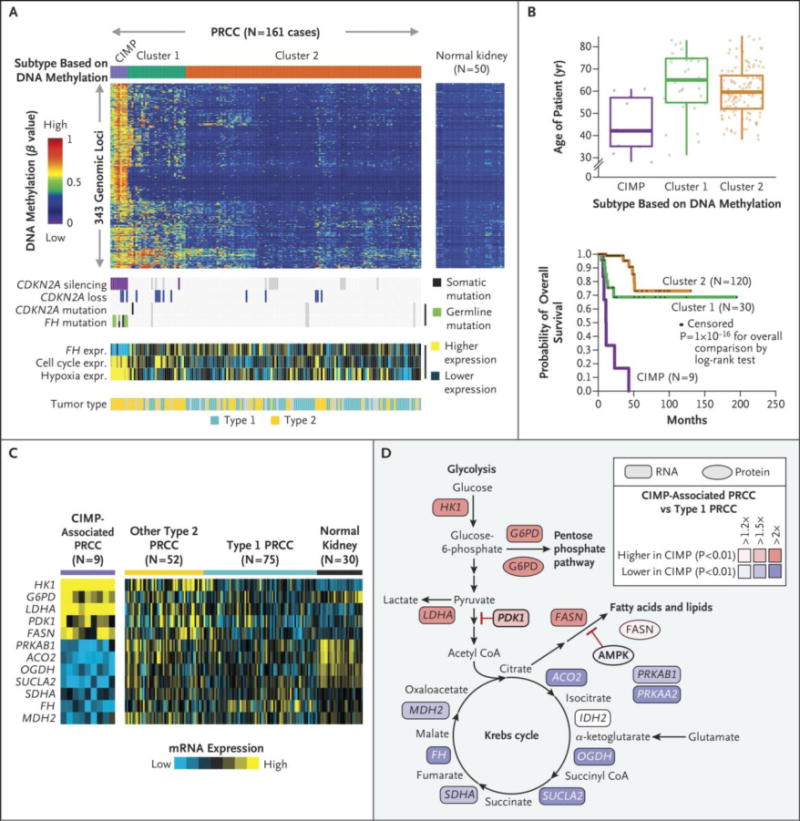
(a) Molecular subtyping by DNA methylation platform revealed three subtypes of papillary renal cell carcinoma, one of which showed widespread DNA hypermethylation patterns characteristic of CIMP tumors. Corresponding data tracks highlight molecular features associated with CIMP tumors (n=9 cases), including CDKN2A silencing, germline or somatic mutations of FH, Type 2 histological status, and expression of both cell cycle-39 and hypoxia-related genes40. (b) Differences in patient age among the three DNA methylation-based subtypes. (c) Differences in patient overall survival among the three DNA methylation-based subtypes (p=1E-16, log-rank). (d) Differential mRNA expression patterns comparing papillary renal cell carcinoma CIMP, Type 1, non-CIMP Type 2, and normal kidney for key genes involved in metabolism. (e) Differential expression patterns of CIMP tumors versus Type 1 in metabolism-related pathways, focused on gene and protein expression patterns previously associated with Warburg-like effects in kidney cancer.19 p-values by t-test.
Multiplatform Subtype Discovery Defines Papillary Renal Cell Carcinoma Subgroups Distinguishable by Histological Type and DNA Methylation
Similar to the chromosomal copy number and methylation analyses, the profiles for mRNA expression, miRNA expression, and protein expression data clustered the papillary renal cell carcinoma cases into separate groups with distinct overall outcomes (Fig. S11-S13 in the Supplementary Appendix). The five data types were combined to perform a “cluster of clusters analysis”23,24 that identified 4 tumor clusters: C1 (Type 1-enriched), C2a and C2b (both Type 2-enriched), and C2c (consisting solely of CIMP papillary renal cell carcinoma) (Fig. 4a)
Figure 4. Multi-platform-based subtype discovery in papillary renal cell carcinoma.
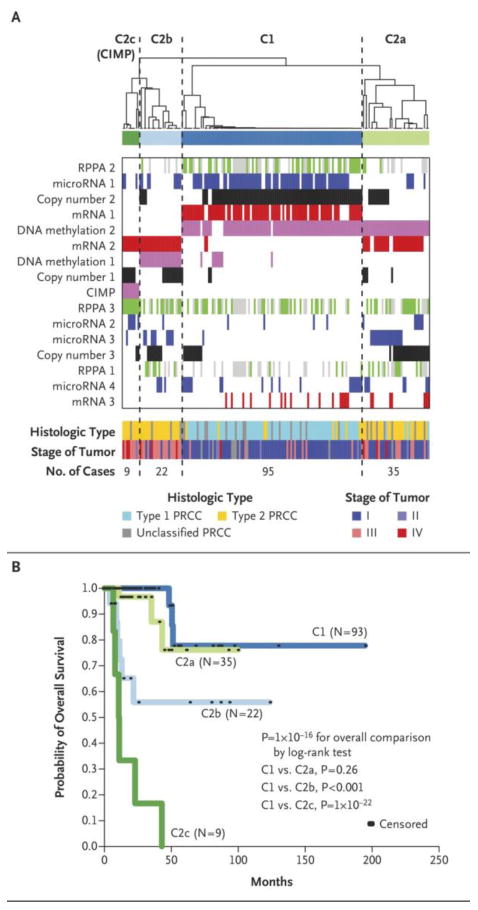
(a) Integration of subtype classifications from five “omic” data platforms using cluster of cluster analysis identified four major papillary renal cell carcinoma groups: C1 (Type 1-enriched), C2a and C2b (Type 2-enriched), and C2c (representing the CIMP papillary renal cell carcinomas). The blue and white heat map displays sample consensus, below which a second heat map displays the subtypes defined independently by DNA methylation (Pink), Chromosomal copy number (CN)(Black), miRNA expression (Red), mRNA expression (Blue), and protein (RPPA) expression (Green, Gray represents samples missing RPPA expression data). Clinical features associated with the multi-platform-based subtypes are shown near the bottom. Histological types are shown with Type 1 in light blue, Type 2 in yellow and unclassified papillary renal cell carcinoma in gray. Stage I and II tumors are shown in dark blue or light blue respectively, and Stage III and IV tumors are shown in light red or dark red respectively. (b) Differences in patient overall survival between COCA defined groups (overall, p<1E-16, log-rank). In comparison to the Type 1-enriched C1group, the overall survival of the Type 2-enriched C2a group was similar, while the C2b (* p=0.003) and CIMP (** p=1E-22) groups had worse overall survival.
Cluster C1 was predominantly Type 1 papillary renal cell carcinoma and strongly associated with gain of chromosome 7, MET mutation, mRNA cluster 1, and low tumor stage (Fig. 4a, Fig. S14 in the Supplementary Appendix). Cluster C2a was predominately Type 2 papillary renal cell carcinoma and was associated with low tumor stage, and DNA methylation cluster 2. Cluster C2b consisted exclusively of Type 2 and unclassified papillary renal cell carcinomas and was associated with DNA methylation cluster 1, higher tumor stage (III/IV), and mutation of SETD2 (Fig. 4a, Fig. S14 in the Supplementary Appendix). The previously observed DNA methylation-based CIMP tumor subtype was preserved as cluster C2c. The best survival was associated with clusters C1 and C2a, while patients in cluster C2b tumors had a poorer survival. The cluster C2c patients were associated with the worst survival (Fig. 4b).
Type 2: NRF2-ARE Pathway
Pathway analysis was performed comparing the miRNA and mRNA signatures of Type 1 and Type 2 papillary renal cell carcinomas (Fig. S15-S17 and Tables S6-S8 in the Supplementary Appendix) and mRNA expression data highlighted the NRF2 antioxidant response (ARE) pathway as a distinguishing feature of Type 2 tumors (Fig. S17a in the Supplementary Appendix). Expression of NQO1, a gene activated by the NRF2-ARE pathway25, was lowest in cluster C1, intermediate in clusters C2a and C2b, and highest in the CIMP cluster C2c (p=1E-18, ANOVA, Fig. S18a in the Supplementary Appendix) and increased NQO1 expression associated with decreased survival (p=0.001, Fig. S18c in the Supplementary Appendix). These findings are consistent with recent studies demonstrating increased activation of the NRF2-ARE pathway in Type 2 tumors and mutations in NRF2-ARE pathway genes (NFE2L2, CUL3, KEAP1 and SIRT1).12,13 Four NFE2L2 (NRF2) mutations in known activating hotspots were identified as well as mutations in both CUL3 (n=5) and KEAP1 (n=1). These NFE2L2/CUL3/KEAP1 mutations correlated with high expression levels of NQO1 (p<1E-6, t-test), but did not solely account for the observed NQO1 expression differences among subtypes (Fig. S18a in the Supplementary Appendix).
Integrated Analysis of low frequency candidate driver mutations
A percentage of mostly smaller tumors lacked high confidence candidate cancer driving events. Manual pathway analysis identified candidate driver mutations in an additional 27% of the cases affecting known cancer-associated genes, such as PTEN, NRAS, KRAS, TP53, TSC2 and those in the MLL and ARID families (Fig. S19a and Table S9 in the Supplementary Appendix). For the remaining thirty-seven (23%) tumors, low frequency somatic events in genes identified by HotNet2 analysis (Fig. S19 in the Supplementary Appendix) or associated with cancer in either the PanCan2126 or COSMIC database were proposed as potential drivers and listed in Table S9 in the Supplementary Appendix. Twenty-six (70%) of these thirty seven PRCCs were Type 1 (p=0.0013, Fisher's exact) and most (21/26, 81%) demonstrated gain of chromosome 7 which includes MET. This gain of chromosome 7, which is seen in a number of tumors such as Wilms' tumor and papillary thyroid cancer, could be considered a driver event, but it does not identify a specific driver. Although gain of chromosome 7 was associated with increased MET expression in papillary renal cell carcinoma (p=0.0002, two-factor ANOVA)(Fig. S20 in the Supplementary Appendix), other potential driver genes on chromosome 7, such as epidermal growth factor receptor (EGFR), could influence tumorigenesis.
Discussion
We used a comprehensive genomics approach to characterize the biologic foundation of papillary renal cell carcinoma and conclude that Type 1 and Type 2 papillary renal cell carcinoma are distinctly different diseases and that type 2 papillary RCC is a heterogeneous disease with multiple distinct sub-groups. Common driver mutations among the different subtypes were relatively rare, as had been observed in two recent studies.7,27 Molecular and phenotypic differences between Type 1 and Type 2 papillary renal cell carcinoma were reflected in individual and combined analyses of various data platforms. The utility of CDKN2A alterations as an independent prognostic marker associated with Type 2 tumors requires validation. This study shows that TFE3/TFEB fusions are underappreciated in adult Type 2 tumors and should be considered in any Type 2 patient. Although TFE3 and TFEB papillary renal cell carcinomas are generally considered diseases of children and young adults16, our mean age was 52, and we found TFEB fusions tumors in patients 64 and 71 years of age.
The most distinct of the three Type 2 subgroups was that defined by the CpG island methylator phenotype (CIMP), which associated with the worst overall survival. The CIMP hypermethylation patterns have been observed in a number of other cancer subtypes, including glioblastoma28, lung adenocarcinoma29, and gastric adenocarcinoma30. The CIMP tumors demonstrated low expression levels of fumarate hydratase mRNA and 5 had germline or somatic mutation of the FH gene. Germline mutation of FH is found in the aggressive Type 2 tumor associated with hereditary leiomyomatosis and renal cell carcinoma.9,31 In this syndrome, the high levels of fumarate accumulating from loss of fumarate hydratase enzyme activity result in impaired function of enzymes such as the TET family of enzymes responsible for maintaining appropriate DNA methylation within the genome.32 The subgrouping of the Type 2 tumors by molecular features and the presence of specific subsets of Type 2 tumors, such as those with TFE3 fusions or CIMP, suggests that sub-stratification of Type 2 papillary renal cell carcinoma by specific molecular markers may provide more accurate diagnosis that could lead to the development of mechanistic, disease-specific targeted therapies.
This classification of papillary renal cell carcinoma may have a significant impact on clinical and therapeutic management and clinical trial design. Alteration of the MET gene or gain of chromosome 7 was found in a large percentage (81%) of Type 1 tumors. Antitumor activity of an agent targeting the MET/VEGFR2 pathways has been demonstrated in a Phase II trial in patients with papillary renal cell carcinoma, with a particularly high response rate in tumors with MET mutations.33 Mutation of the Hippo pathway tumor suppressor, NF2, was observed in a number of papillary renal cell carcinomas. This pathway has been targeted in other cancers with agents such as dasatinib, an inhibitor of the YES1 kinase that interacts with the YAP transcription factor that is upregulated with Hippo pathway dysregulation.34 The CIMP tumors demonstrated a Warburg-like metabolic shift, similar to that observed in fumarate hydratase-deficient hereditary leiomyomatosis and renal cell carcinoma -associated renal tumors11,21,22. A clinical trial targeting this metabolic shift in papillary renal cell carcinoma is currently underway (NCI clinical trial - NCT01130519). Increased expression of the NRF2/ARE pathway has been observed both in hereditary and sporadic Type 2 papillary renal cell carcinomas.12 NQO1 immunohistochemical analysis could provide valuable marker for activation of the NRF2/ARE pathway. Currently, there is intense interest in the NRF2/ARE pathway in cancer35 and novel strategies have recently been developed to target this pathway.36
The identification of altered genes and pathways provides a comprehensive foundation for an understanding of the molecular basis of papillary renal cell carcinoma. This refined classification more accurately reflects the genotypic and phenotypic differences among the different types of these tumors and should lead to more appropriate clinical management and development of more effective forms of therapy.
Supplementary Material
Footnotes
Disclosure: Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Author: This file is the accepted version of your manuscript, and it shows any changes made by the Editor-in-Chief and the Deputy Editor since you submitted your last revision. This is the version that is being sent to Manuscript Editing for further editing in accordance with NEJM style. You will receive proofs of the edited manuscript, by e-mail. The proofs will contain queries from the manuscript editor, as well as any queries that may be present in this file. The proof stage will be your next opportunity to make changes; in the meantime, please do not make any changes or send any new material to us.
References
- 1.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7(5):277–285. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22(11):2089–2100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10(6):537–544. [PubMed] [Google Scholar]
- 4.Zbar B, Tory K, Merino MJ, et al. Hereditary papillary renal cell carcinoma. J Urol. 1994;151(3):561–566. doi: 10.1016/s0022-5347(17)35015-2. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt LS, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genetics. 1997;16(1):68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt LS, Junker K, Nakaigawa N, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18(14):2343–2350. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- 7.Durinck S, Stawiski EW, Pavia-Jimenez A, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2014 doi: 10.1038/ng.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98(6):3387–3382. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grubb RL, Franks ME, Toro J, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177(6):2074–2080. doi: 10.1016/j.juro.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 10.Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30(4):406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 11.Sudarshan S, Sourbier C, Kong HS, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and HIF-1 alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;15:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ooi A, Wong JC, Petillo D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 13.Ooi A, Dykema K, Ansari A, et al. CUL3 and NRF2 Mutations Confer an NRF2 Activation Phenotype in a Sporadic Form of Papillary Renal Cell Carcinoma. Cancer Res. 2013;73(7):2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 14.Jiang F, Richter J, Schraml P, et al. Chromosomal imbalances in papillary renal cell carcinoma: genetic differences between histological subtypes. Am J Pathol. 1998;153(5):1467–1473. doi: 10.1016/S0002-9440(10)65734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kauffman EC, Ricketts CJ, Rais-Bahrami S, et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol. 2014 doi: 10.1038/nrurol.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muratani M, Deng N, Ooi WF, et al. Nanoscale chromatin profiling of gastric adenocarcinoma reveals cancer-associated cryptic promoters and somatically acquired regulatory elements. Nat Commun. 2014;5:4361. doi: 10.1038/ncomms5361. [DOI] [PubMed] [Google Scholar]
- 18.Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J. 2013;280(21):5350–5370. doi: 10.1111/febs.12393. [DOI] [PubMed] [Google Scholar]
- 19.The Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature. 2013;499(7456):43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong WH, Sourbier C, Kovtunovych G, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20(3):315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Lane AN, Ricketts CJ, et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS One. 2013;8(8):e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoadley KA, Yau C, Wolf DM, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93(25):14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovac M, Navas C, Horswell S, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat Commun. 2015;6:6336. doi: 10.1038/ncomms7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19(13):3345–3352. doi: 10.1158/1078-0432.CCR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao M, Yang H, Xu W, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181–186. doi: 10.1200/JCO.2012.43.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13(1):63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12(8):564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sourbier C, Ricketts CJ, Matsumoto S, et al. Targeting ABL1-mediated Oxidative Stress Adaptation in Fumarate Hydratase-Deficient Cancer. Cancer Cell. 2014;26:840–850. doi: 10.1016/j.ccell.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt LS, Junker K, Weirich G, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58(8):1719–1722. [PubMed] [Google Scholar]
- 38.Boezio AA, Berry L, Albrecht BK, et al. Discovery and optimization of potent and selective triazolopyridazine series of c-Met inhibitors. Bioorg Med Chem Lett. 2009;19(22):6307–6312. doi: 10.1016/j.bmcl.2009.09.096. [DOI] [PubMed] [Google Scholar]
- 39.Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13(6):1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris AL. Hypoxia--a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2(1):38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.