

SUMMARY
The canonical function of kinases is to transfer a phosphoryl group to substrates, initiating a signaling cascade; while their non-canonical role is to bind other kinases or substrates, acting as scaffolds, competitors, and signal integrators. Here, we show how to uncouple kinases dual function by tuning the binding cooperativity between nucleotide (or inhibitors) and substrate allosterically. We demonstrate this new concept for the C-subunit of protein kinase A (PKA-C). Using thermocalorimetry and NMR, we found a linear correlation between the degree of cooperativity and the population PKA-C’s closed state. The non-hydrolysable ATP analog (ATPγC) does not follow this correlation, suggesting that changing the chemical groups around the phosphoester bond can uncouple kinases dual function. Remarkably, this uncoupling was also found for two ATP-competitive inhibitors, H89 and balanol. Since the mechanism for allosteric cooperativity is not conserved in different kinases, these results may suggest new approaches for designing selective kinase inhibitors.
Keywords: Protein kinase A, Canonical and non-canonical Function, Cooperativity, Pseudo-kinases, Phospholamban
Graphical Abstract
INTRODUCTION
Protein kinases are ubiquitous phosphoryl transferases that regulate many cellular signaling processes (Fischer and Krebs, 1955). Due to their primary role in cell physiology and pathology, protein kinases have become major drug targets to counteract human diseases, such as heart failure and cancer (Johnson and Lewis, 2001; Manning et al., 2002a). Kinases canonical function is to transfer the γ-phosphate of ATP to Ser/Thr/Tyr residues of substrates, thereby activating or deactivating various signaling pathways (Endicott et al., 2012; Johnson and Lewis, 2001; Manning et al., 2002b; Pearce et al., 2010). About a decade ago, Manning and co-workers identified a non-canonical function for kinases that, in several instances, do not carry out any catalytic function; rather they provide binding scaffolds to modulate, integrate, or compete in signaling cascades, the so-called pseudo-kinases (Manning et al., 2002b). While kinases mediate signaling through phosphoryl transfer and scaffolding (dual function), pseudo-kinases’ function is independent from catalysis (Boudeau et al., 2006; Reiterer et al., 2014). To date, approximately 10% of the 518 members of the mammalian kinases have been identified as pseudo-kinases, with reduced or completely obliterated ability to catalyze phoshoryl transfer (Boudeau et al., 2006; Shaw et al., 2014). Recent site-directed mutagenesis studies suggest that it is possible to uncouple the canonical from the non-canonical function of kinases (Hu et al., 2013; Hu et al., 2011; Iyer et al., 2005). Also, it has been found that small molecules that inhibit kinase phosphorylation in vitro are able to activate kinase pathways in cell (Dar and Shokat, 2011; Hatzivassiliou et al., 2010; Poulikakos et al., 2010). The latter suggests that kinases depleted of their catalytic functions still work as scaffolds and play an active role in cell signaling. Therefore, uncoupling canonical and non-canonical functions of protein kinases with small molecules would enable one to achieve a higher level of control over the kinase-mediated signaling pathways (Shaw et al., 2014).
Although substantial progress has been made for the development of allosteric inhibitors (Arencibia et al., 2013; Cowan-Jacob et al., 2014; Fang et al., 2013), small molecules that bind the ATP binding site (ATP-competitive inhibitors) remain the most common kinase inhibitors (Wu et al., 2015). However, none of these drugs have been engineered to uncouple the dual functions of kinases, rendering them either pseudo-kinases (devoid of catalytic activity) or dead kinases (non-catalytic and non-scaffolding). So, how can we uncouple canonical and non-canonical kinase functions? Since allosteric binding cooperativity (K-type cooperativity, i.e., nucleotide binding affects substrate affinity) is a hallmark for several kinases (Masterson et al., 2012; Masterson et al., 2008), we reasoned that by modulating the chemical moieties of nucleotides and nucleotide-analogs it would be possible to control allosteric binding cooperativity. The latter will make it possible to design inhibitors able to steer kinases’ function toward pseudo-kinases or dead kinases.
As a model system, we chose the catalytic subunit of the cAMP-dependent protein kinase A (PKA-C). This enzyme is one of the most studied and has represented the benchmark for the entire kinase family (Johnson et al., 2001). PKA-C is organized into two lobes, an N-terminal small lobe with 5 β-strands and one helix (αC helix), and a C-terminal large lobe mostly helical that harbors the substrate binding cleft (Knighton et al., 1991a) (Figure 1A). The nucleotide (ATP) binds at a critical junction of the kinase core, which is embedded between the small and large lobes (Knighton et al., 1991a). Through coordination of two Mg2+ ions, the nucleotide positions several amino acids from various catalytic motifs such as the DFG loop, glycine-rich loop, and catalytic loop for phosphoryl transfer (Figure 1C) (McClendon et al., 2014). Structurally, the nucleotide’s adenine ring completes the architecture of the catalytic spine (C-spine), an array of hydrophobic residues that play a key role in intramolecular allosteric signaling and kinase activation (Kornev et al., 2008). The substrate binds the C-lobe, laying on the peptide positioning loop, which provides high-binding affinity for registering the recognition sequence. It has been hypothesized that ATP acts as an allosteric effector priming the kinase structure for substrate binding (Kornev et al., 2008; Masterson et al., 2011a; Whitehouse et al., 1983). The dynamic apo enzyme toggles between three major conformational states along the free energy reaction coordinate (open, intermediate, and closed, Figure 1B). ATP binding shifts the enzyme conformational ensemble from an open to an intermediate state, increasing substrate affinity through a K-type binding cooperativity (Sims et al., 2013; Taylor et al., 2005; Taylor et al., 2004). Binding of substrate further shifts the ensemble toward the enzyme’s closed state. In spite of a plethora of structural (Johnson et al., 2001) and biophysical data (Adams, 2001), it is still unclear how the different chemical moieties of ATP confer positive K-type cooperativity for substrates.
Figure 1. Three-dimensional fold and conformational states of PKA-C.

A) Ribbon diagram of the catalytic subunit of protein kinase A (PDB: 1ATP) shown with the C-spine scaffold (yellow surface), sandwiching the adenine moiety of ATP, and the peptide fragment of the heat stable protein kinase inhibitor (PKI5-24). B) Overlay of the glycine-rich loop of the open (PDB: 1J3H), intermediate (PDB:1BKX) and closed (PDB:1ATP) forms of PKA-C. (C). Electrostatic and hydrophobic contacts with ATP deduced from the 1ATP structure of PKA-C. (D) Chemical structures of the nucleotide analogues used in this study.
By combining isothermal titration calorimetry (ITC) with NMR spectroscopy we show how small molecules (nucleotides or small molecule inhibitors, Figure 1D) are able to modulate the degree of the binding cooperativity for the pseudo-substrate peptide derived from the endogenous protein kinase inhibitor (PKI5-24). For the nucleotide analogs, we found a linear correlation between the degree of cooperativity and the population of the closed state of the enzyme. The highest degree of cooperativity is reached with ATP, where the phosphate groups pre-organize the active site for phosphoryl transfer and prime the substrate binding site for high binding affinity (catalytically competent state). The degree of cooperativity is substantially reduced for ATPγN and completely abrogated for ATPγC. In addition, ATPγC drastically decreases the binding affinity of PKA-C for phospholamban, a natural substrate of this kinase in the heart muscle. For H89 and balanol, two chemically different ATP-competitive inhibitors of PKA-C, we found negative and positive binding cooperativity, respectively. These results demonstrate that it is possible to modulate substrate binding cooperativity by changing the chemical nature of small molecule inhibitors that can decouple the dual functions of kinases, opening new directions for manipulating protein kinases functions in a specific manner.
RESULTS
Different nucleotides provide varying degree of binding cooperativity
To measure substrate binding cooperativity of PKA-C, we utilized ITC to measure the binding affinity of PKA-C for the heat stable protein kinase A inhibitor peptide (PKI5-24). Since PKI5-24 contains the substrate recognition motif for PKA-C (with Arg residues on the P-2 and P-3 positions and an Ala instead of a Ser at the P-site), it is considered a pseudo-substrate(Johnson et al., 2001), recapitulating the high binding affinity of the R-subunits(Cheng et al., 1986; Knighton et al., 1991a; Knighton et al., 1991b; Masterson et al., 2011a). The degree of binding cooperativity between PKI5-24 and the nucleotide was assessed by saturating the kinase with a series of nucleotide analogs: adenine, adenosine, AMP, ADP, ATPγN, ATPγC, and ATP (Figure 2 and S1–2). Under the time scale of our experiments, we did not detect any hydrolysis of ATPγN, which has been detected in the crystallized PKA-C/ATPγN/SP20 complex (SP20 is a substrate peptide derived from PKI5-24 with Asn20Ala and Ala21Ser mutations (Bastidas et al., 2013)). The different nucleotides were chosen to dissect the contribution of each chemical moiety (i.e., adenine ring, ribose, and phosphates) to the K-type cooperativity. All of these nucleotides display very similar binding affinities for the enzyme (Bhatnagar et al., 1983; Ni et al., 2000; Srivastava et al., 2014), with Kd values between 20–50 μM. Note that under our experimental conditions, two Mg2+ ions occupy the binding site, which are required for the high binding affinity of PKI5-24 (Zimmermann et al., 2008). To quantify the extent of cooperativity, the binding thermodynamics were interpreted using a classical heterotropic linkage model (Figure 2A), where the binding of one ligand enhances or reduces the affinity of the second ligand. The degree of cooperativity (σ), which is independent of the order of ligand binding, quantifies the extent of K-type cooperativity (Freire et al., 2009; Masterson et al., 2008). Saturation with most nucleotides increases the binding affinity toward PKI5-24. The highest affinity and degree of cooperativity (σ = 400) was measured for ATP. Specifically, we found a gradual increase of affinity of PKI5-24 and binding cooperativity for the following series: adenine < adenosine < ADP < ATPγN ≪ ATP (Figure 2B). These data indicate that the adenine ring, the ribose and the three phosphates contribute incrementally to the enzyme’s binding affinity for PKI5-24. As previously observed (Bhatnagar et al., 1983), the adenine moiety (Figures 2, S1, S2, and Table 1) is especially important for the binding affinity, since its ring, sandwiched between residues Val57 in the N-Lobe and Leu173 in the C-Lobe, is stabilized by hydrophobic interactions. The ribose moiety and phosphate groups further increase PKI5-24 binding affinity, with a significant increase in cooperativity correlated with the number of phosphate groups. Notice that the affinity of the pseudo-substrate peptide is much greater when the enzyme is bound to ATP than ADP, indicating that ADP facilitates the exit of the enzymatic product. An anomalous behavior is observed for AMP, which has been shown to have a lower binding affinity for PKA compared to other nucleotide analogs (Bhatnagar et al., 1983). In agreement with these previous data, we found a substantially weaker affinity with AMP (Kd ~ 250 μM). These binding experiments repeated at higher AMP concentrations to saturate the kinase; however, we did not observe significant changes in the pseudo-substrate affinity (Figure S3). Since the structure of the kinase in complex with AMP is not available, we speculate that the absence of the β and γ phosphates prevents the closure of the binding cleft, reducing the affinity for the substrate. Interestingly, two non-hydrolysable nucleotide mimics that are commonly used for structural studies, ATPγN and ATPγC, show a decrease in PKI5-24 binding cooperativity. In fact, the presence of a nitrogen atom in place of oxygen at the γ position of the ATP phosphoester dramatically reduces the binding cooperativity (σ = 53). This phenomenon is accentuated for ATPγC, where a methylene group replaces the bridging oxygen. In this case, the binding cooperativity between the non-hydrolysable nucleotide and pseudo-substrate is completely abolished (σ = 1.0).
Figure 2. Binding cooperativity between nucleotide and pseudo-substrate.

A) Two-state heterotropic linkage model for nucleotide and pseudo-substrate binding. B) Plot of the Kd of PKI5-24 to PKA-C in the presence of various nucleotides (See also Figure S1–3).
Table 1.
The affinity, degree of cooperativity (σ), and thermodynamics of PKI5-24 binding with respect to the kinase saturated with the nucleotide determined from ITC. Note that some of the binding isotherms (See also Figure S1,2) do not have a sharp inflection point and the ΔH and ΔS terms are less reliable.
| Kd (μM) | σ | ΔG (kcal/mol) | ΔH (kcal/mol) | TΔS (kcal/mol) | |
|---|---|---|---|---|---|
| Apo | 6.4 ± 0.56 | 1 | −7.14 ± 0.05 | −22.03 ± 0.63 | −14.90 ± 0.68 |
| ATPγC | 6.1 ± 0.27 | 1.0 | −7.17 ± 0.02 | −13.61 ± 0.36 | −6.43 ± 0.38 |
| AMP | 5.7 ± 0.18 | 1.1 | −7.20 ± 0.02 | −20.17 ± 0.07 | −12.97 ± 0.08 |
| Adenine | 1.5 ± 0.03 | 4.3 | −8.00 ± 0.01 | −19.43 ± 0.16 | −11.43 ± 0.18 |
| Adenosine | 0.53 ± 0.02 | 12 | −8.63 ± 0.02 | −21.32 ± 0.18 | −12.69 ± 0.20 |
| ADP | 0.23 ± 0.0097 | 29 | −9.13 ± 0.03 | −17.67 ± 0.35 | −8.54 ± 0.37 |
| ATPγN | 0.12 ± 0.0050 | 53 | −9.49 ± 0.02 | −18.12 ± 0.27 | −8.64 ± 0.30 |
| ATP | 0.016 ± 0.0019 | 400 | −10.72 ± 0.07 | −11.90 ± 0.34 | −1.17 ± 0.38 |
ATPγC prevents substrate binding
To further understand the role of the bridging oxygen of the β and γ phosphates on the binding cooperativity of more realistic substrates that display much lower affinity than the pseudo-substrate, we utilized a 19-amino acid peptide corresponding to the cytoplasmic domain of phospholamban (PLN1-19), a signaling target for PKA-C that regulates cardiac contractility (Masterson et al., 2010; Masterson et al., 2011b; Metcalfe et al., 2005). Although phospholamban contains a transmembrane domain, the cytoplasmic domain alone is recognized and phosphorylated by PKA-C (Masterson et al., 2011b). Remarkably, the binding titrations carried out in the presence of saturating concentration of ATPγC showed a dramatic reduction of the binding affinity. Previous binding studies show that the dissociation constant of PLN1-19 in the ternary complex PKA-C/ATPγN/PLN1-19 is approximately 28 μM (Kim et al., 2015a). In stark contrast, for the PKA-C/ATPγC complex the PLN1-19 peptide has a Kd > 1 mM (Figure S4A). Accordingly, NMR titrations of the PLN1-19 peptide on the PKA-C/ATPγC complex do not show any detectable chemical shift changes in both the amide and methyl group fingerprints (Figure S4B). Although PLN1-19 and PKI5-24 share the same recognition sequence, PKI5-24 contains a high affinity region (Cheng et al., 1986) that is not part of other substrates. Both ITC and NMR titrations indicate that the loss of binding affinity by ATPγC observed in the pseudo-substrate is not only confirmed using PLN1-19, but is accentuated with natural substrates.
Linkage between structural transitions and binding cooperativity
To probe the conformational transitions of the enzyme with the different nucleotides, we mapped the enzyme’s amide fingerprint using [1H,15N]-TROSY-HSQC experiments (Pervushin et al., 1997) as the amide chemical shifts are sensitive reporters of allosteric transitions (Figure S5) (Axe et al., 2014; Cembran et al., 2014; Selvaratnam et al., 2011). Although previous crystallographic structures of these complexes were reported to be identical (Johnson et al., 2001), small changes in chemical shifts have been demonstrated to report on subtle changes in conformation and allostery (Boulton et al., 2014; Selvaratnam et al., 2012a; Selvaratnam et al., 2012b). Resonance assignments for the different free and ligated states of the kinase were previously obtained using triple-resonance experiments (Kim et al., 2015a) and transferred to the spectra of the kinase saturated with the different nucleotides, as well as in the ternary complexes. We found that the addition of the pseudo-substrate to nucleotide-saturated PKA-C shifts the amide resonances of the enzyme along linear chemical shift trajectories (Figure 3A). This indicates that the enzyme predominately interconverts between two states and pseudo-substrate binding shifts the populations of the enzyme toward the closed state to different extents.
Figure 3. CONCISE analysis of the chemical shift changes with nucleotide.

A) [1H,15N]-TROSY-HSQC spectra showing the backbone amide chemical shift changes of PKA-C saturated with different nucleotides upon binding PKI5-24 (See also Figure S4–6) Residues following linear trajectories (blue spheres) plotted on the cartoon representation of the PKA-C crystal structure (PDB: 1ATP).
A succinct view of the process is offered by the analysis of the amide chemical shifts using CONCISE (Cembran et al., 2014), a statistical approach that utilizes principal component analysis to quantify the global coordinated response of the amide chemical shifts upon ligand binding. This method identifies the predominant linear trajectory of all the chemical shifts and the aggregate amide chemical shifts for each state is grouped together by their average position along the linear trajectory (PC score). These changes are displayed as probability density distributions defining a specific conformational state of the kinase along the conformational equilibrium (Cembran et al., 2014). The chemical shifts of each of the three major conformational states (open, intermediate, and closed) were previously defined by performing triple resonance experiments to assign the backbone chemical shifts or the apo, nucleotide bound and PKI bound forms of the enzyme (Kim et al., 2015a). Addition of PKI5-24 to all of the nucleotide-bound forms of the kinase shifts the conformational equilibrium toward the closed state (Figures 3, S6, and Table S1 and S2), reflected in the higher PC score from CONCISE, but to different degrees (Figure S7). In particular, a gradual shift of the probability density distributions of the amide chemical shifts is apparent going from adenine, adenosine, and ADP up to ATP, indicating that the presence of the ribose and the increased number of phosphates gradually shifts the conformational equilibrium toward the kinase closed state. To link the binding thermodynamics of PKI5-24 with the conformational transition, we first compared the free energy of binding of PKI5-24 under saturating conditions of nucleotide with the PC score (Figure S8) and did not find a firm correlation. However, a comparison between the free energy of binding for PKI5-24 and the PC score of the enzyme’s ternary complexes with PKI5-24 (F igure 6B) shows a linear correlation between the extent of the closed state and the free energy of binding (|R| = 0.95, Figure 6B), indicating that the degree of cooperativity of substrate binding strongly depends on the extent of the closed state.
Figure 6. Chemical Shift Trajectories with Nucleotides and ATP-competitive inhibitors.

A) [1H,15N]-TROSY-HSQC spectra showing the backbone amide chemical shift changes of PKA-C saturated with different ATP-competitive inhibits and nucleotides upon binding PKI5-24. B) Linear correlation between PC1 score and degree of cooperativity of the aggregate chemical shifts including the ATP-competitive inhibitors (see also Figure S7).
The CONCISE analysis yields insights into the global opening and closing transitions following only those residues with linear trajectories. Therefore, we followed the chemical shift trajectories of the Ile, Val, and Leu methyl groups. Methyl group chemical shifts also report on allosteric networks particularly in large proteins (Ruschak and Kay, 2012; Shi and Kay, 2014; Velyvis and Kay, 2013). Upon ligand binding most of the methyl group chemical shifts follow predominately linear trajectories along the open to close conformational states, reporting on a two state equilibrium in the fast exchange regime (Figure 4). However, several methyl groups in the PKA-C/ATPγC/PKI5-24 complex located near the binding site as well as in distal regions follow non-linear chemical shift trajectories, with many resonances exhibiting substantial line broadening even at ligand saturation conditions. These features suggest that ATPγC shifts the enzyme into another conformational state that is outside the open-to-closed reaction coordinates traceable with the other nucleotides and that binds substrates with lower affinity.
Figure 4. Effects of the ligand binding on the kinase side chains.

Methyl-TROSY spectra of 13C methyl-labeled PKA-C saturated with different nucleotides. Most of the resonances follow linear chemical shift trajectories. Several resonances for ATPγC do not lay the shared linear trajectory
ATP-competitive inhibitors exhibit positive and negative cooperativity
To understand the allosteric response of commonly used high affinity ATP-competitive inhibitors, we analyzed the binding cooperativity of H89 and balanol (Table 2 and Figure 5). H89 is an isoquinoline sulfonamide-based inhibitor designed to achieve higher selectivity for PKA-C and is commonly used to block PKA-C activity in cells (Chijiwa et al., 1990). Balanol is a natural occurring small molecule isolated from Verticillium balanoides with high inhibitory potency for PKC (Kulanthaivel et al., 1993) and PKA-C (Koide et al., 1995). We found that balanol displays a positive cooperativity (σ = 7.0) for PKI5-24. In contrast, H89, displays negative binding cooperativity (σ = 0.55). Our ITC results reveal that high affinity ATP-competitive inhibitors can modulate allosteric binding cooperativity in a manner similar to the nucleotides.
Table 2.
The affinity, degree of cooperativity (σ), and thermodynamics of PKI5-24 binding with respect to the kinase saturated with the ATP-competitive inhibitor determined from ITC.
| Kd (μM) | σ | ΔG (kcal/mol) | ΔH (kcal/mol) | TΔS (kcal/mol) | |
|---|---|---|---|---|---|
| Balanol | 0.92 ± 0.13 | 7.0 | −8.30 ± 0.09 | −16.14 ± 0.56 | −7.83 ± 0.65 |
| H89 | 11.2 ± 0.75 | 0.57 | −6.81 ± 0.04 | −18.46 ± 0.62 | −11.65 ± 0.60 |
Figure 5. Binding cooperativity between ATP-competitive inhibitors and pseudo-substrate.

A) ITC isotherms for PKI5-24 binding to PKA-C saturated with Balanol (left), and H89 (right). B) Plot of the Kd of PKI5-24 to PKA-C in the presence and absence of ATP-competitive inhibitors. C) Structure of PKA-C with balanol (PDB:1BX6, orange) and H89 (PDB: 1YDT, blue) with key electrostatic interactions (see also Figure S4–6 and Table S1–2).
To map the conformational state of PKA-C complexes with H89 or balanol and the pseudo-substrate PKI5-24, we used [1H,15N]-TROSY-HSQC experiments and performed similar analysis (Figure 6). Several residues of the amide backbone followed linear trajectories along the two-state equilibrium. Several other residues, however, showed marked deviations from these trends and were excluded from the CONCISE analysis (Figures 6, S7 and S8). Many of these non-linear trajectories result in the unique chemical environment around each ligand and not accurately report on the global conformational transition(Selvaratnam et al., 2012b). The probability distribution curves show that these ternary complexes are only partially closed and occupy intermediate position in the conformational equilibrium. When plotted as a function of the degree of binding cooperativity, the ternary complex with H89 falls within the linear correlation, displaying negative cooperativity, while the ternary complex with balanol, which shows a positive binding cooperativity, deviates from this relationship (Figure 6B). Although balanol shows cooperativity with pseudo-substrate binding, it fails to drive the entire enzyme toward a substrate competent state as in the case of ATP.
Analysis of the crystal structures and the NMR spectral signatures of the binary complexes with H89 and balanol explain the differences between their cooperative behaviors. The hydroxybenzophenone moiety together with the ester linkage of balanol form several hydrogen bonds with residues in the Gly-rich loop, i.e., Val57, Gly55, Phe54, Ser53, and Gly52, as well as the side chain groups of the conserved ion pair Lys72 and Glu91, and Asp184 (a residue in the DFG loop), while the bromocinnamoyl group of H89 does not make such electrostatic interactions (Figure 5C). This electrostatic interactions from balanol enable the Gly-rich loop to be clamped down and be coupled to the residues in the C-helix and large lobe. The NMR spectra show that, while both inhibitors caused apparent chemical shift changes in the small lobe, hinge and C-terminal residues, balanol perturbs the chemical shifts of the residues in the catalytically important motifs more significantly than H89 (Figures 7 and S9). These residues include Gly186, Ala188 (DFG loop), Thr197 (activation loop) and Glu203 (residue in the peptide positioning loop which interacts with the P-6 Arg). This allosteric interaction between the residues in the small lobe and substrate binding site may be responsible for the observed contrasting cooperativity effect with the two ATP-competitive inhibitors.
Figure 7. Chemical Shift Changes in the Active Site.

A) [1H,15N]-TROSY-HSQC spectra of the backbone chemical shifts of active site residues in PKA-C. In the ATPγN bound state, many of these resonances experience exchange so they are unobservable (dotted circle). B) Significant chemical shift changes upon binding of balanol and H89. Note that balanol induces more significant chemical shift changes in the active site compared to H89 (see also Figure S7).
DISCUSSION
Although it has been known that PKA-C exhibits K-type allosteric cooperativity (Kivi et al., 2014; Masterson et al., 2008), this work illuminates the structural basis for this phenomenon. The correlation we found between the conformational state of the ternary complex and the free energy of binding for the pseudo-substrate has many implications for kinase substrate recognition and function. The nucleotide emerges not only as carrier of the phosphate group for chemistry at the active site, but also having a structural role for mediating allosteric binding cooperativity. Importantly, high binding affinity of the substrate and pseudo-substrate can only be achieved when the appropriate nucleotide brings together the small and the large lobes, shifting the population of the enzyme toward the intermediate state (Cembran et al., 2014), pre-organizing the active site such that it is complementary for substrate binding and enabling the ternary complex to reach a catalytically competent state (Fersht, 1974; Schramm, 2011). This conformational transition is most closely obtained with native substrates and ATP, leading to the most favorable free energy of binding. Removal of the β- and γ-phosphate or ribose does significantly alter the nucleotide binding affinity, but results in an enzyme conformation progressively less complementary for substrate binding.
Interestingly, for both AMP and ATPγC, they did not fall within the linear relationship between free energy of pseudo-substrate binding and extent of the closed state, showing that these nucleotides do not exhibit positive K-type cooperativity (Figures 2B and 6). Not only AMP has a lower binding affinity, but it is also unable to drive the conformation of the glycine-rich loop in a competent state, abrogating binding cooperativity. Interestingly, the non-hydrolysable nucleotide mimic ATPγC has a similar effect (Figure 2). Our data could be explained by comparing the crystal structures of the kinase in the presence of SP20 and ATPγC (PDB entry 4IAC(Gerlits et al., 2013)) with that in complex with PKI5-24 and ATP (PDB entry 1ATP). Overall, the backbone structures of the two complexes are identical. However, in the 4IAC structure ATPγC does not interact with the backbone amide groups of Phe54 and Gly55, preventing the glycine-rich loop from closing and resulting in a more open conformation (Figure S11). Furthermore, in the presence of ATPγC, the second Mg2+ ion adopts a bi-pyramidal coordination geometry rather than the octahedral geometry found with both ATP and ATPγN (Gerlits et al., 2013). Therefore, the coordination of the metal ion plays a key role in positioning the γphosphate and the substrate for productive phosphoryl transfer. Moreover, the chemical shifts of the methyl groups suggest that the ternary complex PKA-C/ATPγC/PKI5-24 occupies a different state in the conformational landscape of the enzyme, with a loss of binding cooperativity for the pseudosubstrate and complete obliteration of binding for phospholamban. Although all chemical moieties are important for binding cooperativity, our data underscore the central role of the oxygen atom bridging the β and γ phosphate. Structurally, this atom provides the coordination geometry for the second Mg2+ ion, clamping down the glycine-rich loop and shifting the conformational equilibria for the formation of a productive ternary complex with substrates. In fact, the degree of cooperativity is significantly attenuated when nitrogen replaces the oxygen (ATPγN). The latter was also predicted but not proven in the published work by Walsh and coworkers (Whitehouse and Walsh, 1983). If the degree of cooperativity is attenuated by the substitution of the oxygen with nitrogen (ATPγN), it is completely lost with ATPγC, a widely used non-hydrolysable ATP analog for both kinases and ATPases(Toyoshima and Mizutani, 2004). In the case of PKA-C, this compound appears to prevent the formation of a catalytically competent complex with endogenous substrates.
Based on the above considerations, we conclude that the oxygen bridging the β and γ phosphate is a hot spot for modulating binding cooperativity. By changing the chemistry around this hot spot, it is possible to convert a kinase into a completely dead kinase abrogating both its canonical and non-canonical functions. These results may have important implications in the design of new inhibitors of kinases. It is possible to anticipate that newly designed inhibitors may be directed to either the catalytic function (i.e., phosphoryl transfer) or both catalytic and binding functions. In the former case, the kinase would still function as a pseudo-kinase preserving its signaling role as scaffolds, anchors, spatial modulators, traps, and ligand-driven regulators of canonical kinases (Hu et al., 2011; Reiterer et al., 2014). In fact this case has previously been observed with small molecular inhibitors of RAF kinase, activating kinase signaling pathways in a dose dependent manner (Hatzivassiliou et al., 2010; Poulikakos et al., 2010) or in the more recent finding that ATP-competitive inhibitors can block protein kinase recruitments to the Hsp90-Cdc37 system(Polier et al., 2013). In the latter case, the kinase would be totally dead and removed from its signaling pathways (Figure 8). Mutations have been reported to convert active kinases into pseudo-kinases, preserving their non-catalytic functions (Hu et al., 2013; Iyer et al., 2005). In the case of PKA-C, residual catalytic activity enables yeast to survive(Gibbs and Zoller, 1991). On the other hand, a catalytically dead PKA-C fails to be autophosphorylated, but it can still bind ATP as well as the regulatory subunits and is recognized and phosphorylated by PDK-1 (Iyer et al., 2005). Another notable case is with a mutation that fuses the C-spine, blocks ATP binding and allows for dimerization of RAF kinase domains (Hu et al., 2011). ATPγC renders the kinase dead, abolishing both catalysis and binding of substrates, despite assembly of the C-spine.
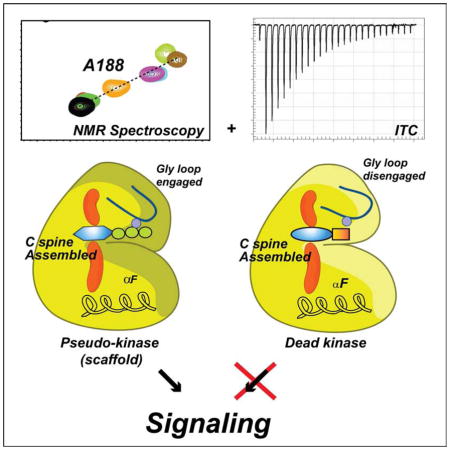
Figure 8. Uncoupling canonical and non-canonical function of kinases.

The apo enzyme (blue) displays both disassembled C spine and disengaged Gly-loop. Binding of non-hydrolyzable ATP analogs or good ATP mimic drug inhibitors (i.e., able to engage Gly-rich loop via coordination of Mg2+ ion) produce a pseudokinase that is unable to carry out phosphoryl transfer but is able to bind substrates cooperatively (yellow). Binding of drug inhibitors that are unable to properly coordinate the Mg2+ ion to engage the Gly-loop (i.e., ATPγC, H89, see also Figure S8) obliterates both catalytic and scaffolding function (dead kinase).
In fact, widely used ATP-competitive inhibitors such as balanol and H89 are able to render PKA-C a pseudo-kinase and a dead kinase, respectively. Although both inhibitors contain aromatic rings that intercalate with the C spine residues, balanol possesses several polar groups that form hydrogen bonds with side-chain groups critical for substrate recognition and catalysis, including the conserved Lys72 and Glu91 in the active site (Narayana et al., 1999), the conserved Asp184 in the DFG loop, and the Gly-rich loop. H89 lacks these hydrophilic groups and the interaction within the binding site is almost exclusively through hydrophobic contacts. The polar interactions in balanol bring down the Gly-rich loop in a quasi-competent state. This is further supported by the crystal structure where the Gly-rich loop in the H89-bound complex is directed away from the catalytic loop, resulting in the loss of interaction between the pseudosubstrate and the Gly-rich loop, resulting in a negative cooperativity. In the committed complex, the movement of the Gly-rich loop coordinated by the nucleotide corresponds to allosteric rearrangements of the residues in the substrate binding site (Masterson et al., 2010; Masterson et al., 2008). Interestingly, both balanol and H89’s electrostatic interactions with the critical Mg2+ ion are not observed in the X-ray structures. Future design of inhibitors exploiting the hot spot for binding cooperativity may lead to greater control of kinase function in vivo and tune the kinase binding cooperativity.
In a recent work on Src kinase, Foda et al. show a negative binding cooperativity between ATP and substrates(Foda et al., 2015); while a positive cooperativity was measured for ADP and phosphorylated substrate. These authors found that the negative cooperativity is mediated by an allosteric network of contacts initiated by a protonation event occurring at the DFG loop (Foda et al., 2015). This contrasts the positive K-type binding cooperativity found for PKA-C (Masterson et al., 2008), where a high degree of cooperativity was observed for ATP with the endogenous protein kinase inhibitor (PKI), regulatory subunits (R-subunits), (Herberg and Taylor, 1993) and substrates (Kim et al., 2015a). These findings suggest that the mechanism for cooperativity is not uniform throughout the kinome and that cooperativity hot spots may become a target for designing inhibitors able to fine-tune specific signaling pathways.
In conclusion, our study identifies a hot spot for tuning binding cooperativity and decoupling canonical and non-canonical functions in kinases, introducing the concept of a possible higher level of control achievable by high affinity competitive inhibitors. By modulating the extent of closure of the Gly-rich loop, newly designed kinase inhibitors would then be able to convert a kinase into a pseudokinase, i.e., creating a scaffold that it is still involved in signaling, or alternatively, subtract kinases from their signaling pathways altogether, impairing both enzymatic activity and substrate binding (dead kinases) (Reiterer et al., 2014; Shaw et al., 2014).
EXPERIMENTAL PROCEDURES
Adenosine 5′-triphosphate (ATP), adenosine 5′-monophosphate (AMP), γ-β-methyleneadenosine 5′-triphosphate (ATPγC), adenine, and N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89) were purchased from Sigma Aldrich (St. Louis, MO, USA). Adenosine 5′-diphosphate (ADP) and adenosine were purchased from Research Products International (Mt. Prospect, IL, USA). Adenosine 5′-(β,γ-imido)triphosphate (ATPγN) was purchased from Roche Diagnostics (Indianapolis, IN, USA). Balanol was purchased from AnalytiCon Discovery, LLC (Rockville, MD, USA).
Sample Preparation
Recombinant catalytic subunit of PKA was expressed in BL21 (DE3) cells as previously described by Studier(Studier, 2005) at 24 °C. Purification of PKA-C was performed as previously described using the His6-RIIα(R213K)(Hemmer et al., 1997) subunit and a second purification step was performed using the HiTrap SP cation exchange column as previously described(Chao et al., 2014). The most abundant isoform, corresponding to phosphorylation at S338, T197 and S10, was used for all experiments. Peptide synthesis was performed on a CEM Liberty microwave synthesizer as described previously(Masterson et al., 2011a). Kinase activity was tested with a gel shift assay and quantified using A280 = 52060 M−1cm−1.
ITC measurements
All ITC measurements were performed with a Microcal VP-ITC instrument or TA NanoITC instrument at 300K. Samples were buffer exchanged into 20 mM MOPS, 90 mM KCl, 10 mM DTT, 10 mM MgCl2, 1 mM NaN3, pH 6.5. Approximately 1.7 mL of 11.4–32μM of PKA-C was used for each experiment and 280 μL of 140–350μM of PKI5-24 in the titrant syringe. For the AMP binding experiment 300 μL of 238μM of PKA-C was used with 50 μL of 3.3mM of AMP. Final concentration of 2mM of nucleotide was used for nucleotide saturated experiments and a concentration of 50 μM for the inhibitor saturated experiments. All experiments were performed in triplicate. The heat of dilution of the ligand to the buffer was taken into account by measuring the heat of dilution of the ligand to the buffer and was subtracted from the experiment accordingly. Binding was assumed to be 1:1 and was analyzed using the NanoAnalyze software (TA instruments New Castle, DE, USA), with the Wiseman Isotherm(Wiseman et al., 1989):
where the change of the total complex, d[MX] with respect to the change of the ligand concentration, d[Xtot] is dependent on r, the ratio of the Kd with respect to the total protein concentration, and Rm, the ratio between the total ligand and total protein concentration. The free energy of binding was determined using the following:
where R is the universal gas constant and T is the temperature at measurement (300 K). The entropic contribution to binding was calculated using the following:
Calculations for the cooperativity constant (σ) were calculated as follows:
where Kd,Apo is the Kd of PKI5-24 binding to the apo enzyme and Kd,nucleotide or Kd,inhibitor is the Kd of PKI5-24 to the nucleotide- or inhibitor-bound enzyme, respectively.
NMR Experiments
Samples for 13C IVL 15N labeled PKA-C were expressed and purified as previously described(Chao et al., 2014; Masterson et al., 2008). Effective final sample concentrations were 0.2–0.25 mM in 20 mM KH2PO4, 90 mM KCl, 10 mM DTT, 10 mM MgCl2 1 mM NaN3 at pH 6.5 with 12 mM of nucleotide. Adenosine and adenine lack solubility in aqueous solution and concentrations of 10 mM and 6mM respectively were used. For ATP-competitive inhibitors concentrations of 0.8 mM of Balanol and 3 mM of H89 were used. Samples with ATP and ATPγN were performed with 60 mM MgCl2 for MgATP. Additions of 4, 8, 12, 18, 24, and 32 μL of 4.0 mM of PKI5-24 were used for a minimum final two fold molar excess of ligand. NMR assignments on the apo, nucleotide bound (ATPγN) and ternary (ATPγN and PKI5-24) were carried out on an 850 MHz Bruker Avance III spectrometer and described elsewhere (Kim et al., 2015b). 1H-15N TROSY-HSQC experiments and 1H-13C HMQC experiments for nucleotide-bound PKA-C were carried out on a Varian Inova 600 MHz spectrometer equipped with a Cold HCN probe operating at 300 K. [1H-15N] TROSY-HSQC experiments of PKA-C with ATP-competitive inhibitors were performed on a Bruker Avance 700 MHz spectrometer equipped with a TXI probe.
Supplementary Material
HIGHLIGHTS.
Linearly between extent of the kinase closed state and cooperativity
Canonical and non-canonical function of the kinases can be uncoupled
Bridging oxygen of ATP is a hot spot for cooperativity
Formation of pseudo-kinase and dead kinase by ATP-competitive inhibitors
Acknowledgments
This work is supported by the NIH (GM100310 and GM72701 to GV and T32AR007612 to JK). We thank Prof. Alessandro Cembran (University of Minnesota-Duluth) and Prof. Larry Masterson (Hamline University) for helpful discussions, and Prof. Robert Geraghty (University of Minnesota) for access to the VP-ITC instrument. NMR Experiments were carried out at the Minnesota NMR Center.
Footnotes
AUTHORS CONTRIBUTIONS
G.V., S.S.T., M.W. conceived, directed and analyzed all experimental research; G.V., J.K. prepared the manuscript; J.K. and G.L. prepared the samples, performed NMR spectroscopy experiments, processed and interpreted the data. All authors discussed the results and implications and commented on the manuscript at all stages.
Supplemental information includes 8 figures and 2 tables and can be found with this article online at ***
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chemical reviews. 2001;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]
- Arencibia JM, Pastor-Flores D, Bauer AF, Schulze JO, Biondi RM. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochimica et biophysica acta. 2013;1834:1302–1321. doi: 10.1016/j.bbapap.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Axe JM, Yezdimer EM, O’Rourke KF, Kerstetter NE, You W, Chang C-eA, Boehr DD. Amino Acid Networks in a (β/α)8 Barrel Enzyme Change during Catalytic Turnover. Journal of the American Chemical Society. 2014;136:6818–6821. doi: 10.1021/ja501602t. [DOI] [PubMed] [Google Scholar]
- Bastidas AC, Deal MS, Steichen JM, Guo Y, Wu J, Taylor SS. Phosphoryl transfer by protein kinase A is captured in a crystal lattice. Journal of the American Chemical Society. 2013;135:4788–4798. doi: 10.1021/ja312237q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar D, Roskoski R, Jr, Rosendahl MS, Leonard NJ. Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: a new fluorescence displacement titration technique for characterizing the nucleotide binding site on the catalytic subunit. Biochemistry. 1983;22:6310–6317. doi: 10.1021/bi00295a042. [DOI] [PubMed] [Google Scholar]
- Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Boulton S, Akimoto M, Selvaratnam R, Bashiri A, Melacini G. A tool set to map allosteric networks through the NMR chemical shift covariance analysis. Sci Rep. 2014;4:7306. doi: 10.1038/srep07306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cembran A, Kim J, Gao J, Veglia G. NMR mapping of protein conformational landscapes using coordinated behavior of chemical shifts upon ligand binding. Phys Chem Chem Phys. 2014;16:6508–6518. doi: 10.1039/c4cp00110a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao FA, Kim J, Xia Y, Milligan M, Rowe N, Veglia G. FLAMEnGO 2.0: An enhanced fuzzy logic algorithm for structure-based assignment of methyl group resonances. Journal of magnetic resonance. 2014;245:17–23. doi: 10.1016/j.jmr.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. The Journal of biological chemistry. 1986;261:989–992. [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. The Journal of biological chemistry. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Cowan-Jacob SW, Jahnke W, Knapp S. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future medicinal chemistry. 2014;6:541–561. doi: 10.4155/fmc.13.216. [DOI] [PubMed] [Google Scholar]
- Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annual review of biochemistry. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, Johnson LN. The structural basis for control of eukaryotic protein kinases. Annual review of biochemistry. 2012;81:587–613. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Fang Z, Grutter C, Rauh D. Strategies for the selective regulation of kinases with allosteric modulators: exploiting exclusive structural features. ACS chemical biology. 2013;8:58–70. doi: 10.1021/cb300663j. [DOI] [PubMed] [Google Scholar]
- Fersht AR. Catalysis, binding and enzyme-substrate complementarity. Proceedings of the Royal Society of London Series B, Biological sciences. 1974;187:397–407. doi: 10.1098/rspb.1974.0084. [DOI] [PubMed] [Google Scholar]
- Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. The Journal of biological chemistry. 1955;216:121–132. [PubMed] [Google Scholar]
- Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA. A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat Commun. 2015;6:5939. doi: 10.1038/ncomms6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire E, Schön A, Velazquez-Campoy A. Chapter 5 Isothermal Titration Calorimetry. 2009;455:127–155. doi: 10.1016/S0076-6879(08)04205-5. [DOI] [PubMed] [Google Scholar]
- Gerlits O, Waltman MJ, Taylor S, Langan P, Kovalevsky A. Insights into the phosphoryl transfer catalyzed by cAMP-dependent protein kinase: an X-ray crystallographic study of complexes with various metals and peptide substrate SP20. Biochemistry. 2013;52:3721–3727. doi: 10.1021/bi400066a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CS, Zoller MJ. Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. The Journal of biological chemistry. 1991;266:8923–8931. [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Hemmer W, McGlone M, Taylor SS. Recombinant strategies for rapid purification of catalytic subunits of cAMP-dependent protein kinase. Analytical biochemistry. 1997;245:115–122. doi: 10.1006/abio.1996.9952. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Taylor SS. Physiological inhibitors of the catalytic subunit of cAMP-dependent protein kinase: effect of magnesium-ATP on protein-protein interactions. Biochemistry. 1993;32:14015–14022. doi: 10.1021/bi00213a035. [DOI] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–1046. doi: 10.1016/j.cell.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, Shaw AS. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6067–6072. doi: 10.1073/pnas.1102554108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer GH, Moore MJ, Taylor SS. Consequences of lysine 72 mutation on the phosphorylation and activation state of cAMP-dependent kinase. The Journal of biological chemistry. 2005;280:8800–8807. doi: 10.1074/jbc.M407586200. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chemical reviews. 2001;101:2243–2270. doi: 10.1021/cr000226k. [DOI] [PubMed] [Google Scholar]
- Johnson LN, Lewis RJ. Structural basis for control by phosphorylation. Chemical reviews. 2001;101:2209–2242. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Kim J, Masterson LR, Cembran A, Verardi R, Shi L, Gao J, Taylor SS, Veglia G. Dysfunctional conformational dynamics of protein kinase A induced by a lethal mutant of phospholamban hinder phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2015a;112:3716–3721. doi: 10.1073/pnas.1502299112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Masterson LR, Cembran A, Verardi R, Shi L, Gao J, Taylor SS, Veglia G. Dysfunctional conformational dynamics of protein kinase A induced by a lethal mutant of phospholamban hinder phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2015b:201502299. doi: 10.1073/pnas.1502299112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivi R, Jemth P, Jarv J. Thermodynamic aspects of cAMP dependent protein kinase catalytic subunit allostery. Protein J. 2014;33:386–393. doi: 10.1007/s10930-014-9570-1. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991a;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991b;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- Koide K, Bunnage ME, Gomez Paloma L, Kanter JR, Taylor SS, Brunton LL, Nicolaou KC. Molecular design and biological activity of potent and selective protein kinase inhibitors related to balanol. Chem Biol. 1995;2:601–608. doi: 10.1016/1074-5521(95)90124-8. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS, Ten Eyck LF. A helix scaffold for the assembly of active protein kinases. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulanthaivel P, Hallock YF, Boros C, Hamilton SM, Janzen WP, Ballas LM, Loomis CR, Jiang JB, Katz B. Balanol: a novel and potent inhibitor of protein kinase C from the fungus Verticillium balanoides. Journal of the American Chemical Society. 1993;115:6452–6453. [Google Scholar]
- Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002a;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002b;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Masterson LR, Cembran A, Shi L, Veglia G. Allostery and binding cooperativity of the catalytic subunit of protein kinase A by NMR spectroscopy and molecular dynamics simulations. Advances in protein chemistry and structural biology. 2012;87:363–389. doi: 10.1016/B978-0-12-398312-1.00012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Cheng C, Yu T, Tonelli M, Kornev A, Taylor SS, Veglia G. Dynamics connect substrate recognition to catalysis in protein kinase A. Nature chemical biology. 2010;6:821–828. doi: 10.1038/nchembio.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Mascioni A, Traaseth NJ, Taylor SS, Veglia G. Allosteric cooperativity in protein kinase A. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:506–511. doi: 10.1073/pnas.0709214104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Shi L, Metcalfe E, Gao J, Taylor SS, Veglia G. Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 2011a;108:6969–6974. doi: 10.1073/pnas.1102701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson LR, Yu T, Shi L, Wang Y, Gustavsson M, Mueller MM, Veglia G. cAMP-dependent protein kinase A selects the excited state of the membrane substrate phospholamban. Journal of molecular biology. 2011b;412:155–164. doi: 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon CL, Kornev AP, Gilson MK, Taylor SS. Dynamic architecture of a protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4623–4631. doi: 10.1073/pnas.1418402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe EE, Traaseth NJ, Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- Narayana N, Diller TC, Koide K, Bunnage ME, Nicolaou KC, Brunton LL, Xuong NH, Ten Eyck LF, Taylor SS. Crystal structure of the potent natural product inhibitor balanol in complex with the catalytic subunit of cAMP-dependent protein kinase. Biochemistry. 1999;38:2367–2376. doi: 10.1021/bi9820659. [DOI] [PubMed] [Google Scholar]
- Ni DQ, Shaffer J, Adams JA. Insights into nucleotide binding in protein kinase A using fluorescent adenosine derivatives. Protein Science. 2000;9:1818–1827. doi: 10.1110/ps.9.9.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole–dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in3solution. Proceedings of the National Academy of Sciences. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polier S, Samant RS, Clarke PA, Workman P, Prodromou C, Pearl LH. ATP-competitive inhibitors block protein kinase recruitment to the Hsp90-Cdc37 system. Nature chemical biology. 2013;9:307–312. doi: 10.1038/nchembio.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiterer V, Eyers PA, Farhan H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 2014;24:489–505. doi: 10.1016/j.tcb.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Ruschak AM, Kay LE. Proteasome allostery as a population shift between interchanging conformers. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3454–3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm VL. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem. 2011;80:703–732. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, Chowdhury S, VanSchouwen B, Melacini G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:6133–6138. doi: 10.1073/pnas.1017311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, Mazhab-Jafari MT, Das R, Melacini G. The Auto-Inhibitory Role of the EPAC Hinge Helix as Mapped by NMR. PloS one. 2012a;7:e48707. doi: 10.1371/journal.pone.0048707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaratnam R, VanSchouwen B, Fogolari F, Mazhab-Jafari MT, Das R, Melacini G. The projection analysis of NMR chemical shifts reveals extended EPAC autoinhibition determinants. Biophysical journal. 2012b;102:630–639. doi: 10.1016/j.bpj.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and pseudokinases: lessons from RAF. Molecular and cellular biology. 2014;34:1538–1546. doi: 10.1128/MCB.00057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Kay LE. Tracing an allosteric pathway regulating the activity of the HslV protease. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2140–2145. doi: 10.1073/pnas.1318476111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims PC, Moody IS, Choi Y, Dong C, Iftikhar M, Corso BL, Gul OT, Collins PG, Weiss GA. Electronic measurements of single-molecule catalysis by cAMP-dependent protein kinase A. Journal of the American Chemical Society. 2013;135:7861–7868. doi: 10.1021/ja311604j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava AK, McDonald LR, Cembran A, Kim J, Masterson LR, McClendon CL, Taylor SS, Veglia G. Synchronous Opening and Closing Motions Are Essential for cAMP-Dependent Protein Kinase A Signaling. Structure. 2014;22:1735–1743. doi: 10.1016/j.str.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein expression and purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, Anand GS. Dynamics of signaling by PKA. Biochimica et biophysica acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochimica et biophysica acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Mizutani T. Crystal structure of the calcium pump with a bound ATP analogue. Nature. 2004;430:529–535. doi: 10.1038/nature02680. [DOI] [PubMed] [Google Scholar]
- Velyvis A, Kay LE. Measurement of active site ionization equilibria in the 670 kDa proteasome core particle using methyl-TROSY NMR. Journal of the American Chemical Society. 2013;135:9259–9262. doi: 10.1021/ja403091c. [DOI] [PubMed] [Google Scholar]
- Whitehouse S, Feramisco JR, Casnellie JE, Krebs EG, Walsh DA. Studies on the kinetic mechanism of the catalytic subunit of the cAMP-dependent protein kinase. The Journal of biological chemistry. 1983;258:3693–3701. [PubMed] [Google Scholar]
- Whitehouse S, Walsh DA. Mg X ATP2-dependent interaction of the inhibitor protein of the cAMP-dependent protein kinase with the catalytic subunit. Journal of Biological Chemistry. 1983;258:3682–3692. [PubMed] [Google Scholar]
- Wiseman T, Williston S, Brandts JF, Lin LN. Rapid Measurement of Binding Constants and Heats of Binding Using a New Titration Calorimeter. Analytical biochemistry. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends in pharmacological sciences. 2015 doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Zimmermann B, Schweinsberg S, Drewianka S, Herberg FW. Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem J. 2008;413:93–101. doi: 10.1042/BJ20071665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.