

Abstract
Known for its distinct metamorphic behavior, XCL1 interconverts between a canonical chemokine folded monomer (XCL1mon) that interacts with the receptor, XCR1, and a unique dimer (XCL1dim) that interacts with glycosaminoglycans and inhibits HIV-1 activity. This study presents the first detailed analysis of the GAG binding properties of XCL1dim. Basic residues within a conformationally selective dimeric variant of XCL1 (W55D) were mutated and analyzed for their effects on heparin binding. Mutation of Arg23 and Arg43 greatly diminished the level of heparin binding in both heparin Sepharose chromatography and surface plasmon resonance assays. To assess the contributions of different GAG structures to XCL1 binding, we developed a solution fluorescence polarization assay and correlated affinity with the length and level of sulfation of heparan sulfate oligosaccharides. It was recently demonstrated that the XCL1 GAG binding form, XCL1dim, is responsible for preventing HIV-1 infection through interactions with gp120. This study defines a GAG binding surface on XCL1dim that includes residues that are important for HIV-1 inhibition.
Chemokines are noted for their ability to orchestrate cell migration and support a vast array of immunological and homeostatic mechanisms that mediate many different biological processes.1 Like other members of the chemokine family, XCL1 stimulates leukocyte chemotaxis through interactions with a cognate G protein-coupled receptor (GPCR) XCR1 and glycosaminoglycans (GAGs) on cell surfaces and within the extracellular matrix.2,3 These interactions are important for cell signaling and forming chemotactic gradients that provide directional cues for target cells, respectively.
Unlike other chemokines that utilize a single folded conformation to interact with both GPCRs and GAGs, XCL1 has evolved to parse GPCR and GAG binding capabilities between two distinct structural states, XCL1mon and XCL1dim. 4,5 XCL1mon binds to the receptor XCR1 using the canonical chemokine fold consisting of three β-strands and a C-terminal α-helix.4,6 The XCL1dim conformation is an unrelated β-sandwich dimer that is responsible for binding to GAGs.5 Both native state structures can be accessed through a dynamic equilibrium unfolding process,7 designated as metamorphic interconversion.
Since its discovery as a metamorphic protein, we have worked to develop a structure–activity map that defines the functional elements of XCL1 responsible for metamorphic interconversion, XCR1 binding, and GAG binding.5,7,8 For example, Tyler et al. identified interactions that allow the folding equilibrium of XCL1mon and XCL1dim to respond to perturbations in ionic strength.8 XCL1mon is destabilized by the electrostatic repulsion existing between Arg23 and Arg43 because of their proximity within the secondary structure. However, this repulsion is mitigated upon coordination with a chloride ion, stabilizing XCL1mon. Conversely, these repulsive forces push the equilibrium toward XCL1dim under low-salt conditions, where inter- and intramolecular salt bridges (Arg9–Glu50 and Glu31–K25) further stabilize XCL1dim. Thus, individual side chains can contribute to XCL1 function by stabilizing a specific conformational state, interacting directly with a binding partner, or both.
Recent findings suggest that XCL1 functions as an “inflammatory specialist” and facilitates interactions between dendritic cells and T cells that result in the propagation of the cytotoxic immune response and maintenance of self-tolerance.9–11 At this point, however, little is known about how the metamorphic behavior of XCL1 contributes to its role in the immune system. An intriguing aspect of XCL1 biology is its ability to bind to GAGs using the XCL1dim conformation that is not found among other members of the chemokine family. Recently, Guzzo et al. demonstrated that XCL1 is able to bind to gp120 and prevent HIV-1 infection in both primary and TZMbl cells.12 Mutational analysis indicated that the anti-HIV-1 activity of XCL1 depends on residues that also participate in GAG binding.13 A link between XCL1–GAG binding and antiviral potency may reveal the evolutionary advantage conferred by the metamorphic XCL1 native state.
Previous efforts to characterize XCL1–GAG interactions preceded the determination of the XCL1dim structure.14 Because this earlier study did not account for the influence of individual amino acid substitutions on the metamorphic native state equilibrium and the results were interpreted in the context of the XCL1mon conformation, the contributions of specific residues to GAG binding remain uncertain. In the study presented here, we define the basis for XCL1–GAG interactions in the context of XCL1dim, the functionally relevant GAG binding conformation. Because of the emerging role of XCL1dim in HIV-1 inhibition, and the enhanced efforts to understand its metamorphic behavior, these studies provide a foundation for future HIV and drug discovery studies.
MATERIALS AND METHODS
Mutagenesis and Purification of Recombinant XCL1 W55D Proteins
The QuikChange site-directed mutagenesis kit (Stratagene) was used to mutate several basic residues in the XCL1 W55D variant, which preferentially adopts the XCL1dim conformation.4 Protein purification was conducted as previously described15 with minor modifications. Upon elution from Ni-NTA resin, W55D proteins were refolded by dialysis into warmed (37 °C) refolding buffer consisting of 4 L of 20 mM Tris (pH 8.0), 10 mM cysteine, and 0.5 mM cystine. The protein was dialyzed at 37 °C overnight while being stirred. After incubation, ULP1 protease was added to the protein solution to cleave the His-SUMO fusion tag from XCL1. The cleavage reaction mixture was incubated for approximately 3 h at 30 °C. Cation exchange chromatography was performed to purify the His-SUMO tag from XCL1. The cation exchange and reverse phase high-performance liquid chromatography (HPLC) steps were performed as previously described.15 The molecular weights of all purified proteins were verified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry.
One-Dimensional (1D) and Two-Dimensional (2D) Nuclear Magnetic Resonance (NMR) Analysis of XCL1 W55D Proteins
Lyophilized protein was dissolved into 20 mM sodium phosphate (pH 6.0) and 10% D2O to a final concentration of 100 μM. 1H NMR and 1H–15N heteronuclear quantum coherence (HSQC) NMR experiments were performed with XCL1 W55D proteins on a Bruker 600 MHz spectrometer. All experiments were performed at 37 °C with the exception of the unfolded variant (CC0), which was performed at 10 °C. HSQC spectra were processed using NMRPipe.16
Heparin and SP-Sepharose Binding Assay
Electrostatic binding and heparin binding were assessed for XCL1 W55D variant proteins using cation exchange SP-Sepharose and heparin Sepharose chromatography, respectively, based on previously described methods.15,17,18 Briefly, ~200 μg of each purified protein was individually loaded onto 1 mL HiTrap SP-Sepharose HP and heparin HP columns (GE Healthcare). A Shimadzu Prominence HPLC system was used to run the columns with a 0 to 1 M NaCl gradient in 20 mM phosphate (pH 6.0) buffer over 90 min. Protein elution was monitored by recording the absorbance at 280 nm. For variants that had two elution peaks (i.e., R23A/R43A) or shoulders (i.e., K8A), the peak that was selected for analysis was the most dominant peak that eluted at the lowest concentration of NaCl or the nonshoulder portion of the peak, respectively. The elution peak for each protein was recorded as a function of NaCl concentration from the heparin Sepharose ([NaCl]H) resin and the cation exchange resin ([NaCl]S). Δ[NaCl]H and Δ[NaCl]S were calculated by subtracting heparin and SP-Sepharose elution values from that of XCL1 W55D. To determine the heparin binding specificity for each protein (ΔΔ[NaCl]), the electrostatic interactions (Δ[NaCl]S) were subtracted from the heparin values (Δ[NaCl]H) in the equation ΔΔ[NaCl] = Δ[NaCl]H − Δ[NaCl]S.
Surface Plasmon Resonance
Lyophilized proteins were dissolved in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 0.05% Tween 20 at concentrations varying from 100 to 1000 nM. The samples were injected onto a BIAcore 3000 instrument (GE Healthcare/BIAcore, Uppsala, Sweden) with a C1 sensor chip prepared with heparin. Chip preparation and validation were conducted as previously described.19,20 The proteins were applied to the heparin-coated chip at 25 °C with a flow rate of 40 μL/min for 5 min, a flow rate typically used for chemokine–GAG interactions that minimizes mass transfer effects.21 Proteins were dissociated from the chip, and the chip surface was regenerated with 0.1 M glycine, 1 M NaCl, and 0.1% Tween 20 (pH 9.5). Data analysis was completed using BIAevaluation software (GE Healthcare/BIAcore) using a 1:1 Langmuir binding model.19,20 Apparent dissociation constant [Kd(app)] values were calculated using both kinetic (koff/kon) and steady state analysis. Dissociation constants are reported as apparent because we speculate that this type of analysis, based on a simple 1:1 binding model, does not account for cooperative interactions or the assembly of chemokine dimers or higher-order chemokine oligomer complexes on the SPR chip. In this case, we are concerned with formation of XCL1dim. This limits accurate determination of binding affinities. For the steady state, the maximal response values generated for each protein concentration tested were recorded at equilibrium. The data were fit using ProFit (QuantumSoft).
Fluorescence Polarization (FP) Assays
Proteins were serially diluted, at concentrations ranging from 0 to 100 μM, in 20 mM sodium phosphate (pH 7.2, the optimal pH for fluorescein fluorescence). Purified fluorescein-tagged heparin with a degree of polymerization of 4 (dp4) was added to each diluted protein sample at a final concentration of 100 nM. Samples were transferred to a quartz cuvette, and FP was measured at 37 °C using a PTI (Birmingham, NJ) fluorometer equipped with FeliX32 software. Excitation and emission of the fluorescein tag were monitored at 492 and 512 nm, respectively. Fluorescein-tagged dp4 was prepared from controlled partial heparin lyase 1 treatment of fluorescein-tagged heparin22 followed by size fractionation in the Linhardt lab (Rensselear Polytechnic Institute). ProFit (QuantumSoft) was used for nonlinear fitting of the binding curves and apparent dissociation constants [Kd(app)] for dp4–XCL1.
Fluorescence Polarization Competition Assays
XCL1 W55D was diluted to a final concentration equivalent to its Kd(app) value calculated from the FP method described above (2.2 μM) in 20 mM sodium phosphate (pH 7.2). Molecules of purified unlabeled/unmodified heparin or heparan sulfate (HS) were titrated at concentrations ranging from 0 to 100 mM. Fluorescein-labeled heparin dp4 was added to each solution at a final concentration of 100 nM. FP was measured for each sample at 37 °C using the same instrumentation and software noted above. ProFit software was used for nonlinear fitting analysis of competition curves. IC50 values were determined using in-house scripts. Ki values were calculated using the BotDB IC50-to-Ki online converter.23 HS was purchased from Glycan Therapeutics Inc. (Chapel Hill, NC): HS-4 = GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-pNP, HS-8 = GlcNS-GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNS-GlcA-pNP, HS-10 = GlcNS-GlcA-GlcNS-Glc2S-GlcNS-Glc2S-GlcNS-GlcA-pNP, HS-14 = GlcNS6S-GlcA-GlcNS6S-Glc2S-GlcNS6S-Glc2S-GlcNS6S-GlcA-pNP, and HS-15 = GlcNS6S-GlcA-GlcNS3S6S-Glc2S-GlcNS6S-Glc2S-GlcNS6S-GlcA-pNP.
RESULTS
Comparison of the Electrostatic Surface of XCL1mon and XCL1dim
Electrostatic attraction occurs between basic chemokine residues and acidic carboxylate and sulfate groups of GAGs.17,18 Because Coulombic attraction plays a role in this interaction, basic residues within XCL1 were chosen as initial targets of mutagenesis to probe for residues that mediate XCL1–GAG binding. A conformationally selective variant of XCL1 was previously designed to eliminate structural interconversion to isolate the dimer species (XCL1dim). Tryptophan 55, a residue that normally stabilizes the hydrophobic core of the XCL1mon conformation, was mutated to an aspartic acid to produce the conformationally selective dimer known as XCL1 W55D.5 Because of the role of the XCL1dim conformation in GAG binding, alanine point mutations of several basic residues were made within the W55D background.
To undergo the transition between the two native states, XCL1 undergoes a large-scale unfolding process that involves the rearrangement of secondary structural elements.7 This restructuring is accompanied by changes in the overall distribution of electrostatic charges on the surface of XCL1 (Figure 1A–D). XCL1mon contains 15 basic and six acidic residues (Figure 1E), resulting in a theoretical isoelectric point (pI) of 10.6. In this conformation, four basic residues are localized to a patch that includes the C-terminal α-helix and parts of the loop regions, while the remaining 11 basic residues reside in the loop regions as well as the unstructured N- and C-termini (Figure 1A,E). By comparison to XCL1dim (Figure 1D), XCL1mon has a smaller electrostatic distribution with basic residues mainly localized to a patch composed of the 20s loop, 40s loop, and part of the C-terminal α-helix (Figure 1C).
Figure 1.

Comparison of the electrostatic surfaces of XCL1mon and XCL1dim. Structural representation of charged amino acid residues in (A) XCL1mon (PDB entry 1J8I) and (B) XCL1dim (PDB entry 2JP1). Aspartic and glutamic acid side chains are shown as red sticks, while arginine and lysine side chains are shown as blue sticks. For the sake of clarity, basic and acidic residues were not labeled on the distal subunit in panel B. Disulfide bonds are colored yellow. Electrostatic potentials were calculated using the APBS plugin in PYMOL and mapped on the surface of (C) XCL1mon and (D) XCL1dim. Blue shading is indicative of basic surfaces, and red shading is indicative of acidic surfaces. (E) Amino acid sequence and illustration of the secondary structural elements of XCL1 for XCL1mon and XCL1dim (β = β-sheet). Residues colored blue are basic and residues colored red are acidic.
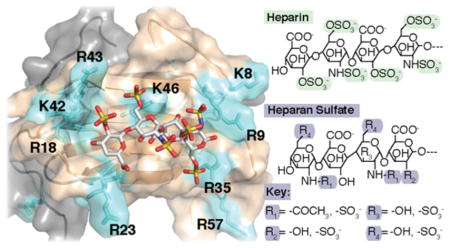
The head-to-tail dimer of XCL1dim carries an overall net charge of +18, resulting in a theoretical pI of 10.8. Many of the basic residues reside within loop and unstructured regions (Figure 1B,E), forming basic patches on the opposing ends of the β-sandwich. These patches are composed of a contribution of basic residues from each subunit, with one monomer providing residues R18, R23, K25, K42, and R43 and the second monomer providing residues K8, R9, R35, and R57 (Figure 1B). With the exception of K46, which resides on the third β-strand, the remaining basic residues (R57, R61, R65, R66, R70, and K77) and acidic residues (E4, D7, D58, and D64) are located in the loop and unstructured N- and C-terminal regions.
Folding of XCL1 W55D Variants
Individual basic residues within W55D were mutated to alanine. These mutants included K8A, R9A, R18A, R23A, K25A, R35A, K42A, R43A, K46A, R61A, and R70A. The R57A protein was expressed but proved to be unstable (data not shown) and was not included in subsequent studies. R61A and R70A were chosen to represent basic residues within the C-terminal helix and tail, while R65A, K66A, and K77A mutations were not probed. An R23A/R43A double mutant was also constructed in the W55D background based on previous studies that indicate its importance for metamorphic interconversion and XCL1mon activity.8,14
All W55D mutants were examined by 1D and 2D NMR spectroscopy to analyze the effect of mutation on the stability of W55D. Figure 2A shows the 1D NMR spectra for all the W55D variants, with the shaded peaks indicative of folded XCL1dim. Spectra for the locked monomer variant of XCL1 (CC3), as well as an unfolded variant (CC0), are shown for comparison. The spectra of a majority of variants match the W55D spectrum, confirming the presence of the XCL1dim conformation. Broadened resonances present in the K25A, R35A, and K46A 1D spectra may be suggestive of conformational exchange. Even though the W55D variant is restricted from undergoing the transition to the XCL1mon state, it retains access to an unfolded state that is on the pathway of XCL1mon–XCL1dim interconversion.7 We speculate that the K25A, R35A, and K46A mutants increase the rates of exchange between the dimeric state and the unfolded intermediate. For instance, a previous report has shown the existence of salt bridge interaction between K25 and E31 in XCL1dim that stabilizes the XCL1dim state, and disruption of this electrostatic interaction (within WT XCL1) shifts the equilibrium toward the XCL1mon state.8 This is evident in the 1H–15N HSQC spectra in Figure 2B where the peaks are much weaker for the K25A spectrum (not observable) even though none of the 1D spectra for K25A, R35A, and K46A resembled the spectrum of the unfolded variant (CC0). It is possible that within the context of W55D, these mutations could bias the formation of an altered dimer or unfolded state; however, further structural analysis of these mutants was not pursued because of increased exchange rates. Because of its role in the metamorphic interconversion of XCL1, the K25A variant was further examined for heparin binding using both heparin chromatography and SPR assays (described below), while R35A and K46A were examined by only heparin chromatography. In comparison, a 1H–15N HSQC spectrum of W55D R23A/R43A (Figure 2B) reveals that these mutations do not alter the structure of the XCL1dim conformation when compared to the W55D spectrum.
Figure 2.

Structural validation of W55D mutants. Mutants were examined by NMR to determine whether a particular mutation impacted the folded state of W55D. Protein samples were prepared in 20 mM sodium phosphate (pH 6.0) and 10% D2O to a final concentration of 100 μM. All spectra were recorded at 37 °C with the exception of CC0 (unfolded variant), which was recorded at 10 °C. 1H one-dimensional spectra are shown in panel A. The boxed area of the spectra highlights resonances that are comparable and indicative of the XCL1dim conformation. The folded states of W55D R9A, W55D K25A, and W55D R23A/R43A were further examined by 2D NMR 1H–15N HSQC analysis (B). The boxed area of the spectra and colors in panel B correlate with the boxed area and colors in panel A.
Heparin Affinity Chromatography of XCL1 Variants
Variants of XCL1 W55D, including a C-terminally truncated variant (W55D 1–72) and an unfolded variant (CC0), were first analyzed for their GAG binding activity by monitoring their elution from heparin Sepharose resin as a function of NaCl concentration (Table 1 and Figure 3A,C). The elution profiles of all variants were compared to that of W55D, which eluted at a NaCl concentration of 920 mM, consistent with previous measurements showing that the XCL1dim conformation of XCL1 binds heparin with an affinity much higher than that for the XCL1mon conformation.5 The K8A, K25A, and K46A variants eluted at similar NaCl concentrations (~800 mM). Removal of the 21 unstructured C-terminal residues had no effect on heparin binding, ruling out the involvement of K77 in heparin binding. Both R61 and R70 reside in the unstructured portion of XCL1dim (Figure 1E), and substitution of these residues had a minor effect on heparin binding with elution occurring at ~800 mM. Residues such as R9A, R18A, R23A, and R35A showed a moderate decrease in the level of heparin binding with elution values of ~600 mM NaCl. These residues reside at the ends of the β-sheets, forming the basic patches seen in panels B and D of Figure 1. Mutations that include K42A, R43A, and R23A/R43A displayed the greatest decrease in the level of heparin binding (<600 mM). These residues also reside in the same regions flanking the β-sheets. The unstructured XCL1 variant (CC0) eluted at the lowest NaCl concentration (460 mM), indicating that despite retaining its electrostatic potential, the removal of secondary and tertiary structure abolishes heparin binding. Data for WT XCL1 and CC3 are shown for comparison and correlate with data from a previous publication.15 NaCl concentrations at which each XCL1 variant eluted from heparin Sepharose were subtracted from the W55D elution concentration (920 mM) and are reported as Δ[NaCl]H values (Table 1 and Figure 3D).
Table 1.
Elution of Proteins from Heparin and SP-Sepharose Resina
| protein | Hep [NaCl] (mM) | Δ[NaCl]H (mM) | S-Seph [NaCl] (mM) | Δ[NaCl]S (mM) | ΔΔ[NaCl] (mM) |
|---|---|---|---|---|---|
| W55D | 920 | – | 590 | – | – |
| K8A | 820 | 100 | 540 | 50 | 50 |
| R9A | 680 | 240 | 610 | −20 | 260 |
| R18A | 620 | 300 | 450 | 140 | 160 |
| R23A | 650 | 270 | 460 | 130 | 140 |
| K25A | 820 | 100 | 850 | −260 | 360 |
| R35A | 640 | 280 | 490 | 100 | 180 |
| K42A | 570 | 350 | 420 | 170 | 180 |
| R43A | 550 | 370 | 420 | 170 | 200 |
| K46A | 810 | 110 | 470 | 120 | −10 |
| R61A | 770 | 150 | 520 | 70 | 80 |
| R70A | 760 | 160 | 530 | 60 | 100 |
| R23A/R43A | 410 | 510 | 330 | 260 | 250 |
| W55D 1–72 | 920 | 0 | 660 | −70 | 70 |
| CC0b | 460 | 460 | 410 | 180 | 280 |
| CC3 | 440 | 480 | 470 | 120 | 360 |
| WT | 760 | 160 | 460 | 130 | 30 |
Protein elution was monitored as a function of NaCl concentration (Hep [NaCl] and S-Seph [NaCl]). Δ[NaCl]H and Δ[NaCl]S values were calculated by subtracting heparin and SP-Sepharose elution from W55D. The value of Δ[NaCl]S was then subtracted from Δ[NaCl]H to remove the electrostatic contribution from the protein–heparin interaction: ΔΔ[NaCl] = Δ[NaCl]H − Δ[NaCl]S. ΔΔ[NaCl] refers to the heparin specificity index. The larger the ΔΔ[NaCl] values, the greater the effect of the mutation on specific heparin binding. All variants with the exception of the following were made in the XCL1 W55D background: CC0, CC3, and WT.
CC0 is an unfolded variant of XCL1. Mutation of C11 and C48 to alanine removes the disulfide bond.
Figure 3.

Determination of heparin binding residues in XCL1dim. W55D and several variants were eluted from a heparin Sepharose and an SP-Sepharose column as a function of increasing NaCl concentration. The raw chromatographs are shown for heparin in panel A and for SP-Sepharose in panel B. The dashed lines are aligned with the maximal peak height for W55D elution and were added as a point of reference. Asterisks are indicative of peaks that were selected for analysis. Graphical representations of values from Table 1. (C) Concentrations of NaCl ([NaCl]) needed to elute proteins from heparin (gray bars) and SP-Sepharose (white bars). (D) Changes in NaCl concentration (Δ[NaCl]) needed to elute proteins from heparin and Sepharose columns compared to W55D.
Mutants were then subjected to cation exchange SP-Sepharose chromatography to measure their nonspecific electrostatic interaction with the negatively charged resin (Table 1 and Figure 3B,C). Because of the nature of these mutations, the net charge of these proteins ranged from +6 (W55D R23A/R43A) to +9 (WT XCL1). It was expected that an increase in the net positive charge of the protein would correspond to an increased NaCl concentration needed for elution from the cation exchange resin. Interestingly, elution of some variants did not correlate with changes in net charge. For example, both R9A and XCL1(1–72) eluted at NaCl concentrations near that used for W55D (~600 mM) despite having a charge of +7 (W55D, charge of +8). On the other hand, the R23A/R43A double mutant had the lowest net charge and eluted at the lowest NaCl concentration. K25A appears to be an outlier and elutes at a NaCl concentration of 850 mM. The change in NaCl concentration needed to elute variants from SP-Sepharose (Δ[NaCl]S) was calculated by subtracting elution values from W55D (Table 1 and Figure 3D).
To assess the heparin binding affinity for each XCL1 protein, the nonspecific electrostatic attractions measured by cation exchange were subtracted from the values measured by heparin Sepharose chromatography (Table 1 and Figure 4A). The variants can be classified into three distinct categories based on the apparent loss of affinity for heparin, as indicated by an increased ΔΔ[NaCl] value: (i) little or no effect (ΔΔ[NaCl] ~ 0 mM), (ii) intermediate effect (ΔΔ[NaCl] = 100–190 mM), and (iii) substantial effect (ΔΔ[NaCl] ≥ 200 mM). Mutations of K8, K46, and R61 have a negligible effect on heparin binding and fall into the first category. In addition, the XCL1(1–72) data suggest that, despite lacking a portion of the unstructured C-terminus including K77, this variant is still able to access the XCL1dim conformation and bind to heparin with an affinity similar to that of W55D. Mutants that are classified into the second category include R18A, R23A, R35A, K42A, and R70A. Both R61 and R70 are part of the unstructured C-terminal tail, suggesting a minor role for C-terminal residues within this region in heparin binding. R9A, K25A, R43A, R23A/R43A, and the unfolded CC0 protein displayed the greatest effect on heparin binding indicating that these residues as well as the overall secondary structure of XCL1 contribute to specific heparin binding. WT and CC3 values are shown for reference and correlated with values from a previously published report.15 All mutations that displayed a defect in heparin binding affinity are mapped on the structure of XCL1dim (Figure 4B).
Figure 4.

Residues that are involved in mediating interactions between XCL1 and heparin Sepharose resin. (A) Graphical representation of the ΔΔ[NaCl] values listed in Table 1. The orange line and bars denote mutations that had the greatest effect on specific heparin binding (ΔΔ[NaCl] ≥ 200 mM). The blue line and bars denote mutations that had an intermediate effect on specific heparin binding (ΔΔ[NaCl] = 100–190 mM). Black bars denote mutations that had little or no effect on specific heparin binding (ΔΔ[NaCl] < 100 mM). (B) Structure of XCL1dim (PDB entry 2JP1) with residues represented as sticks. The coloring of the sticks is based on ΔΔ[NaCl] and corresponds to the graph in panel A.
W55D Binds with High Affinity to Heparin in SPR Analysis
Surface plasmon resonance (SPR) is commonly employed for kinetic measurements of GAG–protein binding24 and has been used to study GAG–chemokine binding interactions.19,20 For comparison with results from heparin affinity chromatography, we conducted SPR studies of XCL1 WT, CC3, W55D, and W55D mutants that demonstrated the largest ΔΔ[NaCl] values (R9A, K25A, and R23A/R43A) mentioned above. Direct fitting using a 1:1 Langmuir model of the kinetic on and off rates for binding of XCL1 proteins to the heparin-coated chip indicated that W55D binds with the highest affinity (Table 2 and Figure 5A). Steady state analysis and nonlinear fitting of the RUmax values as a function of protein concentration (Figure 5B) yielded Kd(app) values that were approximately 10–40-fold higher than those computed from on and off rates (Figure 5C and Table 2).
Table 2.
SPR Analysis of W55D and Variantsa
| protein | kon (M−1 s−1) | koff (s−1) | Koff/kon = Kd(app) (nM) | χ2 | Kd(app) (nM) |
|---|---|---|---|---|---|
| W55D | 5.2 × 104 | 1.6 × 10−3 | 30.0 | 5.4 | 580 ± 94 |
| W55D R9A |
3.8 × 104 | 6.4 × 10−4 | 17.0 | 17.0 | 660 ± 200 |
| W55D K25A |
5.0 × 104 | 1.9 × 10−3 | 38.0 | 7.6 | 1090 ± 140 |
| W55D R23A/R43A |
1.8 × 104 | 2.2 × 10−3 | 120 | 0.4 | 1800 ± 460 |
| CC3 | 2.7 × 104 | 1.7 × 10−3 | 63.0 | 0.7 | 1100 ± 480 |
| XCL1 WT |
2.6 × 104 | 1.2 × 10−3 | 46.1 | 1.9 | 420 ± 40 |
Kinetic on and off rates derived from simultaneous curve fitting of a 1:1 binding model at protein concentrations of 100, 250, 500, 750, and 1000 nM. Kd values were calculated through both kinetic (fourth column) and steady state (sixth column) analysis.
Figure 5.

SPR confirmation of high-affinity binding of XCL1 W55D to heparin. (A) SPR sensorgrams comparing W55D (XCL1dim), W55D R9A, W55D K25A, W55D R23A/R43A, CC3 (XCL1mon), and WT binding and elution from a heparin chip. Proteins were injected at concentrations of 1000, 500, 250, and 100 nM. Kinetic fits for the raw data are shown (red lines). (B) Steady state Kd analysis. Maximal response units were plotted for each concentration of protein tested. Kd(app) values are listed in Table 2. (C) Kinetic plot of kon and koff rates listed in Table 2. The diagonal lines represent KD values. The figure legend applies to both panels B and C.
Consistent with the chromatographic results, significantly weaker heparin binding was observed for the R23A/R43A double mutant and the monomeric CC3 variant. In contrast, SPR responses to the R9A and K25A variants, which exhibited a reduced level of binding to the heparin column, were higher than the RU values of W55D, suggesting that all three proteins retain a high affinity for heparin under “near physiological” conditions of SPR (Figure 5A). While the kinetic and steady state Kd(app) values for R9A are comparable to that of unmodified W55D, the steady state Kd(app) of K25A is comparable to those of CC3 and R23A/R43A variants, consistent with its reduced level of binding to heparin Sepharose. Interestingly, substitution of K25 enhanced binding to the SP-Sepharose column substantially while reducing the level of binding to heparin Sepharose only modestly (Figure 3A). The R9A variant also showed a modest increase in the level of SP-Sepharose binding. R9 and K25 participate in salt bridge interactions that stabilize the XCL1dim conformation necessary for high-affinity heparin binding,5,8 and the NMR spectrum of K25A is altered relative to those of the other variants (Figure 2A). The disparity between the high RU values from SPR and apparent loss of affinity from heparin chromatography may arise from protein conformational changes rather than the loss of specific GAG–protein contact. It is feasible that the high salt content in the heparin chromatography experiments may have altered the structure of these variants to an unfolded state that resulted in a loss of heparin affinity. On the other hand, under the “milder” conditions of SPR, these variants may have been stabilized by the presence of heparin and lower salt content despite the loss of a dimer-stabilizing salt bridge.
Heparin chromatography is a suitable method for screening for potential defective heparin binding mutants, but it relies on increasing NaCl concentration for the assessment of differences in affinity. High ionic strength reduces XCL1dim thermostability, which may already be compromised because of certain mutations, and could contribute to disparities between the chromatography and SPR results for the R9A and K25A variants. SPR analysis is performed under constant solution conditions that are conducive to protein stability. Another advantage of SPR is the ability to analyze kinetic parameters of protein–ligand interactions under conditions that mimic physiological sheer-flow environments that occur in blood and lymphatic vasculature.
Despite deviating from a physiological flow rate and the use of heparin, we believe that our SPR measurements of association and dissociation approximate chemokine–GAG interactions and allow us to examine these interactions in a “near physiological” context. As demonstrated in Figure 5A, the kinetic fits are not ideal, and we speculate that the use of a simple 1:1 binding model fails to account for the intricacies of chemokine–GAG binding, such as the assembly of higher-order chemokine oligomer complexes on the SPR chip. These intricacies also affect the steady state approach. Upon analysis of alternative binding models [bivalent analyte and two-state reaction (data not shown)], we did not observe an improvement in fitting accuracy. Despite the variation in the fits, we are able to obtain a rank ordering of XCL1–heparin binding affinity through analyses of dissociation constants calculated from both kinetic and steady state approaches.
Analysis of GAG Specificity by Fluorescence Polarization (FP)
The measurement of the heparin specificity index (ΔΔ[NaCl]), as shown in Table 1, is a successful method for identifying amino acids that are important for GAG binding. However, this assay does not provide quantitative binding affinities or information about the preference for different GAG types. The most accurate Kd measurements result from working with well-defined experimental conditions that include the smallest number of experimental parameters.25 With this in mind, we developed an FP-based assay to directly measure the affinity of XCL1–GAG interactions using a fluorescein-labeled heparin dp4 molecule (Figure 6A) as a probe. XCL1 W55D was titrated into the probe, and binding was monitored by changes in FP (Figure 6B).
Figure 6.

Fluorescein-labeled heparin dp4 can be used to study XCL1–GAG binding. (A) Schematic of fluorescein-labeled heparin dp4. (B) Kd(app) curves for fluorescein-labeled heparin dp4 binding to W55D [Kd(app) = 2.2 ± 0.2 μM], W55D with 150 mM NaCl [Kd(app) = 67 ± 43 μM], W55D R23A/R43A [Kd(app) = 19.0 ± 3.1 μM], W55D R23A/R43A with 150 mM NaCl [Kd(app) = 390 ± 102 μM], lysozyme [Kd(app) = 190 ± 26 μM], and lysozyme with 150 mM NaCl [Kd(app) = 1200 ± 600 μM]. Lysozyme was selected as a negative binding control, and NaCl was added to all proteins to test for nonspecific electrostatic interactions. (C) Competition assay using fluorescein-labeled heparin dp4. An unlabeled heparin molecule (dp4 and dp8) competed with fluorescein-labeled dp4 for binding sites on W55D. Calculated half-maximal inhibitory concentration (IC50) and inhibitory constant (Ki) values are represented.
Results from these experiments show that the dp4 probe was able to bind to W55D with a Kd(app) of 2.2 ± 0.2 μM. All proteins were tested in the absence and presence of 150 mM NaCl to assess nonspecific interactions between the probe and the proteins. Lysozyme, a basic protein (pI = 11.3) that is not reported to interact with GAGs, was used as a negative control for dp4 probe binding. In the absence and presence of NaCl, Kd(app) values of 190.0 ± 26.0 and 1200 ± 603 μM, respectively, were obtained for lysozyme. These results demonstrate that the dp4-fluorescein probe is able to bind to W55D with a measured affinity that is 1–2 orders of magnitude higher than that of a nonspecific interaction with lysozyme. Even in the presence of NaCl, the level of W55D–probe binding was nearly 20-fold greater than the level of lysozyme–probe binding. W55D R23A/R43A demonstrated an 8-fold increase in Kd(app) (19.0 ± 3.1 μM) that was further increased by the addition of NaCl (390 ± 102 μM). A competition assay was conducted with unlabeled heparin dp4 and dp8 (Figure 6C) to further validate the fluorescent heparin dp4 probe. As expected, unlabeled heparin was able to compete for the binding sites occupied by the dp4-fluorescein molecule with dp8 displaying an IC50 lower than that of dp4.
Although heparin has been used successfully as a model GAG for determining chemokine–GAG interactions, it may not be entirely representative of other types of physiological GAGs.26 Heparan sulfate (HS), dermatan sulfate, keratan sulfate, and chondroitin sulfate comprise the carbohydrate components of proteoglycans, while heparin is mainly secreted from mast cells in a soluble, unattached form.26,27 In addition, heparin is more highly sulfated than the other GAGs and is one of the most negatively charged biomolecules.17 Because of the vast heterogeneity that exists among members of the glycosaminoglycan family, it is necessary to examine different types of GAG fragments to fully understand chemokine–GAG interactions. With this purpose, five differentially sulfated HS octasaccharide molecules were analyzed for XCL1 binding (Figure 7A,B). These HS molecules were unlabeled and exhibited no fluorescence. Using XCL1 W55D and the fluorescein-labeled heparin dp4 probe, described above, each HS molecule was individually titrated into solutions of W55D loaded with the dp4 probe. Analysis of the displacement binding curves to obtain IC50 values showed that the most negatively charged molecule, HS-15, has the highest affinity (Figure 7C). Interestingly, despite a lower net charge, HS-10 was slightly more effective at displacing the heparin probe than HS-14, suggesting that a single 3-O sulfo group contributes more to XCL1–HS binding than multiple 6-O sulfo groups. An unsulfated molecule (HS-4) and a less sulfated molecule (HS-8) were unable to compete for the binding sites occupied by the dp4 probe (charge of −8). Taken together, the heparin and HS FP data demonstrate that both length and sulfation pattern28 influence XCL1–GAG binding interactions.
Figure 7.

XCL1 W55D is able to bind to oligomers of heparan sulfate (HS). Unlabeled HS molecules competed with fluorescein-labeled heparin dp4 for binding sites on W55D. (A) Schematic of a HS 8-mer molecule. Each HS molecule contains a different sulfation pattern at sites R1–R4. Each HS molecule is designated with its corresponding net negative charge (−4, −8, −10, and −15). The first HS ring is numbered to indicate the position of the sulfate attachments. The nitrophenyl group on the end of theses molecules is an artifact of synthesis and has no fluorescence capability. (B) Table with definitions of the R groups for each HS molecule along with their corresponding IC50 and Ki values. (C) IC50 competition curves for each HS molecule.
DISCUSSION
Recent publications have discussed the role of XCL1 in the maintenance of self-tolerance by mediating development of T-regulatory (Treg) cells in the thymus11 as well as promoting immune responses by facilitating interactions between CD8+ T-cells and dendritic cells (DC).10 However, the role of metamorphic behavior of XCL1 within the context of these biological interactions has been largely unexamined. From the results presented here, we see that XCL1–heparin binding is dependent on residues that are also implicated in metamorphic interconversion, mainly R43. It is reasonable to hypothesize that GAG binding may regulate XCL1 activity by keeping XCL1dim sequestered in the extracellular matrix and unable to activate the cognate receptor, XCR1. If we consider the heterogeneity of GAGs throughout tissues and a highly specific interaction between XCL1 and different GAG subsets, this regulation may be extraordinarily complex and allow for highly localized XCL1–XCR1 immune responses. In addition, there may have been evolutionary pressures from microbial agents that pushed XCL1 to become metamorphic to mount immune responses against new pathogens. There is growing evidence in cell culture-based assays that XCL1dim is able to prevent HIV-1 of PBMCs by binding to gp120 of HIV-112,13 (discussed further below). The intriguing metamorphic behavior of XCL15 and its recently described anti-HIV activity12,13 prompted this effort to identify the GAG binding surface of the XCL1dim conformation of XCL1. This study addresses several important aspects of XCL1–GAG interactions, providing both an analysis of the GAG binding surface of the XCL1dim conformation and an assessment of GAG structural features that confer XCL1 binding affinity.
The residues that were identified as making the greatest contribution to heparin binding by chromatography experiments with a ΔΔ[NaCl] of >100 mM were R9, R18, R23, K25, R35, K42, and R43 (Table 1 and Figure 4A). On the basis of their position within the three-dimensional (3D) structure of XCL1dim, these residues form basic patches along the loop regions. These surfaces may facilitate the binding of multiple heparin fragments to the surface or allow longer heparin fragments to “wrap around” the protein as previously proposed for a CCL2 tetramer.29 SPR analysis confirmed that the combination of R23A and R43A substitutions significantly diminished XCL1 heparin binding. These findings are intriguing because both residues also contribute to the metamorphic behavior of XCL1.8
In addition to measuring changes in affinity, SPR data can also reveal differences in the formation of higher-order complexes.30 Comparison of the sensorgrams reveals a nearly 5-fold increase in the equilibrium SPR (RUmax) response of the binding reaction for the W55D dimer relative to the monomeric CC3 and the dimeric R23A/R43A double mutant. WT XCL1 maintains access to the dimeric state and has RU values similar to those of W55D. Taken together, these results confirm XCL1dim as the high-affinity heparin binding conformation and demonstrate that R23 and R43 are important for this interaction.
R23 and R43 are not only essential for heparin binding but also important for other functional activities of XCL1. For example, Guzzo et al. performed an extensive analysis of anti-HIV-1 activity on several of the W55D mutants13 described in this work. They found that upon mutation of K42 and R43, XCL1dim lost the ability to bind to the envelope of gp120 and inhibit HIV-1 infection of PBMCs, while mutation of R18, R35, and K46 demonstrated only diminished HIV-1 inhibition. In comparison, residue R43 appears to be involved in both heparin binding and gp120 binding.
R43 also plays an extensive role in XCL1 interconversion. Tyler et al. probed the basis of electrostatic effects on the XCL1 conformational equilibrium and identified several key interactions that selectively stabilize or destabilize the XCL1mon and XCL1dim states, tuning the thermodynamic balance that allows structural interconversion.8 In the XCL1mon conformation, close positioning of R23 and R43 produces a Coulombic repulsion that favors formation of XCL1dim unless a chloride ion is present to mitigate the unfavorable interaction.8 Conversely, Tunistra et al. showed that addition of low-molecular weight heparin shifted the equilibrium of WT XCL1 toward the XCL1dim conformation.5 In this work, we find that substitutions at R23 and R43 are not disruptive to the XCL1dim conformation (Figure 2A,B) but result in a substantial decrease in the level of heparin binding (Tables 1 and 2 and Figures 4A and 6B). Taken together, previous work and the results of this study demonstrate that R23 and R43 play dual roles in XCL1 structure and function, promoting native state interconversion, GAG binding, and HIV-1 gp120 binding and inhibition.
Fluorescence polarization competition assays are commonly used in drug discovery to screen for inhibitors that block the binding of a probe molecule. We showed that a fluorescein-labeled heparin tetrasaccharide can be used for competition binding assays and measured the relative XCL1 binding affinities of five differentially sulfated HS molecules. Other studies have demonstrated differences in chemokine–GAG selectivity,26,31 and this FP approach can be expected to contribute significantly to our understanding of specificity profiling as a greater diversity of synthetic and purified GAGs becomes commercially available. Profiles of cell membrane-bound heparan sulfate proteoglycans have been described previously,32 and analysis of these glycans would be insightful and could potentially be used as a platform for chemokine-based drug discovery.
Until recently, chemokine–GAG complexes have proven to be largely intractable for high-resolution structure determination,33–35 and excessive XCL1 precipitation upon addition of heparin oligosaccharides precluded our own efforts to analyze an XCL1dim–GAG complex by NMR. The focus of future studies will be to identify a heparan sulfate, chondroitin sulfate, or other GAG molecule that forms a well-behaved XCL1 complex for 3D structure determination and determine whether other glycan types bind to XCL1. For example, Guzzo et al. showed that XCL1dim interacts directly with the HIV-1 envelope, blocking it from attaching to cells and propagating infection.12 As the gp120 envelope protein is heavily glycosylated,36 it is reasonable to hypothesize that XCL1 may neutralize HIV-1 through direct interactions with the carbohydrate components of the envelope glycoprotein. Identifying HIV-1 glycoproteins that bind to XCL1dim would facilitate structural studies and aid in the design and development of new HIV therapeutics.
The data and biophysical tools described above provide the groundwork for future studies elucidating the complex relationship between the metamorphic behavior of XCL1 and GAG binding. By understanding which residues are important for GAG binding and how this binding influences XCL1 metamorphic behavior, we will continue to map the structure–activity relationships of functionally relevant XCL1 residues in increasing detail.
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants AI013225 and AI058072 to B.F.V., HL62244 and HL125371 to R.J.L., and AI037113 to T.M.H.
ABBREVIATIONS
- XCL1mon
XCL1 monomer, formerly known as Ltn10
- XCL1dim
XCL1 dimer, formerly known as Ltn40
- HS
heparan sulfate
- dp
degree of polymerization
- GPCR
G protein-coupled receptor
- GAG
glycosaminoglycan
- FP
fluorescence polarization
- PDB
Protein Data Bank
Footnotes
Notes
The authors declare the following competing financial interest(s): B.F.V. and F.C.P. have significant financial interest in Protein Foundry, LLC.
References
- 1.Mellado M, Rodriguez-Frade JM, Manes S, Martinez-A C. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. 2001;19:397–421. doi: 10.1146/annurev.immunol.19.1.397. [DOI] [PubMed] [Google Scholar]
- 2.Hoogewerf AJ, Kuschert GS, Proudfoot AE, Borlat F, Clark-Lewis I, Power CA, Wells TN. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- 3.Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- 4.Tuinstra RL, Peterson FC, Elgin ES, Pelzek AJ, Volkman BF. An engineered second disulfide bond restricts lymphotactin/XCL1 to a chemokine-like conformation with XCR1 agonist activity. Biochemistry. 2007;46:2564–2573. doi: 10.1021/bi602365d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF. Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci U S A. 2008;105:5057–5062. doi: 10.1073/pnas.0709518105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuloglu ES, McCaslin DR, Kitabwalla M, Pauza CD, Markley JL, Volkman BF. Monomeric solution structure of the prototypical ‘C’ chemokine lymphotactin. Biochemistry. 2001;40:12486–12496. doi: 10.1021/bi011106p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyler RC, Murray NJ, Peterson FC, Volkman BF. Native-state interconversion of a metamorphic protein requires global unfolding. Biochemistry. 2011;50:7077–7079. doi: 10.1021/bi200750k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tyler RC, Wieting JC, Peterson FC, Volkman BF. Electrostatic optimization of the conformational energy landscape in a metamorphic protein. Biochemistry. 2012;51:9067–9075. doi: 10.1021/bi300842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med. 2010;207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei Y, Takahama Y. XCL1 and XCR1 in the immune system. Microbes Infect. 2012;14:262–267. doi: 10.1016/j.micinf.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, Bosl MR, Hollander GA, Hayashi Y, de Waal Malefyt R, Nitta T, Takahama Y. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med. 2011;208:383–394. doi: 10.1084/jem.20102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guzzo C, Fox J, Lin Y, Miao H, Cimbro R, Volkman BF, Fauci AS, Lusso P. The CD8-Derived Chemokine XCL1/Lymphotactin Is a Conformation-Dependent, Broad-Spectrum Inhibitor of HIV-1. PLoS Pathog. 2013;9:e1003852. doi: 10.1371/journal.ppat.1003852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzzo C, Fox JC, Miao H, Volkman BF, Lusso P. Structural Determinants for the Selective Anti-HIV-1 Activity of the All-beta Alternative Conformer of XCL1. J Virol. 2015;89:9061–9067. doi: 10.1128/JVI.01285-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson FC, Elgin ES, Nelson TJ, Zhang F, Hoeger TJ, Linhardt RJ, Volkman BF. Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J Biol Chem. 2004;279:12598–12604. doi: 10.1074/jbc.M311633200. [DOI] [PubMed] [Google Scholar]
- 15.Fox JC, Nakayama T, Tyler RC, Sander TL, Yoshie O, Volkman BF. Structural and agonist properties of XCL2, the other member of the C-chemokine subfamily. Cytokine+ 2015;71:302–311. doi: 10.1016/j.cyto.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 17.Hamel DJ, Sielaff I, Proudfoot AE, Handel TM. Chapter 4. Interactions of chemokines with glycosaminoglycans. Methods Enzymol. 2009;461:71–102. doi: 10.1016/S0076-6879(09)05404-4. [DOI] [PubMed] [Google Scholar]
- 18.Handel TM, Johnson Z, Crown SE, Lau EK, Sweeney M, Proudfoot AE. Regulation of protein function by glycosaminoglycans–as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- 19.Salanga CL, Dyer DP, Kiselar JG, Gupta S, Chance MR, Handel TM. Multiple glycosaminoglycan-binding epitopes of monocyte chemoattractant protein-3/CCL7 enable it to function as a non-oligomerizing chemokine. J Biol Chem. 2014;289:14896–14912. doi: 10.1074/jbc.M114.547737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyer DP, Salanga CL, Volkman BF, Kawamura T, Handel TM. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology. 2015:cwv100. doi: 10.1093/glycob/cwv100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlsson R, Falt A. Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J Immunol Methods. 1997;200:121–133. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- 22.Glabe CG, Harty PK, Rosen SD. Preparation and properties of fluorescent polysaccharides. Anal Biochem. 1983;130:287–294. doi: 10.1016/0003-2697(83)90590-0. [DOI] [PubMed] [Google Scholar]
- 23.BotDB IC50-to-Ki converter. 2009 https://botdb.abcc.ncifcrf.gov/toxin/kiCalPL.jsp.
- 24.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem, Int Ed. 2002;41:390–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 25.Sanders CR. Biomolecular Ligand-Receptor Binding Studies: Theory, Practice, and Analysis. Vanderbilt University; Nashville, TN: 2010. pp. 1–42. structbio.vanderbilt.edu/sanders/Binding_Principles_2010.pdf. [Google Scholar]
- 26.Esko JD, Linhardt RJ. Proteins that Bind Sulfated Glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. Cold Spring Harbor Laboratory Press; Plainview, NY: 2009. [PubMed] [Google Scholar]
- 27.Martin L, Blanpain C, Garnier P, Wittamer V, Parmentier M, Vita C. Structural and functional analysis of the RANTES-glycosaminoglycans interactions. Biochemistry. 2001;40:6303–6318. doi: 10.1021/bi002670n. [DOI] [PubMed] [Google Scholar]
- 28.Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16:625–636. doi: 10.1016/j.cytogfr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 29.Lau EK, Paavola CD, Johnson Z, Gaudry JP, Geretti E, Borlat F, Kungl AJ, Proudfoot AE, Handel TM. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279:22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- 30.Stravalaci M, Bastone A, Beeg M, Cagnotto A, Colombo L, Di Fede G, Tagliavini F, Cantu L, Del Favero E, Mazzanti M, Chiesa R, Salmona M, Diomede L, Gobbi M. Specific recognition of biologically active amyloid-beta oligomers by a new surface plasmon resonance-based immunoassay and an in vivo assay in Caenorhabditis elegans. J Biol Chem. 2012;287:27796–27805. doi: 10.1074/jbc.M111.334979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Y, Sweeney MD, Saad OM, Crown SE, Hsu AR, Handel TM, Leary JA. Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J Biol Chem. 2005;280:32200–32208. doi: 10.1074/jbc.M505738200. [DOI] [PubMed] [Google Scholar]
- 32.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harbor Perspect Biol. 2011;3:a004952. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw JP, Johnson Z, Borlat F, Zwahlen C, Kungl A, Roulin K, Harrenga A, Wells TN, Proudfoot AE. The X-ray structure of RANTES: heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure. 2004;12:2081–2093. doi: 10.1016/j.str.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem. 2007;282:10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshauer C, Morgan AM, Ryan EO, Handel TM, Prestegard JH, Wang X. Interactions of the Chemokine CCL5/RANTES with Medium-Sized Chondroitin Sulfate Ligands. Structure. 2015;23:1066–1077. doi: 10.1016/j.str.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433:834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]