

Abstract
Tyrosine-based and trimeric G protein-based signaling are the two most widely studied and distinct mechanisms for signal transduction in eukaryotes. How each of them relay signals across the plasma membrane independently of each other has been extensively characterized; however, an understanding of how they work together remained obscure. Recently, a rapidly emerging paradigm has revealed that tyrosine based signals are relayed via G proteins, and that the crosstalk between the two hubs are more robustly and sophisticatedly integrated than was previously imagined. More importantly, by straddling the two signaling hubs that are most frequently targeted for their therapeutic significance, the tyrosine-based G-protein signaling pathway has its own growing list of pathophysiologic importance, both as therapeutic target in a variety of disease states, and by paving the way for personalized medicine. The fundamental principles of this emerging paradigm and its pharmacologic potential are discussed.
Signaling at the crossroads
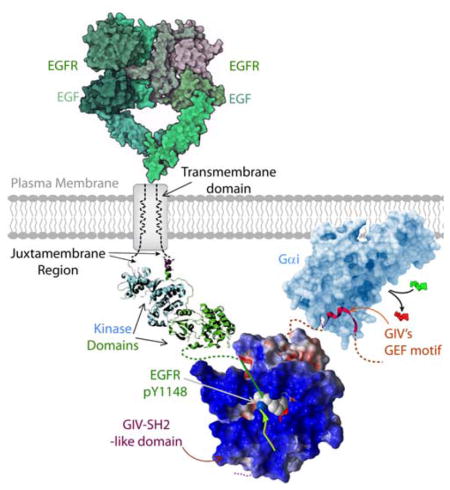
A unique modular makeup, comprised of a SH2-like module in tandem with a GEF module allows GIV (also known as Girdin) to bind autophosphorylated cytoplasmic tails of ligand-activated growth factor receptor tyrosine kinases, such as EGFR (shown here) and to bind and activate Gαi proteins by triggering nucleotide exchange [GDP, (red)-to-GTP(green)]. In doing so, GIV serves as a platform for tyrosine-based signals to transactivate G proteins.
INTRODUCTION
The flow of information from environmental cues to the interior of the cell is controlled by a complex array of proteins at the plasma membrane. Although signal transduction is traditionally studied in a reductionist fashion by dissecting a single pathway/cascade at a time, it is well-known that these distinct signaling pathways cross-talk at multiple levels. Such cross-talks between multiple pathways generate larger complex signaling networks that ultimately control cell fate.
In eukaryotes, the two most widely studied and distinct signaling pathways are the growth factor receptor tyrosine kinases (RTKs) and heterotrimeric G proteins. Upon binding of growth factors like EGF or insulin, RTKs either directly, or indirectly via other non-RTKs phosphorylate a variety of proteins to propagate signals to the cell’s interior (1). Finiteness of signaling is maintained by the antagonistic actions of protein tyrosine kinases (PTKs; both RTKs and non-RTKs) that trigger tyrosine-based signals and protein tyrosine phosphatases (PTPs) that terminate those signals (Figure 1), likened to ‘writers’ and ‘erasers’ of signals, respectively. Signals are propagated by yet another set of adaptor proteins that recognize and bind the phosphotyrosine (pTyr)-containing protein segments, called ‘readers’, which in turn set off downstream signaling cascades via activation of other enzymes or transcriptional factors. Two well-defined modules that enable specific recognition of pTyr-sequences by ‘reader’ proteins are Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains (Figure 1).
Figure 1. Design principles of tyrosine-based G protein signaling.

Schematic displays the fundamental principles that govern signal transduction within the tyrosine-based (left) and G protein-based (right) signaling pathways. Within each pathway, signals are triggered, propagated or terminated by an independent set of ‘writers’, ‘readers’ and ‘erasers’, respectively. The unusual coexistence of a GEF motif (which triggers G protein signaling) and a SH2-like domain (which propagates tyrosine-based signals) in GIV’s C-terminus allows GIV to trigger G protein signaling in response to tyrosine-based signals. The schematic on the right is adapted after significant modification from Pincus D., et al. (89).
Heterotrimeric (henceforth trimeric) G proteins, on the other hand, are traditionally viewed as molecular switches that transmit signals from a different class of membrane receptors, the 7 transmembrane (7TM) receptors, also known as, G protein-coupled receptors (GPCRs) (2). Canonical G protein signaling is initiated when inactive trimeric G proteins (i.e., GDP-bound Gα-subunits in complex with Gβγ-heterodimers) are activated by ligand-occupied GPCRs, which are guanine nucleotide exchange factors (GEFs) and promote the exchange of GDP for GTP on the α subunit (2). Signaling is terminated by the intrinsic GTPase activity of the Gα-subunit, and further accelerated by GTPase accelerating proteins (GAPs), leading to re-association of Gα with Gβγ. Therefore, by analogy, GPCRs serve as ‘writers’ and GAPs serve as ‘erasers’ of signals within the G protein pathway (Figure 1). These sequential “on” and “off” events regulate the so-called “G protein cycle”, and represent the core steps in canonical signal transduction via GPCRs. Such cyclical activation is spatially and temporally restricted-- activation is triggered exclusively at the plasma membrane (PM) by agonist activation of GPCRs via a process that is terminated within a few hundred milliseconds. Signals are propagated when active Gα monomers and Gβγ-heterodimers specifically bind to and activate their respective effector proteins, which serve as ‘readers’ within the pathway.
Multiple studies have unraveled a complex array of cross-talk between these two pathways-- such that one pathway transactivates the other pathway either by directly activating the receptors (3) or by indirectly activating the downstream intermediates or adaptor proteins (4). For example, transactivation of RTKs by GPCRs via scaffolding proteins such as β-arrestins (5) is a well-documented and widely-accepted phenomenon. However, the reverse concept, i.e., transactivation of trimeric G proteins by RTKs remained controversial and its biological significance remained ambiguous. Despite numerous clues that support the concept that growth factors can activate heterotrimeric G proteins (6), the fundamental question as to how such activation occurs in cells remained poorly understood and the concept itself drew skepticism. This is largely because there was neither any evidence that G proteins come within close proximity of ligand-activated RTKs, nor that RTKs, or any member of the growing family of signal transducing adaptors used by RTKs can serve as GEFs.
SIGNALING AT THE CROSSROADS OF PHOSPHOTYROSINES AND G PROTEINS
The discovery of Gα-Interacting Vesicle-associated protein (GIV; also known as Girdin) (7), a non-receptor GEF, has provided insights into tyrosine-based G protein signaling in a way that had never been possible before. GIV is a multidomain signal transducer that has a unique modular makeup (Figure 2)-- Within its ~200 aa C-terminal segment GIV features three unlikely modules/motifs in-tandem: 1) a GEF motif that accelerates nucleotide exchange on Gαi subunits, 2) a SH2-like module that binds pTyr ligands on the cytoplasmic tails of ligand-activated RTKs with high degrees of specificity, and 3) two pTyr-containing sequences that serve as docking sites for other SH2 adaptors. While GIV’s GEF module serves as a ‘writer’ of G protein signals, GIV’s SH2-like module and the two pTyr-containing motifs serve as “readers” that propagate tyrosine-based signals (Figure 1, 2).
Figure 2. The modular make-up of GIV’s C-terminus links G proteins to tyrosine-based signal transducers.

Top: Bar diagram showing the various known functional modules of full length GIV. Bottom: The various signaling interfaces assembled by the C-terminal ~200 aa of GIV are shown. Anti-clockwise from upper left: A previously validated (7, 20) homology model of the GIV•Gαi interface is shown). GIV’s GEF module (red) binds and activates Gαi proteins (light blue) by triggering nucleotide exchange (GTP, red for GDP, green). A previously validated (25) homology model of GIV’s SH2-like domain bound to an autophosphorylated sequence on EGFR tail is shown on the lower left. GIV’s SH2-like module folds upon binding phosphotyrosine ligands on the cytoplasmic tails of multiple RTKs. A previously reported (32) homology model of GIV’s phosphotyrosines (purple) bound to the N- and C-terminal SH2-domains (green and beige, respectively) of p85α-subunit of PI3K is shown on the lower right. Phosphorylation of GIV at two sites (Y1764 and Y1798) by multiple receptor and non-receptor protein tyrosine kinases generates two docking sites for SH2-adaptor containing proteins. Red box = interface that triggers G protein based signals; Blue boxes = interfaces that propagate tyrosine-based signals.
GIV, a ‘writer’ of G protein signals
Much like GPCRs, GIV serves as an activator of Gαi. However, the similarities between the two end there. GIV belongs to a newly emerging family of guanine nucleotide exchange modulators (GEMs (8)). GEMs, like other non-receptor GEFs, GAPs and guanine nucleotide dissociation inhibitors (GDIs) are accessory proteins that fine-tune G protein signaling (9–13). GEMs are a homogeneous group of proteins and synthetic peptides, all of which bind Gα-subunits and accelerate nucleotide exchange on Gαi subunits via an evolutionarily conserved short (~ 30 aa long) motif. This is the fundamental difference between non-receptor GEFs that belong to the GEM family and other receptor (i.e., GPCRs) or non-receptor GEFs [such as AGS1 (14), Ric-8A (15), Ric-8B (16), Arr4 (17), CSPα (18), etc.], all of which activate Gα subunits via unknown or non-conserved motifs. Multiple studies (7, 19, 20) have provided structural insights into the assembly of the GIV-Gαi complex by using a combination of homology modeling (Figure 2) and site-directed mutagenesis (7, 21). These studies revealed that conserved hydrophobic residues that align on one side of a short aliphatic helix in GIV engages with a hydrophobic cleft between the switch II and the α3 helix of Gαi, a mechanism distinct from how Gα-subunits engage with GPCRs (22).
With regard to the impact of GIV-dependent G protein activation on downstream signaling pathways, multiple studies [summarized in (8)] using a selective GEF-deficient GIV mutant (F1685A) have demonstrated that the signaling network (Figure 2) triggered in cells with wild-type GIV is a mirror image of the network in cells expressing a GEF-deficient mutant GIV: signals that are enhanced in cells that are GEF-proficient are suppressed in cells that are GEF-deficient, and vice versa. It is because cells can alter (increase or decrease) the levels of GIV mRNA/protein or selectively modulate GIV’s GEF activity to modulate growth factor signaling pathways across a range of intensities (23), we likened GIV to a cellular “rheostat” for signal transduction (24). Consistent with its ability to integrate signals downstream of multiple receptors, GIV modulates diverse cellular processes, e.g., cell migration, survival, autophagy, secretion, cell polarity, endocytosis, exocytosis, cell adhesion, etc [summarized in (8)]. Similarly, GIV-dependent signaling has been implicated in a number of pathophysiologic conditions, e.g., cancer progression, organ fibrosis, insulin resistance/type II diabetes, vascular injury, neuronal plasticity and memory [summarized in (8)].
GIV, a ‘reader’ of pTyr signals
Within the carboxyl terminus (CT) of GIV, adjacent to the aforementioned motif via which GIV binds and activates Gαi (7) there is a stretch of ~110 aa which “reads” pTyr signals with high degree of specificity. It does so by folding into a SH2-like domain in the presence of phosphotyrosine ligands (25); the domain is necessary and sufficient to recognize and bind sites of autophosphorylation on the receptor tail (25). There are several unique features of GIV’s SH2-like module: No conventional programs predict its existence, mostly because GIV’s SH2-like module, unlike the remaining ~140 or more SH2 domains, is intrinsically disordered or partially structured at resting state. By that token, GIV resembles other eukaryotic proteins that are intrinsically disordered or partially structured under physiological conditions and fold into functional modules, especially in the context of signal transduction (26–31); in many of these cases, binding and folding are coupled (29). Much like those intrinsically disordered proteins, the ~110 aa stretch within GIV-CT folds into a SH2-like module upon binding to its biological target, that is, activated RTKs to assemble the RTK•GIV signaling interface. Consequently, GIV-CT serves as a platform that links RTKs to G proteins within RTK-GIV-Gαi ternary complexes only when both its GEF and SH2-like modules are intact (25), and when either module is absent, RTKs can neither engage with, nor transactivate G proteins (25).
Another feature within GIV’s C-terminus that helps it propagate pTyr-based signals are two C-terminally located tyrosines, Y1764 and Y1798, which are phosphorylated by multiple receptor and non-receptor TKs (Figure 2)(32). Once phosphorylated, the two sequences serve as docking sites for the two SH2-domains of p85α (PI3K), which leads to maximal activation of PI3K (32). That GIV’s phosphotyrosines could bind both SH2 domains of p85α is in accordance with the fact that the N- and C-terminal SH2 domains of p85α are structurally related (33, 34), and both recognize and bind similar phosphotyrosine sequences (34). Of note, GIV’s sequences flanking these two tyrosines differ from the canonical, p85α-binding YXXMX consensus (34) and instead resemble other non-canonical p85α-binding sequences: While the pY1798ATLP peptide shares homology with the pY343LVL peptide from EPO receptor (35), the pY1764FISS peptide shares homology with the pYEPTG peptide from Syk kinase (36) or the pYVNTT peptide in Tie2 (37) that bind p85α-CSH2 with high affinity and specificity. At least in the case of the pY1764FISS motif of GIV, this sequence joins Syk and Tie2 to represent a non-canonical class of peptides with a pYXX[ST] consensus. It is noteworthy that the spacing of GIV’s phosphotyrosines and the distance between the two SH2 domains of p85α (PI3K) (38) is compatible with the possibility that the tandem phosphotyrosines of GIV’s C-terminus simultaneously occupy the tandem SH2 domains of p85α. Previous work has established that such double-occupancy of p85α-SH2 domains in-tandem is required for full activation of the catalytic p110(PI3K) subunit (39), presumably via one of the two proposed mechanisms—by triggering allosteric conformational changes (40) and/or by promoting kinase oligomerization (41). As compared to single occupancy, in-tandem double occupancies in the tyrosine-SH2 signaling pathway is known to confer substantially higher affinity and enhanced biological specificity (42). Whether the biological specificity and efficiency of PI3K activation by GIV’s phosphotyrosines we observe stems from similar in-tandem interactions of PI3K with GIV remain to be investigated. Regardless, what is clear is that binding of tyrosine phosphorylated GIV to the p85α-SH2 domains provides a mechanism by which pTyr-based signals are transmitted directly from activated tyrosine kinases to PI3K. Last, but not least, GIV’s phosphotyrosines could serve as docking sites for not just p85α(PI3K), but also other SH2 adaptors. Ongoing work indicates such is the case.
The aforementioned findings have fundamentally enriched our knowledge of GIV’s unique C-terminal stretch and provide many clues into what might be the molecular mechanism(s) behind GIV’s ability to engage, directly or indirectly, with multiple upstream receptors/pathways. The fact that GIV-CT has two-states, one that is intrinsically disordered and another that is folded, perhaps provide the biggest clue. Intrinsically disordered proteins, while structurally poor, are functionally rich by virtue of the flexibility of their modular structures, as recently described in the case of phosphatase and tensin homologue (PTEN) (26, 43, 44). Post-translational modifications, burial or exposure of conserved linear motifs and molecular recognition features present in the CT of PTEN during folded- vs disordered states directly regulate PTEN’s interactions with other proteins, which are required for executing diverse cellular functions. It has also been shown that PTEN’s disordered and folded-state interactomes are further enriched in proteins that are intrinsically disordered, revealing how PTEN functions are regulated by the disordered CT to nucleate flexible network-hubs and orchestrate ‘on-demand’ modes of signaling. Based on the diversity of pathways and receptors that GIV modulates, we suspect that the evolution of structural plasticity in the intrinsically disordered GIV-CT is likely to assemble distinct interactomes in the disordered versus SH2-folded states, thereby contributing to functional enrichment. Because GIV’s C-terminus is enriched in Ser/Thr residues, some of which are heavily phosphorylated (www.phosphosite.org), it is possible that additional phosphoevents on GIV’s C-terminus trigger and/or regulate folding of GIV’s SH2-like domain and binding to RTKs. Because phosphotyrosine-binding domains (PTB) are another major ‘reader’ of tyrosine-based signals (Figure 1), it is possible that GIV’s C-terminus may either fold into PTB domain or harbor PTB-binding motifs. Further biochemical, structural and biophysical studies are essential to understand how plasticity of the GIV-CT influences when and where it may engage receptors/proteins within signaling pathways, depending on whether it is in folded- vs disordered state, to nucleate distinct signaling hubs.
TYROSINE-BASED G PROTEIN SIGNALING: A new paradigm governed by new rules
As a protein that can dually function as a ‘reader’ of pTyr-based signals as well as a ‘writer’ of G protein-based signals, studies on GIV have put forward a concrete mechanism for tyrosine-based G protein signaling. Some of these studies, carried out in living cells, have revealed how tyrosine-based non-canonical G protein signaling is governed by a new set of rules that differ from GPCR-dependent canonical signaling. These rules are discussed in detail elsewhere (8), and stated briefly here.
First, the rules of receptor engagement is different. Unlike the canonical GPCR/G protein pathway, in which the G proteins engage exclusively with ligand-activated receptor (GPCR)-GEFs, the non-receptor GIV-GEF engages with a diverse array of receptors, including GPCRs, Integrins and RTKs (7, 8, 45) and, thereby, enables transactivation of G proteins in response to a wide variety of stimuli. While the molecular mechanisms that govern how GIV couples G proteins to receptors are best understood in the context of ligand activated RTKs (discussed above), efforts are underway to decipher how GIV couples G proteins to receptors of other classes, and whether the GIV-receptor interactions in each case is direct or indirect.
Second, the temporal aspects of tyrosine-based G protein signaling are distinct. Förster resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) studies using genetically encoded fluorescent proteins have revealed that GPCRs engage G proteins with a T1/2 of 30–50 msec, which coincides with rapid onset of canonical G protein activation (T1/2 300–500 msec) that is short-lived (few secs-min) (46–48). By contrast, the temporal profile of non-canonical G protein activation by GIV is unique with regard to both receptor-G protein engagement and the onset and duration of G protein activation: 1) The timing of recruitment of GIV to RTKs such as EGFR at the PM is ~3–5 min, which resembles the kinetics demonstrated in the case of multiple other SH2 adaptor proteins, e.g., Grb2 (49). 2) Although some GIV:Gαi complexes are pre-formed (i.e., in serum starved cells), the assembly of such complexes are triggered maximally within ~5 to ~15 min after growth factor stimulation and disassembled by ~ 30 min. Assembly is triggered by phosphoactivation of GIV (at Ser 1674 within the GEF motif) by cyclin dependent kinase, CDK5, a step that is essential for GIV to initiate non-canonical G protein signaling downstream of growth factor RTKs (50). Because CDK5 is activated within seconds after growth factor stimulation (51), it is likely that once activated, CDK5 can promptly phosphoactivate GIV’s GEF function before or during the latter’s recruitment to the activated receptor at the PM, ensuring subsequent maximal binding and activation of Gαi. As for the mechanism of disassembly of GIV-Gαi complexes, it is likely to be brought on by a negative feedback loop initiated by PKCθ which phosphoinhibits GIV’s GEF motif (at Ser1689) and selectively terminates GIV’s ability to bind or activate Gαi (23). 3) G proteins engage with ligand-activated RTKs maximally by ~5 min. The combined synergy of bimolecular fluorescence complementation (BiFC) and FRET studies further confirmed that GIV’s C-terminus serves as bona fide platform for assembling RTK-GIV-Gαi complexes at the PM in response to growth factors. RTK-GIV-Gαi complexes are assembled at the PM within ~5–10 min after ligand stimulation; assembly of such complexes is impaired if GIV and Gαi cannot bind each other. 4) G protein activation is delayed and prolonged. FRET assays using fluorescent G proteins [Gαi3-YFP (internal tag), CFP-Gβ1 (N-terminal tag) and Gγ2], as originally developed and validated by the groups led by Gilman, Lohse (52, 53) and by others (54, 55) to study the canonical G protein pathway revealed the unusual temporal dynamics of transactivation of G proteins by growth factors. Although the extent of Gi activation downstream of RTKs (EGFR; (56)) and GPCRs (α2 AR; (52)) appear similar, the temporal dynamics of non-canonical G protein activation by GIV represent a clear deviation from the dynamics of canonical G protein signaling that is triggered by GPCRs [summarized in (8)]. Transactivation of Gi and dissociation of the trimer in response to growth factors starts at ~5 min and lasts several minutes, and depletion of GIV abrogates such response. 5) Consistent with the contrasting temporal patterns of canonical vs non-canonical Gi activation, suppression of cAMP by Gi-coupled GPCRs within the canonical pathway occurs rapidly (i.e., within seconds) (57), but non-canonical transactivation of Gi by RTKs leads to a delayed suppression of cAMP (i.e., ~6–10 minutes). The fundamental molecular basis that governs such delayed activation of Gi and suppression of cAMP is the dynamics of binding of GIV’s SH2-like domain to ligand-activated RTKs; the latter is a prerequisite for facilitating the proximity between G proteins and RTKs. These FRET studies helped nucleate a new paradigm in which tyrosine-based signals initiated by RTKs can transactivate G proteins utilizing GIV as a platform (see legend for graphic abstract).
Third, FRET studies have revealed that the spatial pattern of tyrosine-based G protein activation by GIV also poses a stark contrast to canonical G protein signaling. Canonical G protein activation is initiated primarily at the PM exclusively by ligand-activated GPCRs. Although recent studies using nanobodies have revealed that some signaling continues also within endosomes (58), to date, no such activation on internal membranes that are discontinuous with the PM has ever been observed. By contrast, GIV-dependent signaling has been described at multiple intracellular compartments, including centrosomes, focal adhesions, cell-cell junctions, early endosomes, exocytic vesicles, autophagosomes, and more recently, on Golgi membranes [summarized in (8)]. The specific role of activation of G proteins by GIV has been investigated in the context of autophagy (19), secretory functions of the Golgi (50), and during the establishment of cell polarity (59), and these 3 studies have accomplished 3 key goals: 1) they prove that G proteins are active at internal locations; 2) that such activation can be brought on by cytosolic non-receptor GEF, GIV; and finally, 3) they provide valuable clues into how the same GEF, i.e., GIV may coordinate G protein signaling at the PM and on internal membranes. It is noteworthy that while all three aforementioned studies primarily investigated how GIV activates G proteins on intracellular locations, they also revealed the complexity and variation of the interactome at each location. For example, GIV straddles the evolutionarily conserved partitioning defective (PAR) protein-3 and G proteins during the establishment of cell polarity, somewhat analogous to the way it straddles ADP-ribosylation factor 1 (Arf1) and G proteins on Golgi membranes, or the proautophagic LC3, AGS3 and G protein complexes on autophagosomes. Because GIV serves many other roles at a variety of other intracellular sites, it is likely that those roles also require GIV’s GEF function at those locations and may involve novel protein-protein interactions unique to those locations. As to how GIV coordinately activates G proteins at the PM as well as on internal membranes, it is tempting to speculate that signaling intermediates/pathways that are initiated/facilitated by GIV at the PM, e.g., calcium, cAMP, kinases, phosphatases, etc. rapidly diffuse within the cytoplasm and initiate GIV-dependent Gi activation on internal membranes. A cross-talk between G proteins at two locations may serve as a key mechanism that makes intracellular organelles or structures responsive to tyrosine-based signals triggered by external environmental cues. Further studies are underway to dissect how such cross-talk is orchestrated.
THERAPEUTIC POTENTIAL OF TYROSINE-BASED G PROTEIN SIGNALING
The unparalleled importance of canonical signal transduction via GPCRs/G protein and tyrosine-based signaling in modern medicine have been recognized for long. Dysregulation of either pathway influences the pathogenesis of a myriad of human diseases, from cancer, through fibrosis, neurodegeneration, diabetes and cardiovascular diseases, etc. More importantly, efforts to target these pathways has been rewarded with tremendous success: GPCRs represent the target for ~40% of currently marketed drugs (60), whereas PTKs, both receptor and non-receptor TKs are the targets for another ~15% of drugs (61, 62). Therefore, it is not surprising that a signaling pathway that functions at the crossroads of tyrosine-based signaling and G proteins impacts a rapidly growing list of diseases, e.g., multiple aspects of cancer progression [tumor cell migration, invasion and metastasis (50, 56, 63–71); stemness (72), drug resistance (73), tumor-stroma interplay (74), tumor angiogenesis (75)], organ (liver) fibrosis (76), dermal wound healing (68), nephrotic syndrome (77), insulin resistance/type II diabetes (78, 79), disorders of the blood vessels (80–83), and neuronal plasticity and memory formation (84). The role of GIV’s GEF function has been investigated in some, but not all of these disease states [summarized in (8)], and where investigated, a clear therapeutic goal has emerged (see Figure 3). For example, transcriptional upregulation of GIV and activation of GIV’s GEF function fuels cancer progression by enhancing signaling pathways that are initiated by multiple oncogenic receptors that increase tumorigenic potential of cancer cells by empowering them with key phenotypic characteristics (Figure 3A); downregulating GIV and turning ‘OFF’ GIV’s GEF function is a clear therapeutic goal. Similarly, transcriptional upregulation of GIV and activation of GIV’s GEF function in myofibroblasts responding to chronic injuries enhances the pro-fibrogenic signals and suppresses anti-fibrogenic signals downstream of multiple fibrogenic receptors (Figure 3B); once again, downregulating GIV and turning ‘OFF’ GIV’s GEF function emerged as a clear therapeutic goal. Finally, upregulation of GIV and activation of GIV’s GEF function in myotubes enhances the metabolic insulin response, whereas selective phosphoinhibition of GIV’s GEF function via the fatty acid→PKCθ pathway suppresses the same (Figure 3C); in this case, upregulating GIV and turning its GEF function ‘ON’ is the therapeutic goal.
Figure 3. The untapped pharmacologic potential of GIV in chronic, multi-receptor driven disease states.

A) Schematic summarizing our and others’ work on how GIV modulates several parameters, e.g., invasiveness, stemness, angiogenesis, survival, and drug resistance, all of which together constitute the tumorigenic potential of cancer cells. Each of these parameters, that can be triggered by diverse classes of oncogenic receptors, are driven more efficiently and consequently, the tumorigenic potential is highest when GIV expression is elevated and its GEF function is ‘ON’ [summarized in (8, 85)]. GIV levels are transcriptionally upregulated in invasive cancer cells during cancer progression via the transcription factor, STAT3 (63)., whereas its GEF function is turned ‘ON’ via a single phosphoevent that is triggered by CDK5 (50). Because GIV serves as molecular rheostat, i.e., signaling intensity via GIV’s GEF function is closely related to the two variables, i.e., number of GIV molecules in cells and whether their GEF function is ‘ON’, it is expected that by altering both variables one can alter the tumorigenic potential of cancer cells from low to high. Thus, the pharmacologic goal to halt cancer progression is to reduce the copies of GIV (by tumor-targeted si/shRNA) and/or to selectively turn ‘OFF’ GIV’s GEF function (using small molecule inhibitors). Antagonistic pathways, such as SHP1 phosphatase (90) and PKCθ kinase (23), which terminate tyrosine-based and G protein-based signals, respectively, may also be potentiated to accomplish a similar goal. B) Schematic summarizing our work (76) on how GIV regulates several parameters, e.g., proliferation, chemotaxis, survival, metalloprotease and collagen production, all of which together constitute the fibrogenic potential of myofibroblasts in response to chronic injury/inflammation. Each of these parameters, that can be triggered by diverse classes of fibrogenic receptors, are driven more efficiently and consequently, the fibrogenic potential is highest when GIV expression is elevated and its GEF function is ‘ON’. Upon chronic fibrogenic injury, GIV levels are transcriptionally upregulated and GIV’s GEF function is turned ‘ON’ in myofibroblasts. When GIV’s GEF function is ‘ON’, pro-fibrogenic (red arrows) signals are potentiated, whereas anti-fibrogenic (green arrows) signals are downregulated; the opposite occurs when GIV’s GEF function is turned ‘OFF’. Thus, the pharmacologic goal to halt and reverse fibrogenic progression is to selectively turn ‘OFF’ GIV’s GEF function (using small molecule inhibitors). C) Schematic summarizing our and others’ work (78, 86, 91) on how GIV regulates several steps of the insulin signaling cascade, e.g., receptor autophosphorylation, the recruitment and activation of IRS1, activation of the PI3K-Akt signaling pathway, exocytosis of the glucose transporter, GLUT4 and glucose uptake into skeletal muscles, all of which together constitute the metabolic insulin response. While an intact and efficient metabolic insulin response is required for physiology (i.e., insulin sensitivity), aberrant signals in response to circulating free fatty acids (among other causes) dampen such metabolic response and contribute to insulin resistance, the hallmark of type II diabetes. The metabolic insulin response is driven more efficiently and consequently, the insulin sensitivity is highest when GIV expression is elevated (78) and its GEF function is ‘ON’ (86). Fatty acids trigger insulin resistant states by phosphoinhibiting GIV’s GEF function via the DAG→PKCθ pathway, whereas insulin sensitizers like Pioglitazone act by potentiating tyrosine phosphorylation of GIV and by reversing phosphoinhibition and restoring GIV’s GEF function. Thus, the pharmacologic goal to reverse insulin resistance and reinstate insulin sensitivity is to selectively turn ‘ON’ GIV’s GEF function (by using positive allosteric modulators of GIV’s GEF function or by using specific inhibitors of PKCθ). D) Schematic summary of plausible therapeutic strategies to regulate GIV-dependent signaling. Among the currently available options are inhibitors (shown in yellow boxes) of the various upstream factors which coordinately potentiate GIV-dependent signaling, e.g., receptor and non-receptor tyrosine kinases that trigger tyrosine phosphorylation of GIV, or STAT3 which activates GIV transcription in diseased states, or the Ser/Thr kinase CDK5 which phosphoactivates GIV’s GEF function. Although selective inhibition of GIV’s GEF function using small molecules still remains unrealized, the therapeutic relevance of GIV depletion (by si/shRNA) or targeted disruption of its GEF function using cell permeable peptides has been experimentally validated (blue boxes).
With the emergence of a new paradigm of signal transduction, one that broadly impacts a variety of pathophysiologic states [summarized in (8)], the next hurdle was to devise a way to target it. In this regard, the unusual spatiotemporal features of tyrosine-based G protein signaling via GIV (discussed earlier) posed a unique set of challenges as well as advantages. For example, it is challenging to target a ubiquitously expressed protein that serves as a point of convergence downstream of multiple receptors [summarized in (8)], performs a broad array of physiologic functions, and is frequently deregulated in multiple pathologic states. Another challenge is the absence of high-resolution structures. In the absence of such structural information, computational modeling has provided some clues into the ‘druggability’ of the GIV•Gαi interface (7, 20). Experimentally validated models have revealed that GIV’s binding site on Gαi does not overlap with the binding site of GPCRs, gave birth to the notion that the GIV-Gαi interface could be selectively disrupted without affecting the Gαi-GPCR interface. Biochemistry and enzymology studies on multiple G protein mutants that disrupt the GIV•Gαi3 interface (7, 20) also support the notion that it is possible to selectively abolish tyrosine-based G protein signaling via GIV without affecting canonical signaling via GPCRs. It is also conceivable that inhibition of known upstream modulators of GIV, i.e., TKs that phosphorylate GIV (32, 67), STAT3 which transcriptionally upregulates GIV expression (63), and CDK5 which phosphoactivates GIV’s GEF function (50) will also inhibit tyrosine-based G protein signaling via GIV (Figure 3D); however, those inhibitors are unlikely to be specific. Based on the broad range of receptor-initiated signals that converge on GIV and the variety of signaling pathways within ‘disease networks’ that are modulated via GIV’s GEF function [summarized in (8)], it is predicted that disrupting the GIV•Gαi interface will be the most specific and effective approach for modulating multi-receptor signaling via GIV. Such an approach is expected to have the tremendous advantage of allowing ‘network-based therapy’ irrespective of the receptor of origin (85). Recently, we showed just that in a proof-of-concept study using cell-permeable peptides (68). Selective modulation of the GIV•Gαi interface using cell-permeable GIV-CT peptides fused to a TAT-peptide transduction domain (TAT-PTD) containing the minimal modular elements of GIV that are necessary and sufficient for activation of Gi downstream of RTKs can effectively engineer signaling networks and alter cell behavior (68). In the presence of an intact GEF motif, TAT-GIV-CT peptides enhanced cellular processes in which GIV’s GEF function has previously been implicated; e.g., 2D cell migration after scratch-wounding, invasion of cancer cells through the matrigel, myofibroblast activation and collagen production in response to a fibrogenic stimuli and finally, metabolic signaling and glucose uptake in response to insulin (68, 86). Furthermore, topical application of TAT-GIV-CT peptides enhanced dermal wound repair in mice in a GEF-dependent manner (68). The impact of these findings is two-fold. First, the findings described here using TAT-GIV-CT peptides represent a significant advancement in our ability to access, interrogate and manipulate the GIV platform, and thereby, modulate the cross-talk between RTKs and G proteins they facilitate. Second, G proteins are an ideal target for therapeutic intervention because they serve as signal amplification switches. Therefore, potent and pathway-selective activators/inhibitors of G proteins like TAT-GIV-CT peptides can serve multiple purposes ranging from being a research tool to pharmacologic probe for use in experimental and clinical therapeutics (87). The cell-permeable peptides allow for exogenous manipulation of the RTK-GIV-Gαi pathway by enhancing or suppressing transactivation of G proteins by RTKs in a dose dependent manner while minimizing the risk of tampering with other physiologic functions/interactions of G proteins/or other components within the network of modulators of G protein signaling.
Although cell-permeable peptides allowed exogenous modulation of the fundamental function of GIV, i.e., transactivate Gi downstream of ligand-activated RTKs, it is unlikely that these TAT-appended peptides will serve as marketable drugs. But the lessons we learned are invaluable. For example, it appears that GIV-CT peptides may be optimal for potential gene therapy applications to manipulate Gαi activation downstream of multiple growth factors in diverse cell types and in a variety of pathophysiologic conditions. While the therapeutic potential of these peptides is expected to grow with the rapidly growing list of pathophysiologic processes that GIV modulates, it is likely that blocking the GIV-Gαi signaling pathway without targeting the desired tissues/cells may result in a narrow therapeutic window and potential for undesired side effects.
TYROSINE-BASED G PROTEIN SIGNALING OFFERS A UNIQUE OPPORTUNITY FOR PERSONALIZED MEDICINE
Besides its therapeutic potential, what became clear early on was the potential for GIV to serve as a portal for the practice of personalized medicine in several disease states. For example, in the context of cancer progression, others and us have reported that expression of GIV at high levels correlates with tumor aggressiveness and poor survival across a variety of solid tumors (85). A consensus has emerged that patients with GIV-positive tumors are at highest risk for cancer progression and may maximally benefit from systemic chemotherapy. Ongoing clinical trials assessing the expression of GIV in primary tumors as well as on tumor cells isolated from the peripheral circulation are likely to provide a more complete assessment of the prognostic and predictive impact of GIV as biomarker for cancer progression.
Similarly, in the context of liver fibrosis, GIV is expressed in the interstitial cells of the liver after a chronic fibrogenic insult to the liver, and exclusively detected in patients with fibrosis (76). A proof-of-concept study showed that high levels of GIV in the liver could distinguish patients with vital hepatitis C who had progressive fibrosis from those who did not (76). These findings suggest that expression of GIV could be used as a marker for identification of those patients at highest risk for fibrogenic progression and therefore, candidates for aggressive anti-viral and/or anti-fibrotic therapy.
Finally, in the case of type II diabetes, levels of GIV expression correlate with insulin sensitivity: high GIV, equals better metabolic insulin response (78). Additionally, the degree of phosphoinhibition of GIV’s GEF function (by PKCθ at S1674) in skeletal muscles biopsies of patients with insulin resistance correlated with clinical response to treatment with insulin sensitizer Thiazolidinediones (86): high degrees of pre-treatment inhibition or post-treatment residual inhibition was associated with little or no clinical response to the anti-diabetic drug. These studies indicate that several readouts of the tyrosine-based G protein signaling pathway that is set up via GIV could serve as biomarkers, and their presence or absence may help detect diseases, or assess its severity, in order to guide medical decision-making.
CONCLUSIONS AND FUTURE PERSPECTIVES
The insights gained just within the past half-decade has shaped the paradigm of tyrosine-based G protein signaling via GIV. Perhaps the biggest surprise was that such signaling is triggered by receptors that are typically not believed to signal via G proteins. Despite these insights, it is clear that a lot remains unknown. For example, although we now know how RTKs transactivate G proteins, how do other classes of receptors, such as integrins, Toll-like receptors (TLRs), Transforming growth factor (TGFβ) receptors also use GIV to transactivate G proteins remains unclear. Because GIV’s C-terminus offers structural/conformational plasticity, which should directly impact protein-protein interactions, it is possible that such structural plasticity provides context-dependent engagement with a variety of receptors, some directly and others indirectly. Knowing how GIV engages other receptors is of utmost importance because an in-depth insight into that mechanism(s) will fundamentally revolutionize our understanding of the new rules of tyrosine-based non-canonical G protein signaling via GIV.
As for the newly revealed temporal and spatial features of non-canonical G protein signaling, several interesting questions remain unanswered. One such unanswered question is how does tyrosine-based signals initiated at the PM coordinately trigger G protein activation by GIV on internal membranes. Last, but not least, although homology modeling has proven insightful thus far, obtaining high resolution structures of GIV bound to ligand-activated RTKs and Gα-subunits is an urgent and an unmet need. Insights into the dynamics of nucleotide exchange that is brought about by GIV on Gαi proteins will help understand how tyrosine-based indirect transactivation of G proteins by GIV differs from direct activation of G proteins that is brought about by GPCRs. Such insights are expected to greatly facilitate the development of small molecules that can selectively target the tyrosine-based G protein pathway.
Last, but not least, although tyrosine-based G protein signaling may appear to be a linear connection between input (the TKs) and output (G-proteins) elements (Figure 1), experimental data shows that it is rather an integral part of a network that links many receptors to many signaling pathways [summarized in (8)], and links multiple cellular organelles to events at the PM (19, 88). The behavior of such complex systems is hard to grasp by intuition. Because multiple feed-forward/feedback cycles modulate GIV-dependent signaling and orchestrate it in separate time and space, mathematical simulation, not intuition, is expected to be more reliable. Such an approach will help generate more comprehensive models to illuminate how GIV drives diverse cellular processes. By the same token, it will integrate experimental knowledge into a coherent picture so we can test, support, or falsify our hypotheses of molecular mechanisms. Thus, it is clear that solving the unanswered questions will need the engagement of more groups in the scientific community to systematically dissect this emerging paradigm of tyrosine-based G protein signaling from the atomic level to pathway modeling.
Acknowledgments
This work was funded by NIH (R01CA160911, R01CA100768 and DK099226) to P.G. P.G. was also supported by the UC San Diego Moores Cancer Center.
Abbreviations
- BiFC
Bimolecular fluorescence complementation
- cAMP
Cyclic adenosine monophosphate
- CDK5
Cyclin-Dependent Kinase 5
- CT
Carboxyl Terminus
- FRET
Förster resonance energy transfer
- GAP
GTPase-Accelerating Protein
- GDI
Guanosine nucleotide dissociation inhibitor
- GDP
Guanosine diphosphate
- GEF
Guanine Nucleotide Exchange Factors
- GIV
Gα-interacting vesicle-associated
- Girdin
Girders of actin
- GPCR
G protein-coupled receptor
- GTP
Guanosine triphosphate
- PI3K
phosphoinositide 3-kinase
- PKC
protein kinase C
- PM
plasma membrane
- PTD
protein transduction domain
- PTEN
Phosphatase and tensin homolog
- RTK
Receptor tyrosine kinase
- SH2
Src-homology 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schlessinger J. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol. 2014;6(3):a008912. doi: 10.1101/cshperspect.a008912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilman AG. G proteins: transducers of receptor-generated signals. Annual review of biochemistry. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 3.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379(6565):557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 4.Natarajan K, Berk BC. Crosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinases. Methods in molecular biology. 2006;332:51–77. doi: 10.1385/1-59745-048-0:51. [DOI] [PubMed] [Google Scholar]
- 5.Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20(13):1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- 6.Marty C, Ye RD. Heterotrimeric G protein signaling outside the realm of seven transmembrane domain receptors. Molecular pharmacology. 2010;78(1):12–18. doi: 10.1124/mol.110.063453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(9):3178–3183. doi: 10.1073/pnas.0900294106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aznar N, Kalogriopoulos N, Midde K, Lo IC, Ghosh P. Heterotrimeric G Protein Signaling via GIV/Girdin: Breaking the rules of engagement, space and time. BioEssays. 2016 doi: 10.1002/bies.201500133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato M, Blumer JB, Simon V, Lanier SM. Accessory proteins for G proteins: partners in signaling. Annual review of pharmacology and toxicology. 2006;46:151–187. doi: 10.1146/annurev.pharmtox.46.120604.141115. [DOI] [PubMed] [Google Scholar]
- 10.Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1(2):51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annual review of biochemistry. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 12.Blumer JB, Oner SS, Lanier SM. Group II activators of G-protein signalling and proteins containing a G-protein regulatory motif. Acta physiologica (Oxford, England) 2012;204(2):202–218. doi: 10.1111/j.1748-1716.2011.02327.x. [DOI] [PubMed] [Google Scholar]
- 13.De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG. The regulator of G protein signaling family. Annual review of pharmacology and toxicology. 2000;40:235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- 14.Cismowski MJ, Ma C, Ribas C, Xie X, Spruyt M, Lizano JS, Lanier SM, Duzic E. Activation of heterotrimeric G-protein signaling by a ras-related protein. Implications for signal integration. The Journal of biological chemistry. 2000;275(31):23421–23424. doi: 10.1074/jbc.C000322200. [DOI] [PubMed] [Google Scholar]
- 15.Tall GG, Krumins AM, Gilman AG. Mammalian Ric-8A (synembryn) is a heterotrimeric Galpha protein guanine nucleotide exchange factor. The Journal of biological chemistry. 2003;278(10):8356–8362. doi: 10.1074/jbc.M211862200. [DOI] [PubMed] [Google Scholar]
- 16.Chan P, Gabay M, Wright FA, Tall GG. Ric-8B is a GTP-dependent G protein alphas guanine nucleotide exchange factor. The Journal of biological chemistry. 2011;286(22):19932–19942. doi: 10.1074/jbc.M110.163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MJ, Dohlman HG. Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Curr Biol. 2008;18(3):211–215. doi: 10.1016/j.cub.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Natochin M, Campbell TN, Barren B, Miller LC, Hameed S, Artemyev NO, Braun JE. Characterization of the G alpha(s) regulator cysteine string protein. The Journal of biological chemistry. 2005;280(34):30236–30241. doi: 10.1074/jbc.M500722200. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Marcos M, Ear J, Farquhar MG, Ghosh P. A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol Biol Cell. 2011;22(5):673–686. doi: 10.1091/mbc.E10-08-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Marcos M, Ghosh P, Ear J, Farquhar MG. A structural determinant that renders G alpha(i) sensitive to activation by GIV/girdin is required to promote cell migration. The Journal of biological chemistry. 2010;285(17):12765–12777. doi: 10.1074/jbc.M109.045161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Marcos M, Kietrsunthorn PS, Pavlova Y, Adia MA, Ghosh P, Farquhar MG. Functional characterization of the guanine nucleotide exchange factor (GEF) motif of GIV protein reveals a threshold effect in signaling. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(6):1961–1966. doi: 10.1073/pnas.1120538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Sanchez I, Garcia-Marcos M, Mittal Y, Aznar N, Farquhar MG, Ghosh P. Protein kinase C-theta (PKCtheta) phosphorylates and inhibits the guanine exchange factor, GIV/Girdin. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(14):5510–5515. doi: 10.1073/pnas.1303392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh P, Garcia-Marcos M, Farquhar MG. GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh Migr. 2011;5(3):237–248. doi: 10.4161/cam.5.3.15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin C, Ear J, Midde K, Lopez-Sanchez I, Aznar N, Garcia-Marcos M, Kufareva I, Abagyan R, Ghosh P. Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol Biol Cell. 2014;25(22):3654–3671. doi: 10.1091/mbc.E14-05-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malaney P, Uversky VN, Dave V. Identification of intrinsically disordered regions in PTEN and delineation of its function via a network approach. Methods. 2015;77–78:69–74. doi: 10.1016/j.ymeth.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 28.Dunker AK, Uversky VN. Signal transduction via unstructured protein conduits. Nat Chem Biol. 2008;4(4):229–230. doi: 10.1038/nchembio0408-229. [DOI] [PubMed] [Google Scholar]
- 29.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12(1):54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 30.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 31.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323(3):573–584. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, Ear J, Pavlova Y, Mittal Y, Kufareva I, Ghassemian M, Abagyan R, Garcia-Marcos M, Ghosh P. Tyrosine phosphorylation of the Galpha-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Science signaling. 2011;4(192):ra64. doi: 10.1126/scisignal.2002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell SJ, Jackson RM. Diversity in the SH2 domain family phosphotyrosyl peptide binding site. Protein engineering. 2003;16(3):217–227. doi: 10.1093/proeng/gzg025. [DOI] [PubMed] [Google Scholar]
- 34.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72(5):767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 35.He TC, Zhuang H, Jiang N, Waterfield MD, Wojchowski DM. Association of the p85 regulatory subunit of phosphatidylinositol 3-kinase with an essential erythropoietin receptor subdomain. Blood. 1993;82(12):3530–3538. [PubMed] [Google Scholar]
- 36.Moon KD, Post CB, Durden DL, Zhou Q, De P, Harrison ML, Geahlen RL. Molecular Basis for a Direct Interaction between the Syk Protein-tyrosine Kinase and Phosphoinositide 3-Kinase. Journal of Biological Chemistry. 2005;280(2):1543–1551. doi: 10.1074/jbc.M407805200. [DOI] [PubMed] [Google Scholar]
- 37.Kontos CD, Stauffer TP, Yang WP, York JD, Huang L, Blanar MA, Meyer T, Peters KG. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Molecular and cellular biology. 1998;18(7):4131–4140. doi: 10.1128/mcb.18.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science (New York, NY. 2007;318(5857):1744–1748. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 39.Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM. Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. The Journal of biological chemistry. 1995;270(8):3662–3666. doi: 10.1074/jbc.270.8.3662. [DOI] [PubMed] [Google Scholar]
- 40.Shoelson SE, Sivaraja M, Williams KP, Hu P, Schlessinger J, Weiss MA. Specific phosphopeptide binding regulates a conformational change in the PI 3-kinase SH2 domain associated with enzyme activation. The EMBO journal. 1993;12(2):795–802. doi: 10.1002/j.1460-2075.1993.tb05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Layton MJ, Harpur AG, Panayotou G, Bastiaens PI, Waterfield MD. Binding of a diphosphotyrosine-containing peptide that mimics activated platelet-derived growth factor receptor beta induces oligomerization of phosphatidylinositol 3-kinase. The Journal of biological chemistry. 1998;273(50):33379–33385. doi: 10.1074/jbc.273.50.33379. [DOI] [PubMed] [Google Scholar]
- 42.Ottinger EA, Botfield MC, Shoelson SE. Tandem SH2 domains confer high specificity in tyrosine kinase signaling. The Journal of biological chemistry. 1998;273(2):729–735. doi: 10.1074/jbc.273.2.729. [DOI] [PubMed] [Google Scholar]
- 43.Malaney P, Uversky VN, Dave V. The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy. Mol Biosyst. 2013;9(11):2877–2888. doi: 10.1039/c3mb70267g. [DOI] [PubMed] [Google Scholar]
- 44.Malaney P, Pathak RR, Xue B, Uversky VN, Dave V. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep. 2013;3:2035. doi: 10.1038/srep02035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh P, Beas AO, Bornheimer SJ, Garcia-Marcos M, Forry EP, Johannson C, Ear J, Jung BH, Cabrera B, Carethers JM, Farquhar MG. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol Biol Cell. 2010;21(13):2338–2354. doi: 10.1091/mbc.E10-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lohse MJ, Hein P, Hoffmann C, Nikolaev VO, Vilardaga JP, Bunemann M. Kinetics of G-protein-coupled receptor signals in intact cells. Br J Pharmacol. 2008;153(Suppl 1):S125–132. doi: 10.1038/sj.bjp.0707656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lohse MJ, Hoffmann C, Nikolaev VO, Vilardaga JP, Bunemann M. Kinetic analysis of G protein-coupled receptor signaling using fluorescence resonance energy transfer in living cells. Adv Protein Chem. 2007;74:167–188. doi: 10.1016/S0065-3233(07)74005-6. [DOI] [PubMed] [Google Scholar]
- 48.Ross EM. Coordinating speed and amplitude in G-protein signaling. Curr Biol. 2008;18(17):R777–R783. doi: 10.1016/j.cub.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorkin A, McClure M, Huang F, Carter R. Interaction of EGF receptor and grb2 in living cells visualized by fluorescence resonance energy transfer (FRET) microscopy. Curr Biol. 2000;10(21):1395–1398. doi: 10.1016/s0960-9822(00)00785-5. [DOI] [PubMed] [Google Scholar]
- 50.Bhandari D, Lopez-Sanchez I, To A, Lo IC, Aznar N, Leyme A, Gupta V, Niesman I, Maddox AL, Garcia-Marcos M, Farquhar MG, Ghosh P. Cyclin-dependent kinase 5 activates guanine nucleotide exchange factor GIV/Girdin to orchestrate migration-proliferation dichotomy. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(35):E4874–4883. doi: 10.1073/pnas.1514157112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee HY, Jung H, Jang IH, Suh PG, Ryu SH. Cdk5 phosphorylates PLD2 to mediate EGF-dependent insulin secretion. Cell Signal. 2008;20(10):1787–1794. doi: 10.1016/j.cellsig.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 52.Gibson SK, Gilman AG. Gialpha and Gbeta subunits both define selectivity of G protein activation by alpha2-adrenergic receptors. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(1):212–217. doi: 10.1073/pnas.0509763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bunemann M, Frank M, Lohse MJ. Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(26):16077–16082. doi: 10.1073/pnas.2536719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Janetopoulos C, Jin T, Devreotes P. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science (New York, NY. 2001;291(5512):2408–2411. doi: 10.1126/science.1055835. [DOI] [PubMed] [Google Scholar]
- 55.Yi TM, Kitano H, Simon MI. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(19):10764–10769. doi: 10.1073/pnas.1834247100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Midde KK, Aznar N, Laederich MB, Ma GS, Kunkel MT, Newton AC, Ghosh P. Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(9):E937–946. doi: 10.1073/pnas.1420140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO reports. 2004;5(12):1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495(7442):534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sasaki K, Kakuwa T, Akimoto K, Koga H, Ohno S. Regulation of epithelial cell polarity by PAR-3 depends on Girdin transcription and Girdin-Galphai3 signaling. J Cell Sci. 2015;128(13):2244–2258. doi: 10.1242/jcs.160879. [DOI] [PubMed] [Google Scholar]
- 60.Hopkins AL, Groom CR. The druggable genome. Nature reviews. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 61.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 62.Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nature reviews. 2010;9(12):956–970. doi: 10.1038/nrd3297. [DOI] [PubMed] [Google Scholar]
- 63.Dunkel Y, Ong A, Notani D, Mittal Y, Lam M, Mi X, Ghosh P. STAT3 protein up-regulates Galpha-interacting vesicle-associated protein (GIV)/Girdin expression, and GIV enhances STAT3 activation in a positive feedback loop during wound healing and tumor invasion/metastasis. The Journal of biological chemistry. 2012;287(50):41667–41683. doi: 10.1074/jbc.M112.390781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9(3):389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 65.Jiang P, Enomoto A, Jijiwa M, Kato T, Hasegawa T, Ishida M, Sato T, Asai N, Murakumo Y, Takahashi M. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer research. 2008;68(5):1310–1318. doi: 10.1158/0008-5472.CAN-07-5111. [DOI] [PubMed] [Google Scholar]
- 66.Leyme A, Marivin A, Perez-Gutierrez L, Nguyen LT, Garcia-Marcos M. Integrins activate trimeric G proteins via the non-receptor protein GIV/Girdin. J Cell Biol. 2015 doi: 10.1083/jcb.201506041. Accepted, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lopez-Sanchez I, Kalogriopoulos N, Lo I, Kabir F, Midde K, Wang H, Ghosh P. Focal Adhesions are Foci for Tyrosine-Based Signal Transduction via GIV/Girdin and G proteins. Mol Biol Cell. 2015 doi: 10.1091/mbc.E15-07-0496. Accepted, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma GS, Aznar N, Kalogriopoulos N, Midde KK, Lopez-Sanchez I, Sato E, Dunkel Y, Gallo RL, Ghosh P. Therapeutic effects of cell-permeant peptides that activate G proteins downstream of growth factors. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(20):E2602–2610. doi: 10.1073/pnas.1505543112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohara K, Enomoto A, Kato T, Hashimoto T, Isotani-Sakakibara M, Asai N, Ishida-Takagishi M, Weng L, Nakayama M, Watanabe T, Kato K, Kaibuchi K, Murakumo Y, Hirooka Y, Goto H, Takahashi M. Involvement of Girdin in the determination of cell polarity during cell migration. PLoS One. 2012;7(5):e36681. doi: 10.1371/journal.pone.0036681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang C, Lin J, Li L, Wang Y. Expression and clinical significance of girdin in gastric cancer. Mol Clin Oncol. 2014;2(3):425–428. doi: 10.3892/mco.2014.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Y, Yuan L, Yang XM, Wei D, Wang B, Sun XX, Feng F, Nan G, Wang Y, Chen ZN, Bian H. A chimeric antibody targeting CD147 inhibits hepatocellular carcinoma cell motility via FAK-PI3K-Akt-Girdin signaling pathway. Clin Exp Metastasis. 2015;32(1):39–53. doi: 10.1007/s10585-014-9689-7. [DOI] [PubMed] [Google Scholar]
- 72.Natsume A, Kato T, Kinjo S, Enomoto A, Toda H, Shimato S, Ohka F, Motomura K, Kondo Y, Miyata T, Takahashi M, Wakabayashi T. Girdin maintains the stemness of glioblastoma stem cells. Oncogene. 2012;31(22):2715–2724. doi: 10.1038/onc.2011.466. [DOI] [PubMed] [Google Scholar]
- 73.Zhang YJ, Li AJ, Han Y, Yin L, Lin MB. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol. 2014;20(25):8229–8236. doi: 10.3748/wjg.v20.i25.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yamamura Y, Asai N, Enomoto A, Kato T, Mii S, Kondo Y, Ushida K, Niimi K, Tsunoda N, Nagino M, Ichihara S, Furukawa K, Maeda K, Murohara T, Takahashi M. Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer research. 2015;75(5):813–823. doi: 10.1158/0008-5472.CAN-14-1317. [DOI] [PubMed] [Google Scholar]
- 75.Kitamura T, Asai N, Enomoto A, Maeda K, Kato T, Ishida M, Jiang P, Watanabe T, Usukura J, Kondo T, Costantini F, Murohara T, Takahashi M. Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nature cell biology. 2008;10(3):329–337. doi: 10.1038/ncb1695. [DOI] [PubMed] [Google Scholar]
- 76.Lopez-Sanchez I, Dunkel Y, Roh YS, Mittal Y, De Minicis S, Muranyi A, Singh S, Shanmugam K, Aroonsakool N, Murray F, Ho SB, Seki E, Brenner DA, Ghosh P. GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat Commun. 2014;5:4451. doi: 10.1038/ncomms5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang H, Misaki T, Taupin V, Eguchi A, Ghosh P, Farquhar MG. GIV/girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J Am Soc Nephrol. 2015;26(2):314–327. doi: 10.1681/ASN.2013090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hartung A, Ordelheide AM, Staiger H, Melzer M, Haring HU, Lammers R. The Akt substrate Girdin is a regulator of insulin signaling in myoblast cells. Biochim Biophys Acta. 2013;1833(12):2803–2811. doi: 10.1016/j.bbamcr.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 79.Ma GS, Lopez-Sanchez I, Aznar N, Kalogriopoulos N, Pedram S, Midde K, Ciaraldi T, Henry RR, Ghosh P. Activation of G proteins by GIV-GEF is a Pivot Point for Insulin Resistance and Sensitivity. Mol Biol Cell. 2015 doi: 10.1091/mbc.E15-08-0553. Accepted, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ito T, Komeima K, Yasuma T, Enomoto A, Asai N, Asai M, Iwase S, Takahashi M, Terasaki H. Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. Am J Pathol. 2013;182(2):586–596. doi: 10.1016/j.ajpath.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 81.Miyachi H, Mii S, Enomoto A, Murakumo Y, Kato T, Asai N, Komori K, Takahashi M. Role of Girdin in intimal hyperplasia in vein grafts and efficacy of atelocollagen-mediated application of small interfering RNA for vein graft failure. J Vasc Surg. 2014;60(2):479–489. e475. doi: 10.1016/j.jvs.2013.06.080. [DOI] [PubMed] [Google Scholar]
- 82.Miyake H, Maeda K, Asai N, Shibata R, Ichimiya H, Isotani-Sakakibara M, Yamamura Y, Kato K, Enomoto A, Takahashi M, Murohara T. The actin-binding protein Girdin and its Akt-mediated phosphorylation regulate neointima formation after vascular injury. Circ Res. 2011;108(10):1170–1179. doi: 10.1161/CIRCRESAHA.110.236174. [DOI] [PubMed] [Google Scholar]
- 83.Miyachi H, Takahashi M, Komori K. A Novel Approach against Vascular Intimal Hyperplasia Through the Suppression of Girdin. Ann Vasc Dis. 2015;8(2):69–73. doi: 10.3400/avd.ra.14-00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nakai T, Nagai T, Tanaka M, Itoh N, Asai N, Enomoto A, Asai M, Yamada S, Saifullah AB, Sokabe M, Takahashi M, Yamada K. Girdin phosphorylation is crucial for synaptic plasticity and memory: a potential role in the interaction of BDNF/TrkB/Akt signaling with NMDA receptor. J Neurosci. 2014;34(45):14995–15008. doi: 10.1523/JNEUROSCI.2228-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ghosh P. Heterotrimeric G proteins as emerging targets for network based therapy in cancer: End of a long futile campaign striking heads of a Hydra. Aging (Albany NY) 2015;7(7):469–474. doi: 10.18632/aging.100781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma GS, Lopez-Sanchez I, Aznar N, Kalogriopoulos N, Pedram S, Midde K, Ciaraldi TP, Henry RR, Ghosh P. Activation of G proteins by GIV-GEF is a pivot point for insulin resistance and sensitivity. Mol Biol Cell. 2015;26(23):4209–4223. doi: 10.1091/mbc.E15-08-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iiri T, Farfel Z, Bourne HR. G-protein diseases furnish a model for the turn-on switch. Nature. 1998;394(6688):35–38. doi: 10.1038/27831. [DOI] [PubMed] [Google Scholar]
- 88.Lo IC, Gupta V, Midde KK, Taupin V, Lopez-Sanchez I, Kufareva I, Abagyan R, Randazzo PA, Farquhar MG, Ghosh P. Activation of Galphai at the Golgi by GIV/Girdin imposes finiteness in Arf1 signaling. Dev Cell. 2015;33(2):189–203. doi: 10.1016/j.devcel.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pincus D, Letunic I, Bork P, Lim WA. Evolution of the phospho-tyrosine signaling machinery in premetazoan lineages. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(28):9680–9684. doi: 10.1073/pnas.0803161105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mittal Y, Pavlova Y, Garcia-Marcos M, Ghosh P. Src homology domain 2-containing protein-tyrosine phosphatase-1 (SHP-1) binds and dephosphorylates G(alpha)-interacting, vesicle-associated protein (GIV)/Girdin and attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway. The Journal of biological chemistry. 2011;286(37):32404–32415. doi: 10.1074/jbc.M111.275685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lopez-Sanchez I, Ma GS, Pedram S, Kalogriopoulos N, Ghosh P. GIV/girdin binds exocyst subunit-Exo70 and regulates exocytosis of GLUT4 storage vesicles. Biochem Biophys Res Commun. 2015;468(1–2):287–293. doi: 10.1016/j.bbrc.2015.10.111. [DOI] [PMC free article] [PubMed] [Google Scholar]