

SUMMARY
Glucose levels in mammals are tightly controlled through multiple mechanisms to meet systemic energy demands. Down-regulation of hepatic glucokinase (GCK) during fasting facilities the transition of the liver from a glucose-consuming to a gluconeogenic organ. Here we report the transcriptional regulation of hepatic GCK by a long non-coding RNA (lncRNA) named liver GCK repressor (lncLGR). LncLGR is induced by fasting, and physiological overexpression of lncLGR to mimic fasting levels effectively suppresses GCK expression and reduces hepatic glycogen content in mice. Consistently, lncLGR knockdown enhances GCK expression and glycogen storage in fasted mice. Mechanistically, lncLGR specifically binds to heterogenous nuclear ribonucleoprotein L (hnRNPL), which is further confirmed to be a transcriptional repressor of GCK in vivo. Finally, we demonstrate that lncLGR facilitates the recruitment of hnRNPL to GCK promoter and suppresses GCK transcription. Our data establishes an lncRNA-mediated mechanism that regulates hepatic glucokinase expression and glycogen deposition in a physiological context.
Graphical Abstract
INTRODUCTION
Although only 1.5% of the human genome encodes proteins, major sequencing efforts in the past decade have revealed that there is a vast repertoire of uncharacterized non-coding RNAs in the human transcriptome (Djebali et al., 2012; Harrow et al., 2012). Among all non-coding RNA species, the most abundant, and possibly also the least understood, is that comprised of long non-coding RNAs (lncRNAs), which are transcripts that are at least 200nt long and have no coding potential. LncRNAs have been demonstrated to regulate diverse cellular processes ranging from gene transcription, RNA stability and translation control (Moran et al., 2012; Wang and Chang, 2011), but only a small fraction of them have been investigated in a physiologically relevant context. Conceptually, it is straightforward to envision that some of these lncRNAs might function as regulators of energy metabolism in vivo, which is essentially connected to all major biological processes (Kornfeld and Bruning, 2014). Indeed, we have recently identified that a liver-enriched lncRNAs, lncLSTR, robustly regulates triglyceride uptake in mice (Li et al., 2015).
As a central metabolic organ, the liver also plays an important role in maintaining glucose homeostasis. The liver produces glucose through glycogenolysis and gluconeogenesis during fasting, while promoting glucose uptake and glycogen storage during feeding. A key enzyme in the liver, glucokinase (GCK), dictates the direction of hepatic glucose flux, and GCK expression and activity are subject to exquisite regulation (Massa et al., 2011). In the postprandial period, the rise in glucose and insulin increases GCK activity, whereas in the fasting state, the combined decrease in glucose and insulin concentrations and increase in glucagon concentrations, decrease GCK activity. The underlying molecular mechanisms regulating GCK expression during feeding cycles are complex at both transcriptional and post-transcriptional levels (Massa et al., 2011). After a meal, insulin up-regulates GCK transcription through a PI3K-PKB pathway, and several transcription factors including HNF4a, HIF1a, SREBP1c and LRH-1 have been implicated in this process (Foretz et al., 1999; Roth et al., 2004). However, much less is known about how GCK expression is down regulated during fasting, and one assumption is that reduced insulin levels during fasting lead to the suppression of GCK transcription.
In this report, we characterize a fasting-induced lncRNA in the liver that we have named Liver Glucokinase Repressor (lncLGR), which suppresses GCK transcription in vivo by interacting with hnRNPL, an RNA-binding protein that has no previously known role in regulating glucose metabolism. Our results provide an lncRNA-mediated mechanism for the regulation of GCK activity and hepatic glycogen storage and further solidify the functional significance of lncRNAs in maintaining metabolic homeostasis.
RESULTS
Hepatic overexpression of lncLGR suppresses glucokinase expression and decreases glycogen content in mouse liver
LncLGR is a full-length cDNA clone deposited in Fantom3 database as 4632424N07 and in GenBank as AK028540. It is an intergenic lncRNA located on chromosome 13q in the mouse genome, and lncLGR transcripts could be detected in multiple tissues in mice with low abundance (Figure S1A). The copy number of lncLGR in isolated primary hepatocytes is about 3.6 copies per cell (Figure S1B). We initially found that lncLGR expression in mouse liver was significantly induced by fasting and recovered after refeeding (Figure 1A). Stability analysis showed that lncLGR has a half-life time of approximate 8 hours in primary hepatocytes (Figure S1C). To further study the regulation of lncLGR by metabolic hormones and nutrients, we treated mouse primary hepatocytes with insulin, glucagon, or glucose and quantified lncLGR expression levels. As shown in Figure S1D, while glucose or glucagon had no significant effect, insulin alone could suppress lncLGR expression by nearly 50%. Thus, lncLGR appears to be a fasting-induced and insulin-regulated lncRNA, suggesting a functional role in glucose and lipid metabolism. To identify the potential metabolic functions of lncLGR in vivo, we first overexpressed lncLGR in mouse liver using an adenoviral system, which increased the hepatic lncLGR levels by 80% (Figure 1B) in mice with a 4-hour food withdrawal, resembling the levels under fasting conditions (Figure 1A). While there was no significant difference in plasma glucose between lncLGR overexpression (OE) and control mice, plasma triglyceride (TG) levels were moderately but significantly lower (~10% reduction) in the lncLGR OE group (Figures S1E and 1F). Further biochemical analyses revealed that glycogen and TG contents in the liver were both decreased in lncLGR OE mice compared with controls (Figures 1C and 1D). The simultaneous decrease of glycogen and TG contents in the liver led us to hypothesize that lncLGR overexpression could limit the availability of glucose-6-phosphate (G6P) for glycogen synthesis and de novo lipogenesis. Consistently, hepatic G6P levels were significantly lower in lncLGR OE mice compared with controls (Figure 1E). Since hepatic glucokinase (GCK) could directly determine G6P levels in the liver, we next analyzed GCK expression and found that the GCK mRNAs were significantly decreased in lncLGR OE mice, while the expression levels of all three remaining hexokinases were not changed (Figure 1F). Expression levels of key genes in lipogenesis, glucose and glycogen metabolism were also similar between the two groups of mice as well (Figures S1G and S1H). We further confirmed that there were decreased GCK protein levels as well as GCK enzymatic activity in lncLGR OE mice (Figures 1G and 1H). Taken together, these data suggest that lncLGR may regulate hepatic glucose metabolism by down-regulating GCK expression.
Figure 1. Overexpression of lncLGR suppresses GCK expression and glucose metabolism in mouse liver.

(A) Expression levels of lncLGR in the livers of mice (n=4) fed ad libitum (Ad Libitum), subject to a 24-hour fast (Fast), or subject to a 24-hour fast followed by a 4-hour refeeding (Refeed).
(B) Expression levels of lncLGR in the livers of control (Ad-vector) and lncLGR overexpression (Ad-lncLGR) mice (n=6) after a 4h food withdrawal.
(C) Liver TG content, (D) liver glycogen content, (E) liver G6P content and (F) liver gene expression levels in control and lncLGR overexpression mice (n=5–7) after a 4h food withdrawal.
(G) GCK protein levels in the livers of control and lncLGR overexpression mice (n=4). Intensities of bands were quantified and normalized to actin levels as shown on the right.
(H) Relative GCK activity in the livers of control and lncLGR overexpression mice (n=6).
(I) Relative GCK activity and (J) glycogen levels in the livers of mice receiving control, lncLGR overexpression, or both lncLGR and GCK overexpression adenoviruses (Ad-GCK) after a 4h food withdrawal (n=6).
Error bars represent SEM, *p<0.05.
To definitively test whether GCK is the primary mechanism responsible for the reduced hepatic glycogen content in lncLGR OE mice, we co-administered GCK adenoviruses into these mice at a does that is sufficient to rescue the reduced GCK activity (Figure 1I), and demonstrated that the decreased liver glycogen content caused by lncLGR overexpression was also completely reversed in these mice (Figure 1J). These data strongly support that the GCK effects are the primary mechanism by which lncLGR regulates hepatic glycogen storage.
Depletion of hepatic lncLGR results in increased glucokinase expression and glycogen storage in fasted mice
Since GCK expression is up-regulated by feeding and down-regulated by fasting in vivo, it is plausible that one of the physiological roles for fasting-induced lncLGR is to suppress GCK expression. We used a loss-of-function approach to further interrogate this hypothesis. Mice injected with recombinant adenoviruses that carry a lncLGR-targeting shRNA showed a ~40% reduction in hepatic lncLGR levels after an overnight fast (Figure 2A). This system allows us to investigate hepatic glucose metabolism in the fasting condition when lncLGR is physiologically induced. Consistent with the effects seen in the gain-of-function model, lncLGR knockdown (KD) significantly increased GCK expression at both mRNA and protein levels (Figures 2A and 2B). Consistently, hepatic G6P levels were also significantly increased in lncLGR KD mice compared with control mice receiving lacZ-targeting shRNA adenoviruses (Figure 2C). In addition, while mice in the control group exhibited almost complete depletion of glycogen storage in the liver after an overnight fast, lncLGR KD mice retained significantly higher hepatic glycogen levels (Figure 2D). These findings support that suppression of GCK expression by lncLGR might play an essential role in regulating hepatic glucose metabolism during fasting.
Figure 2. LncLGR Knockdown results in increased GCK expression and glycogen storage in mouse livers during fasting.

(A) Gene expression levels in the livers of control (Ad-sh lacZ) and lncLGR KD (Ad-sh lncLGR) mice (n=6) after a 16h food withdrawal.
(B) GCK protein levels in the livers of control and lncLGR KD mice (n=4). Intensities of bands were quantified and normalized to actin levels as shown on the right.
(C) Total G6P content in the livers (n=5) of control and lncLGR KD mice was quantified using a colorimetric assay system.
(D) Total glycogen content in the livers (n=6) of control and lncLGR KD mice was quantified using a colorimetric assay system.
Error bars represent SEM, *p<0.05.
LncLGR binds to hnRNPL which functions as a repressor of GCK expression
We next sought to explore the molecular mechanism by which lncLGR regulates GCK expression. We fractionated mouse liver tissue samples and found that lncLGR is mainly localized in the nucleus (Figure 3A), suggesting a potential role in gene transcription. To identify protein-binding partners of lncLGR in the nucleus, we performed an RNA pull-down assay using nuclear extracts from liver tissue samples and analyzed proteins specifically bound to lncLGR using mass spectrometry (Figure 3B). One of these proteins was hnRNPL, which we could further confirm to bind specifically to lncLGR by anti-hnRNPL immunoblotting (Figure 3B). Subsequently, we performed a reciprocal pull-down of hnRNPL using nuclear extracts of liver tissue samples and quantified RNAs in the immunoprecipitates by quantitative real-time PCR. In this experiment, lncLGR was enriched by 23-fold as compared to an IgG control or lncLSTR (Li et al., 2015), a nuclear lncRNA in mouse liver that we have recently demonstrated to be a regulator of systemic lipid metabolism in mice (Figure 3C). Furthermore, in vitro binding assay using transcribed lncLGR RNAs and purified hnRNPL proteins also support that they directly interact to form an RNP complex (Figure 3D). To determine if hnRNPL regulates GCK expression in vivo, we knocked down hnRNPL in mouse liver and found that a 50% reduction of hnRNPL could significantly increase GCK expression at both the mRNA and protein levels (Figures 3E and 3F) indicating that hnRNPL could function as a repressor of GCK expression in mouse liver.
Figure 3. LncLGR binds to hnRNPL which functions as a repressor of GCK transcription.

(A) Levels of lncLGR in whole cell, cytosolic, or nuclear fractions of liver tissue samples pooled from 4 mice (result of an independent experiment is also shown in figure S2A).
(B) Left: silver stained SDS-PAGE gel analysis of proteins in nuclear extract of mouse liver tissue samples that are bound to biotinylated lncLGR or a reversed YFP (YFP-NC). The highlighted regions were analyzed by mass spectrometry, identifying hnRNPL as a protein unique to lncLGR. Right: immunoblotting analysis of proteins in nuclear extract of liver tissue samples that are bound to biotinylated lncLGR or a reversed YFP using an anti-hnRNPL antibody.
(C) Left: anti-hnRNPL immunoblotting analysis of proteins in immunoprecipitates of mouse liver tissue samples using an anti-hnRNPL antibody. Right: Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), lncLSTR and lncLGR RNA levels in immunoprecipitates of liver tissue samples using an anti-hnRNPL antibody (result of an independent experiment is also shown in figure S2B).
(D) Left: Coomassie Blue Staining of purified recombinant hnRNPL proteins expressed in HEK293T cells. Right: immunoblotting analysis of purified hnRNPL that are bound to biotinylated lncLGR or a reversed YFP using an anti-hnRNPL antibody.
(E) Gene expression levels in the livers of control (Ad-sh lacZ) and hnRNPL KD (Ad-sh hnRNPL) mice (n=5) after a 4h food withdrawal.
(F) HnRNPL and GCK protein levels in the livers of control and hnRNPL KD mice (n=4). Intensities of bands were quantified and normalized to actin levels as shown on the right.
(G) Immunoblotting analysis of nuclear extract of mouse liver tissue samples that are bound to biotinylated lncLGR, lncLGR Δ1-335 or a reversed YFP using an anti-hnRNPL antibody.
(H) Gene expression and (I) glycogen levels in the livers of control (Ad-vector) and lncLGR Δ1-335 overexpression (Ad-lncLGR Δ1-335) mice (n=6) after a 4h food withdrawal.
Error bars represent SEM, *p<0.05.
Next we set up experiments to test if the effects of lncLGR on glucose metabolism are dependent on its binding with hnRNPL. It has been established that hnRNPL often binds to RNAs through specific interactions with CA repeats in the transcripts (Zhang et al., 2013). Interestingly, lncLGR contains a 21-CA repeat near its 5′ end, which we hypothesized is required for its binding with hnRNPL. To test this, we first deleted 5′ 335 nucleotides of lncLGR including the CA repeat, and demonstrated by a RNA pull-down assay that the truncated lncLGR without the CA repeat exhibited drastically decreased, although not completely abolished, binding with hnRNPL compared to the full-length one (Figure 3G). Next, we tested if this shorter lncLGR with impaired hnRNPL binding capacity could still suppress GCK expression in mice. Adenovirus-mediated overexpression of the truncated lncLGR clearly lost the ability to suppress GCK expression as well as the effects on glycogen levels (Figures 3H and 3I). These data unambiguously support that lncLGR-mediated regulation of GCK expression and glycogen content depends on the functional complex formed by lncLGR and hnRNPL.
LncLGR and hnRNPL coordinately suppress glucokinase transcription
Since lncLGR interacts with hnRNPL, and either of them suppresses GCK expression, we next attempted to understand if and how they coordinate this regulation. We first performed a GCK promoter-driven luciferase reporter assay in HEK293 cells and, found that under the condition when overexpression of either lncLGR or hnRNPL alone at a dose with no or moderate but significant effects on GCK promoter activity respectively, their co-overexpression strongly suppresses GCK promoter activity by 50% suggesting that lncLGR and hnRNPL function synergistically to regulate GCK transcription (Figure 4A). Consistent with this observation, HnRNPL has been recently demonstrated to bind DNA and function as a transcription factor (Li et al., 2014). The physical and functional interactions between lncLGR and hnRNPL we observed here also prompted us to hypothesize that hnRNPL could bind to the GCK promoter and suppress its transcription, while lncLGR binding would further enhance this regulation. Consistently, further fractionation of liver nuclei showed that lncLGR is significantly enriched in chromatin fraction (Figure 4B). Direct binding assay using purified hnRNPL protein and biotinylated GCK promoter fragments also demonstrated that hnRNPL directly interact with the GCK promoter DNA within the region from around −1500bp to −700bp (Figure 4C). To further validate this model and examine the specific binding of endogenous hnRNPL on the mouse GCK promoter and its regulation by lncLGR in vivo, we simultaneously knocked down hnRNPL and overexpressed lncLGR in mouse livers by co-injecting two adenoviruses. We then performed chromatin immunoprecipitation (ChIP) analysis using an anti-hnRNPL antibody and found that hnRNPL specifically binds to the GCK promoter in mouse liver (Figure 4D and 4E). Intriguingly, we demonstrated that while hnRNPL knockdown alone reduced its binding to the GCK promoter, this effect was completely reversed by a simultaneous lncLGR overexpression (Figure 4D) despite similar levels of total hnRNPL proteins (Figure 4E). Furthermore, the increased GCK expression caused by hnRNPL knockdown was also completely reversed by lncLGR overexpression (Figure 4F), a pattern that precisely mirrors the hnRNPL binding levels on the GCK promoter observed in these mice (Figure 4D). These results strongly support a model in which lncLGR and hnRNPL operate synergistically to regulate GCK expression, such that lncLGR suppresses GCK transcription by facilitating hnRNPL binding to the GCK promoter.
Figure 4. LncLGR and hnRNPL coordinately suppress GCK transcription.

(A) Mouse liver GCK promoter-driven luciferase activities in HEK293 cells transfected with vectors expressing hnRNPL, lncLGR, or both. Negative control for hnRNPL is a pcDNA 6.2 vector expressing a YFP, and negative control for lncLGR is the empty pcDNA 6.2 vector. (n=3, and the result is representative of at least two independent experiments).
(B) Levels of lncLGR in nucleoplasm and chromatin fractions of mice liver tissue samples (result of an independent experiment is also shown in figure S2C).
(C) Immunoblotting analysis of purified hnRNPL proteins that are bound to biotinylated mouse GCK liver promoter fragments using an anti-hnRNPL antibody.
(D) Chromatin immunoprecipitation of pooled liver tissue samples from mice (n=5) receiving control, hnRNPL KD, or both hnRNPL KD and lncLGR overexpression adenoviruses after a 4h food withdrawal. Bands were amplified with specific primers for liver GCK, neuroendocrine GCK, or β-actin promoters.
(E) Anti-hnRNPL immunoblotting analysis of proteins in immunoprecipitates of cross-linked mouse liver tissue samples shown in Figure 4D using an anti-hnRNPL antibody.
(F) Hepatic gene expressions in mice as described in Figure 4D (n=7).
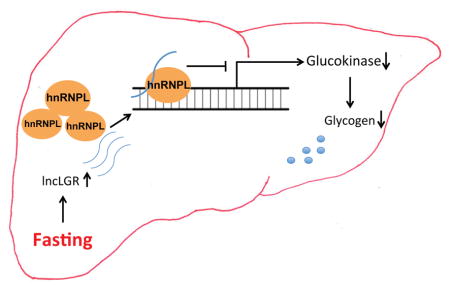
(G) A graphic model depicting the mechanism of hepatic GCK regulation by lncLGR and hnRNPL.
Error bars represent SEM, *p<0.05.
In summary, our results support that during fasting, lncLGR is induced in the liver and forms a functional complex with hnRNPL, which facilitates the recruitment of hnRNPL to the GCK promoter and subsequently suppresses transcription of GCK (Figure 4G).
DISCUSSION
As a key enzyme controlling liver glucose metabolism, GCK is subject to robust regulation by hormonal signals associated with feeding-fasting rhythms, such as insulin and glucagon. Multiple insulin- and nutrient-activated signaling cascades have been identified to enhance GCK expression upon feeding (Massa et al., 2011). In contrast, it remains elusive how GCK is down-regulated during fasting, which is a major physiological step for the liver to shift from a glucose-consuming state to a gluconeogenic one. In this study, we identify an lncRNA-mediated regulatory mechanism of GCK expression in which an lncRNA complex formed by lncLGR and hnRNPL suppresses GCK transcription in animals, and this regulation may allow the liver to precisely control GCK levels during fasting.
Our work also provides critical insight into the function of hnRNPL, which belongs to the hnRNP protein family comprised of over 20 RNA-binding proteins of diverse structure and binding specifies (Alvarez-Dominguez et al., 2015). In general, hnRNPs regulate key steps of RNA metabolism, including pre-mRNA processing, RNA transportation, and mRNA degradation (Alvarez-Dominguez et al., 2015; Chaudhury et al., 2010). Our findings that liver-specific expression of hnRNPL represses GCK expression suggest that hnRNPL could carry out tissue-specific functions in energy metabolism. Our data also indicate that hnRNPL regulates GCK expression through a nonconventional mechanism by directly binding to DNA and functioning as a transcriptional repressor. It has been reported that hnRNPs can regulate transcription in cells and tissues (Chaudhury et al., 2010). However, hnRNPs are often abundantly and ubiquitously expressed thus begging the question of how they achieve their specificities in these transcriptional events. LncRNAs that are restricted to specific tissues or are inducible only by specific stimuli might provide the answers. These lncRNAs may bind to a subset of hnRNPs only under specific conditions, conferring temporal and spatial regulation that allows hnRNPs to bind to a set of promoters and control transcription in a tissue- and context-dependent manner. In addition to the lncLGR-hnRNPL complex shown here, multiple lncRNAs have been reported to engage in transcriptional regulation by recruiting hnRNPs. For example, lincRNA-p21 interacts with hnRNPK, which collectively represses p53-dependent transcriptional responses in trans (Huarte et al., 2010), while activates p53-depedent p21 transcription in cis (Dimitrova et al., 2014). It was also demonstrated that hnRNPL could regulate TNFα transcription via binding to lncRNAs in immune cells (Li et al., 2014). Therefore, guiding an hnRNP to a promoter region might be a general function for lncRNAs in transcriptional regulation.
Of note, the lncLGR locus in the mouse genome lies very closely downstream a protein-coding gene Itga2 suggesting that the lncLGR transcript could be an isoform of Itga2 transcripts with an extended untranslated region (UTR). However, several lines of evidence suggest that lncLGR is an independent transcript. First, Itga2 expression is barely detectable in mouse liver. Second, there is no change in Itga2 expression during fasting and refeeding (data not shown), whereas LncLGR is up-regulated in mouse liver by fasting. Third, knockdown of lncLGR does not change Itga2 expression levels. Finally, we performed RACE using RNAs from mouse hepatic cell line hepa1-6, and identified that the 5′ end of lncLGR is 40 nucleotides shorter than the recorded one, thus placing the transcription initiation site of lncLGR further downstream of Itga2 than the GenBank one does. Interestingly, while lncLGR shares some sequence similarity with 3′ UTR of human ITGA2, the CA repeat required for the efficient interaction between hnRNPL and lncLGR (Figure 3G) is absent in the 3′UTR of human ITGA2, indicating that it might not represent the orthologous human lncLGR. Nevertheless, we tested the effect of hnRNPL on the promoter of human liver GCK and found hnRNPL also suppressed its transcription activity (data not shown).
It should be noted that the copy number of lncLGR is only 3.6 copies per cell in isolated primary hepatocytes at the basal level. Considering that hnRNPL is an abundant protein, this low level of lncLGR seems not able to strongly modify hnRNPL function. However, our results suggest that lncLGR promotes rather than sequestering or blocking the recruitment of hnRNPL to the GCK promoter, where comparable levels of lncLGR to hnRNPL proteins might not be necessary in order to function. Instead, the complex formed by lncLGR and a specialized pool of hnRNPL significantly regulates GCK to generate a functional impact. Although the very low copy number of lncRNAs is usually linked with cis regulation of gene expression (Dimitrova et al., 2014), our results support lncLGR regulates GCK expression in trans. First, the change of lncLGR expression during fasting and refeeding does not associate with expression changes in its neighbor genes, and neither does the lncLGR knockdown. Second, lncLGR overexpression reduces GCK expression. Furthermore, the observation that lncLGR is expressed at a low level but has a functional effect is consistent with reported function of another lncRNA THRIL. The expression level of THRIL in immune cells is very low, about 8 copies per cell, and it regulates TNFα expression via binding hnRNPL as well (Li et al., 2014). Nevertheless, lncRNA biology is a relatively new research area and the relationship between lncRNA expression levels and their functional importance has not been fully defined, it remains to be determined if there are more low copy lncRNAs that function in trans or lncLGR and THRIL are just among the few exceptions.
Based on our findings here and a previous report showing the interaction between lncLSTR and TDP-43 in lipid metabolism (Li et al., 2015), recruitment of hnRNPs could be a general mechanism for lncRNAs to transcriptionally regulate metabolic genes. Cells could utilize lncRNAs induced by metabolic cues to recruit a subset of abundant RNA-binding proteins to control specific metabolic pathways. It is equally possible that some of the abundant hnRNPs constitutively bind to DNA to generate a poised state, whereby complexing with an inducible lncRNA serves as a trigger to activate or suppress gene transcription. Thus, lncRNAs might allow cells and organs to respond to environmental or intrinsic signals by fine-tuning critical metabolic genes like GCK.
METHODS
Adenovirus production and in vivo adenovirus administration
ShRNAs for lncLGR and hnRNPL were designed to act against mouse sequences (lncLGR shRNA: GCACAGCTGTATTAGAATTGT; hnRNPL shRNA: GCCTACGCG TTTAAATGTA). Hairpin template oligonucleotides were synthesized by Integrated DNA Technologies and were subsequently cloned into the adenovirus vector of the pAD/Block-it system (Invitrogen) according to the manufacturer’s protocols. Overexpression constructs of lncLGR, lncLGR Δ1-335 and mouse liver GCK were generated by PCR-amplifying from mouse liver cDNA using the primers as: lncLGR-f: TCTAAAAGCAAAGGAAGAAATGA-3, lncLGR-r: CACTGTCAAAACACTTTTAA TGA; lncLGR Δ1-335-f: ATTCCAGGTGTTGAGCTGAGAAAG, GCK-f: ATGGCTGT GGATACTACAAG, GCK-r: TCACTGGCCCAGCATGCAAC. PCR products were subsequently cloned into the pAdv5 adenovirus vector for virus packaging. Adenoviruses were amplified in HEK293A cells and purified by CsCl gradient centrifugation. Purified viruses were desalted with PD10 columns (GE Healthcare Life Sciences) and titered with Adeno-X Rapid Titer Kit (Clontech). Adenoviruses were delivered into mice intravenously at 1–2×109 pfu/mouse. After seven to twelve days, animal experiments were performed, and tissue samples and plasma were harvested for further analyses.
Plasmid constructs and reporter assay
Full-length hnRNPL expression clone (Myc-Flag-tagged) was purchased from OriGene (OriGene, MR208796). LncLGR was sub-cloned into pcDNA6.2, and a yellow fluorescent protein (YFP) cDNA in pcDNA6.2 was used as a control. Mouse GCK promoters were amplified by PCR using genomic DNA (2kb upstream), and cloned into a promoter-less pcDNA6.2 vector with a firefly luciferase reporter. HEK293A cells were maintained in DMEM medium supplemented with 10% Cosmic Calf Serum. Cells were transfected with the GCK reporter, lncLGR, and hnRNPL or YFP vectors using Lipofectamine 2000 (Invitrogen), and luciferase assays were performed 24 hours later using the Dual-Luciferase Reporter Assay Kit (Promega). Transfection efficiency was measured by normalization to Renilla luciferase activity expressed from a co-transfected pTK-RL vector (Promega).
RNA pull-down assay and native RNA immunoprecipitation (RIP)
RNA pull-down was performed as described previously (Rinn et al., 2007). Biotin-labeled RNAs were transcribed in vitro using the Biotin RNA Labeling Mix and T7 RNA polymerase (Ambion) and purified with the RNeasy Mini Kit (Qiagen). Folded RNAs (1ug) were added into 2mg pre-cleared nuclear lysates (supplemented with 0.2mg/ml heparin, 0.2mg/ml yeast tRNA and 1mM DTT) and incubated at 4°C for one hour. Sixty microliters of washed Streptavidin-coupled Dynabeads (Invitrogen) were added to each binding reaction and further incubated at 4°C for one hour. Beads were washed five times with RIP buffer and heated at 70°C for ten minutes in 1x LDS loading buffer, and retrieved proteins were visualized by SDS-PAGE and silver staining. The unique protein bands shown in the sense RNA pull-down were identified by mass spectrometry. For native RIP, 5ug anti-hnRNPL antibody or mouse IgG were added into 3mg precleared liver nuclear lysates and incubated at 4°C for two hours. 50ul Dynabeads® Protein G were added and incubated for one hour at 4°C with gentle rotation. Beads were washed for five times with RIP buffer and resuspended in 1 ml of Trizol. Co-precipitated RNAs were isolated and analyzed by RT-PCR.
Statistical analysis
Values represent mean ± SEM. Statistical significance of differences was determined by Student’s t test or One-way ANOVA with Bonferroni’s post-hoc comparison where appropriate. P values less than 0.05 were considered to be significant.
Supplementary Material
Highlights.
LncLGR is a fasting-induced long non-coding RNA in mouse liver
LncLGR regulates glycogen storage by suppressing glucokinase activity
LncLGR and hnRNPL coordinately suppress glucokinase transcription
Acknowledgments
This work was supported by the Division of Intramural Research of the National Heart Lung and Blood Institute, NIH (HL006103 and HL006159). We thank Yong Chen and Marjan Gucek from NHLBI Proteomics Core for their helps with mass spectrometry analysis.
Footnotes
AUTHOR CONTRIBUTIONS
X.B.R and P.L designed the study, performed the experiments, analyzed the data, wrote the manuscript and contributed equally. A.C and L.Y performed the experiments. H.C designed, conceived and supervised the study, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Dominguez JR, Bai Z, Xu D, Yuan B, Lo KA, Yoon MJ, Lim YC, Knoll M, Slavov N, Chen S, et al. De Novo Reconstruction of Adipose Tissue Transcriptomes Reveals Long Non-coding RNA Regulators of Brown Adipocyte Development. Cell metabolism. 2015;21:764–776. doi: 10.1016/j.cmet.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. Rna. 2010;16:1449–1462. doi: 10.1261/rna.2254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA, et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Molecular cell. 2014;54:777–790. doi: 10.1016/j.molcel.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Guichard C, Ferre P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12737–12742. doi: 10.1073/pnas.96.22.12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld JW, Bruning JC. Regulation of metabolism by long, non-coding RNAs. Frontiers in genetics. 2014;5:57. doi: 10.3389/fgene.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, Chen Y, Gucek M, Zhu J, Cao H. A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell metabolism. 2015;21:455–467. doi: 10.1016/j.cmet.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, Head SR, Burns JC, Rana TM. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:1002–1007. doi: 10.1073/pnas.1313768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa ML, Gagliardino JJ, Francini F. Liver glucokinase: An overview on the regulatory mechanisms of its activity. IUBMB life. 2011;63:1–6. doi: 10.1002/iub.411. [DOI] [PubMed] [Google Scholar]
- Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, Arnes L, Nakic N, Garcia-Hurtado J, Rodriguez-Segui S, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell metabolism. 2012;16:435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth U, Curth K, Unterman TG, Kietzmann T. The transcription factors HIF-1 and HNF-4 and the coactivator p300 are involved in insulin-regulated glucokinase gene expression via the phosphatidylinositol 3-kinase/protein kinase B pathway. The Journal of biological chemistry. 2004;279:2623–2631. doi: 10.1074/jbc.M308391200. [DOI] [PubMed] [Google Scholar]
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zeng F, Liu Y, Zhao Y, Lv H, Niu L, Teng M, Li X. Crystal structures and RNA-binding properties of the RNA recognition motifs of heterogeneous nuclear ribonucleoprotein L: insights into its roles in alternative splicing regulation. The Journal of biological chemistry. 2013;288:22636–22649. doi: 10.1074/jbc.M113.463901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.