

Abstract
It has been well established that peripheral inflammation resulting from microbial infections profoundly alters brain function. This review focuses on experimental systems that model cerebral effects of peripheral viral challenge. The most common models employ the induction of the acute phase response (APR) via intraperitoneal injection of a viral mimetic, polyinosinic-polycytidylic acid (PIC). The ensuing transient surge of blood-borne inflammatory mediators induces a “mirror” inflammatory response in the brain characterized by the upregulated expression of a plethora of genes encoding cytokines, chemokines and other inflammatory/stress proteins. These inflammatory mediators modify the activity of neuronal networks leading to a constellation of behavioral traits collectively categorized as the sickness behavior. Sickness behavior is an important protective response of the host that has evolved to enhance survival and limit the spread of infections within a population. However, a growing body of clinical data indicates that the activation of inflammatory pathways in the brain may constitute a serious comorbidity factor for neuropathological conditions. Such comorbidity has been demonstrated using the PIC paradigm in experimental models of Alzheimer's disease, prion disease and seizures. Also, prenatal or perinatal PIC challenge has been shown to disrupt normal cerebral development of the offspring resulting in phenotypes consistent with neuropsychiatric disorders, such as schizophrenia and autism. Remarkably, recent studies indicate that mild peripheral PIC challenge may be neuroprotective in stroke. Altogether, the PIC challenge paradigm represents a unique heuristic model to elucidate the immune-to-brain communication pathways and to explore preventive strategies for neuropathological disorders.
Keywords: inflammation, sickness behavior, neuropathologies, viral infections, comorbidity, neuroprotection
1. Introduction
The existence of active communication pathways between the immune system and the brain has been well established. A stellar example is the assemblage of behavioral symptoms, such as, fever, cognitive dysfunction, anxiety, depression, anhedonia, malaise, anorexia, adipsia, lethargy and fatigue elicited by peripheral infections and collectively referred to as “sickness behavior” (1-3). Sickness behavior is thought to promote optimal recovery and survival by altering the priorities of the affected individuals to conserve resources and to forfend the spread of infection within the population. Mechanisms of this immune-to-brain communication involve relaying inflammatory signals from the site of infection to the brain whereby they activate cerebral innate immunity via several humoral and neural pathways (1, 3). Thus, the blood-borne inflammatory mediators can be directly transported through the blood-brain barrier (BBB) or through circumventricular organs (CVO) to the brain parenchyma. Alternatively, the circulating mediators may be transduced by activating the BBB and/or CVO cells resulting in a secretion of secondary mediators into the cerebral parenchyma. Within the cerebral parenchyma, the peripherally-generated and/or BBB/CVO-generated mediators activate cerebral innate immune cells, chiefly astrocytes and microglia that generate tertiary mediators. Tertiary mediators can also be generated by neurons that express receptors cognate to a slew of inflammatory mediators. Consequently, the brain is exposed to the peripherally-generated inflammatory agents, the cerebrally-generated agents or a combination thereof. The interplay of these agents creates an intricate, yet poorly understood, network of autocrine/paracrine and intracellular signaling pathways that interact with neurotransmitter, neuropeptide and neuroendocrine systems. This neuroinflammatory response alters brain function leading to the behavioral symptoms of sickness. In addition, vagal as well as other afferent pathways may detect inflammatory signals in the periphery and relay them directly to specific brain regions that orchestrate sickness behavior.
This review focuses on cerebral response to peripheral viral challenge. The main thrust is on the characterization of the most frequently used experimental model in which the acute phase response (APR) is instigated by a viral mimetic, polyinosinic-polycytidylic acid (PIC). Moreover, the applicability of this paradigm to study several neuropathological conditions as well as the most representative findings is presented.
2. Viral model
2.1. dsRNA
Studies addressing the mechanism by which the immune system affects brain function have largely employed the stimulation of the peripheral innate immunity with pathogen-associated molecular patterns (PAMPs) to mimic microbial infections. The most widely employed experimental paradigms induce APR to bacterial infection via systemic challenge with lipopolysaccharide (LPS), a ubiquitous component of the cell wall of gram negative bacteria. Relatively fewer studies address the effects of anti-viral APR on the brain. This experimental paradigm involves immune challenge with double stranded RNA (dsRNA). dsRNA is a signature PAMP of viral infection, because the vast majority of viruses either contain dsRNA in their genomes or generate dsRNA species during their lifecycle (4, 5). For example, viruses with dsRNA genomes may expose this PAMP in defectively packaged virions. Substantial amounts of dsRNA intermediates are generated during the replication of positive-strand ssRNA viruses, while DNA viruses generate dsRNA intermediates during overlapping convergent transcription. However, no detectable quantities of dsRNA are generated by negative-strand ssRNA viruses (5). Mammalian cells express several receptors that detect the presence of extra- and intracellular dsRNA in a sequence-independent fashion, e.g., Toll-like receptor 3 (TLR3), retinoic acid-inducible gene 1 (RIG-1), melanoma differentiation-associated protein 5 (MDA-5) and protein kinase R (PKR)(6). dsRNA released from infected cells is recognized by these receptors on neighboring cells and rapidly triggers an antiviral response characterized by the expression of type I interferons (IFNα and IFNβ), as well as a plethora of other cytokines and inflammatory mediators. This early event in viral infection represents the first line of anti-viral defense critical for containing the spread of viral infections long before the adaptive immunity against viral antigens becomes effective.
2.2. PIC
A synthetic dsRNA, PIC, has been widely used as a potent inducer of antiviral APR (7-9). Systemic administration of PIC has been shown to induce symptoms of sickness behavior in laboratory rodents (9-16) that is congruent with behavioral effects of peripheral viral infections in humans (17-19). These studies have also demonstrated that PIC challenge models APR to generic viral infections, and that this APR is the major inducer of cerebral response. Although models using bona fide viral infection represent more clinically relevant approach, the PIC-based model offers several heuristic advantages for mechanistic analysis of immune-to-brain communication. For example, in contrast to viral inoculation that entails a prolonged and nonsynchronous incubation time before eliciting innate immune response, the inflammatory response to dsRNA is very rapid, allowing hour-by-hour kinetic analyses. There are no comorbid effects of pathogen-associated tissue damage as the biological activity of PIC is restricted to the stimulation of the innate immune response. Furthermore, the possibility for confounding involvement of adaptive immunity such as antibodies and T cells resultant from prior exposure of animals to viral or cross-reactive proteins is eliminated. Finally, PIC is noninfectious, and thus, the experiments can be conveniently performed under standard laboratory conditions. Consequently, peripheral PIC challenge provides a unique model of peripheral viral infection that allows analysis of humoral communication along the immuno-neural axis. The inflammatory pathways instigated by PIC challenge are depicted in Fig. 1.
Fig. 1.
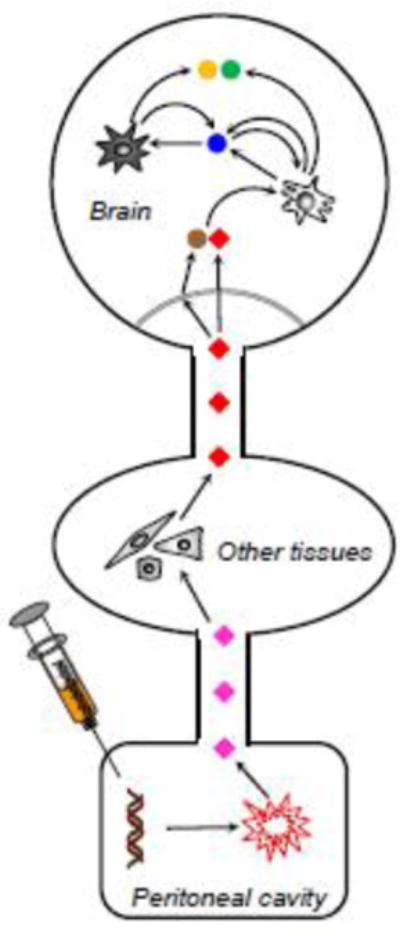
Inflammatory events in the PIC model. Intraperitoneally injected PIC elicits a fulminant response of the peritoneal macrophages and mesothelial cells leading to the generation of cytokines and other inflammatory agents (pink diamonds) that are released into the circulation. PIC itself does not reach the circulation and is rapidly degraded. The blood-borne inflammatory mediators stimulate cells in other organs, e.g., liver, muscles, to generate additional inflammatory mediators and the resulting assortment of mediators (red diamonds) reaches the brain. Some mediators gain access to the brain parenchyma via BBB of CVO (double line), while others activate the endothelial cells that release secondary mediators into the parenchyma (brown diamond). Both the peripherally-generated (transported) and endothelium-generated (transduced) agents activate resident cerebral cells (light cell) to produce additional mediators (blue circle) that can either act in an autocrine (light cell) or paracrine (dark cell) manner. Both the paracrine and autocrine stimulation may either amplify the production of the same mediator (blue circle) or induce generation of additional mediators (orange and green circles). These new mediators, in turn, may instigate paracrine/autocrine stimulation of other parenchymal cells. Altogether, these signaling cascades generate an assortment of inflammatory mediators that specifically alters the activity of neuronal circuits leading to changes in brain function. Of note, some of the cerebrally-generated mediators may enter the circulation, and thus, affect the assortment of peripherally-generated mediators. These brain-to-periphery feedback pathways would further contribute to the complexity of the cerebral response. For specifics see the text.
2.3. PIC vs. LPS
Although both the LPS and PIC paradigms of intraperitoneal immune stimulation provide valuable experimental models, they differ in several aspects. LPS activates Toll-like receptor (TLR) 4 while PIC acts primarily through TLR3. These two receptors employ different signaling pathways that generate inflammatory mediators germane to antibacterial and antiviral APR, respectively. These diverse APR profiles may in turn elicit dissimilar inflammatory responses in the brain. Also, the pharmacokinetics of LPS and PIC are quite dissimilar. LPS is a stable amphipathic molecule that rapidly passes from the peritoneal cavity into the bloodstream (20, 21), and thus, can activate innate immune cells throughout the body including the BBB and CVO cells. Thus, in the LPS paradigm the brain is challenged by a combination of LPS and LPS-induced peripheral inflammatory mediators. In contrast, PIC is a large, charged molecule that is rapidly degraded by ubiquitous RNases in the body fluids (22). Intraperitoneally injected PIC does not reach the circulation and its cerebral effects are mediated by blood-borne inflammatory factors (23).
In line with the above disparities in pharmacokinetics and pharmacodynamics between LPS and PIC, notable differences in behavioral effects of these two inflammagens have been reported. For example, Hopwood and collaborators found PIC to be less potent in inducing anorexia and lethargy that LPS in rats (24). They also observed PIC to be a less effective pyrogen than LPS.
Recently, cerebral response to LPS and PIC has been analyzed at the transcriptome level. At 48 h after LPS challenge, 71 genes were found dysregulated (25), while 260 genes were dysregulated by PIC (26). Only 23 genes were commonly affected by both inflammagens, whereas 48 and 237 genes were dysregulated exclusively by LPS and PIC, respectively. This comparison further substantiates the dissimilarity in neuroinflammatory response of the brain instigated by LPS vs. PIC.
3. Cerebral response
3.1. Behavior
Cunningham and colleagues have provided the first comprehensive assessment of cerebral response of mice to PIC challenge at the behavioral and molecular levels (27). Their findings show the burrowing activity to be the most severely reduced by intraperitoneal injection of PIC. This transient reduction reaches a nadir at 6 h and returns to control level by 48 h after PIC challenge. The animals also display a transient decrease in locomotor activity. These behavioral changes are accompanied by a mild hyperthermia and a loss of body weight. All these effects are dependent on PIC dose in a range of 2 to 12 mg/kg body weight. Moreover, successive challenge at one or three weeks does not induce behavioral tolerance to PIC. Also, no inflammatory tolerance has been shown in mice receiving multiple (seven) daily injections of PIC (28). An earlier study in rabbits, found systemic administration of PIC to increase body temperature and to activate the hypothalamo-pituitary-adrenocortical axis (29).
The burrowing test that provides a sensitive assessment of behavioral dysfunction in rodents (30), is the most discriminatory assessment of PIC-induced sickness behavior as the burrowing activity is reduced by approximately 95% at 6 h (27, 31). We have recently shown that the rearing activity of mice challenged with PIC is equally reduced at 6 h (26). In contrast to the burrowing test that is rather intricate and time-consuming (2 h testing time), the rearing test is a simple procedure that can be completed within 15 min. Consequently, the rearing test provides a convenient and sensitive procedure for gauging sickness behavior.
3.2. Molecular level
PIC challenge triggers a robust surge of peripheral cytokines in mice. This is manifested as a transient elevation of blood levels of interferon β (IFNβ), interleukin 6 (IL-6), tumor necrosis factor α (TNFα) and IL1β (26, 27). Blood levels of these cytokines peak at 3 h post-injection, rapidly decline by 6 h and gradually dwindle to the basal level. The cytokine surge is concomitant with transient upregulation of genes encoding IL-1β, IL-6, TNFα and IFNβ in the hippocampus and hypothalamus (27). We have extended this study and showed that the cerebral response is global rather than regional as it is featured in all major parts of the brain, i.e., the cerebellum, forebrain and brainstem (23, 31, 32). In all brain regions, the expression of a multitude of inflammatory and stress genes is robustly upregulated from several- to several thousand-fold. Also, injection of blood plasma from PIC-challenged into naïve mice mimics the effect of PIC by instigating the upregulation of cerebral genes (23). Because PIC does not reach the circulation (23), these results indicate that the cerebral neuroinflammatory response is mediated by peripherally-generated, blood-borne inflammatory factors. The kinetics of blood-borne cytokines and cerebral gene expression is presented in Fig. 2.
Fig. 2.
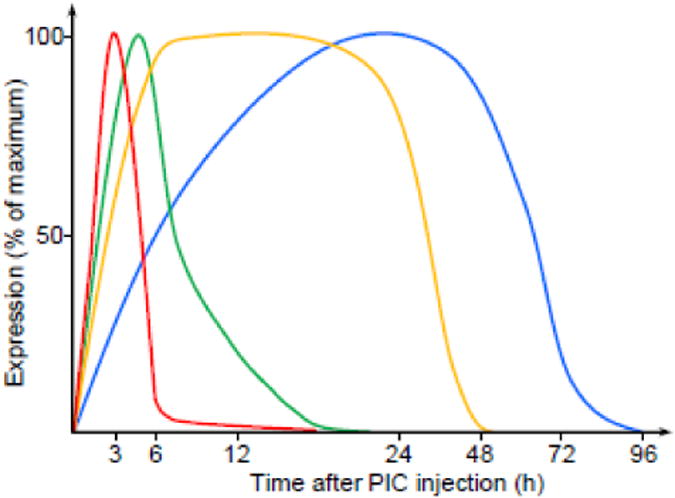
Time course of neuroinflammatory response following PIC challenge. Intraperitoneal injection of PIC elicits a robust but transient surge in blood cytokines (red), e.g., IFNβ, IL-6 and TNFα. This cytokine surge upregulates the transcription of inflammatory genes in the brain. Three major patterns of mRNA upregulation can be distinguished. Some genes, e.g., the Ifnb, Il6, Cxcl10 and Cxcl1 genes, feature a rapid upregulation that subsides by 24 h (green). Other genes, e.g., the Tnfa, Ccl7 and Ccl12 genes, are also rapidly upregulated but display a protracted course (orange). The genes encoding complement proteins, e.g., the Cfb, C3 and C2 genes, exhibit more gradual upregulation that lasts for three days (blue). Generalized from (23, 26, 27, 31, 33).
Our recent whole genome transcriptome analysis revealed a robust polygenic response in the hippocampus of PIC-challenged mice (33). Thus, 625 genes were found to undergo dynamic dysregulation between 0 and 48 h after the challenge. As expected, the majority of these differentially expressed genes were related to immune and inflammatory processes of both the innate and acquired immunity. However, a plethora of genes related to neurotransmission were also dysregulated. Moreover, ten microRNAs that modulate both neural and immune functions or are associated with seizure pathology also featured differential expression following PIC challenge (26). Altogether, these results indicate an extensive genomic reprogramming in the hippocampal cells instigated by peripheral antiviral response.
4. Mechanistic considerations
The major inflammatory mediators generated either in the periphery or in the brain in response to PIC challenge are potent effectors of neurotransmission. These effects are likely to be mediated or at least contribute, to the development of behavioral alterations as exemplified below.
4.1. Cytokines
IL-1β is the primary cytokine responsible for the induction of fever following PIC injection (10). Intraventricular injection of IL-1β also induces anorexia (34). IL-1β inhibits voltage-dependent calcium channel currents in cultured hippocampal and cortical neurons (35, 36), resulting in the reduction of presynaptic glutamate release (37) and impaired long term potentiation (LTP) (38, 39). At low concentrations, IL-1β potentiates NMDA response in the hippocampal neurons (40, 41), whereas the opposite effects is elicited at high concentrations (42). In a similar concentration-dependent fashion, IL-1β may enhance (43) or reduce (44) the GABAergic activity in hippocampal neurons. Neuronal excitability may be additionally impacted by IL-1β-engendered impairment of glutamate uptake (45) by astrocytes. IL-6 also has a biphasic modality effect on neuronal excitability (46-48). IL-6 inhibits LTP (49) and impairs cognitive functions (50). Moreover, systemic injection of IL-6 decreases interstitial dopamine level in the nucleus accumbens (51). TNFα increases surface expression of AMP A receptors (52, 53), leading to the enhancement of excitatory synaptic efficacy. TNFα also promotes endocytosis of GABAA receptors, and thus, decreases the strength of inhibitory synapses (53). This cytokine also reduces the outward potassium current in cortical neurons (54), facilitates synaptic scaling (55), and inhibits LTP (38). In astrocytes, TNFα increases glutamate release (56) and reduces the capacity to buffer extracellular potassium (57). IFNβ was recently shown to mediate several traits of sickness behavior, i.e., hypothermia, anhedonia and anorexia (58). This antiviral cytokine enhances the excitability of neocortical neurons in culture (59), CA3 pyramidal neurons in hippocampal slice cultures (60) and CA1 pyramidal neurons in acute hippocampal slices (61). However, in striatal medium spiny neurons, IFNβ attenuates glutamatergic neurotransmission (62), raising the intriguing possibility that IFNβ action depends on the type of targeted neurons. IFNβ also reduces expression of the glutamate/aspartate transporter (GLAST) in astrocytes (61).
4.2. Chemokines
Nearly half of the genes encoding mouse chemokines are upregulated by PIC challenge (23). Of a particular interest are the genes encoding eight chemokines, i.e., CXCL1, CXCL2, CXCL9, CXCL10, CCL2, CCL4, CCL5 and CCL7 genes. These genes feature upregulation from 60 to 4000 fold over the baseline. PIC challenge also upregulates three genes encoding receptors that bind most of these chemokines, i.e., the Cxcr2, Ccr1 and Ccr5 genes. Signaling pathways triggered by the cognate chemokines are potent modulators of neuronal activity and behavior. For example, CXCL9/10 are ligands of the CXCR3 receptor, whose activation inhibits long-term potentiation, elevates intracellular calcium and increases electrical activity of hippocampal neurons (63, 64). CXCR3 ligation also alters the expression of several glutamatergic and GABAergic receptors (65). CXCL1 and CXCL2, the agonists of CXCR2, enhance glutamatergic receptor activity and modulate electrical activity of Purkinje neurons (66-68). The activation of CXCR2 also reduces calcium currents and the excitability of septal neurons (69). The ligation of CCR2, the receptor of CCL2 and CCL7, inhibits GABAergic responses of spinal neurons (70), increases dopaminergic neuron discharge and dopamine release in the substantia nigra (71). The ligation of CCR1 that binds CCL4 was reported to increase NMDA-evoked Ca2+ signals and NMDA receptor levels in the hippocampal neurons (72). Behaviorally, the ligation of CCR1 and CCR5 evokes a febrile response (73-75). Ligands of CXCR3, CCR1, CCR2 and CCR5 suppress food intake (35, 76, 77), while the ligation of CCR2 increases locomotor activity (71). Neuronal as well as glial expression of the chemokines and their cognate receptors (78, 79) implicates the existence of intricate paracrine/autocrine pathways that affect neuronal activity. Consequently, complex intercellular chemokine signaling networks in the brain are likely to underscore the development of sickness behavior induced by peripheral inflammatory signals generated in response to PIC challenge.
4.3. COX2
The inducible cyclooxygenase (COX2) is the central enzyme in the biosynthesis of important inflammatory mediators, viz. prostaglandins and prostacyclin. COX2 upregulation has been demonstrated in endothelial cells in the mouse brain following systemic PIC challenge at both the message and protein levels (27). This is congruent with similar studies in the guinea pig brain (80). Moreover, the inhibition of COX2 has been shown to obliterate febrile response induced by PIC challenge in rabbits (29) and in guinea pigs (80). Together, these results support the role of endothelial COX2 as a transducer of blood-borne inflammatory signals to the brain parenchyma. In addition, prostaglandins have detrimental effects on cognitive functions by the impairment of learning and memory processes (81, 81). In vitro, the PGE2 prostaglandin increases excitability and synaptic transmission of hippocampal neurons (82) and enhances astrocytic glutamate release (56).
4.4. NO and CX3CL1
The upregulation of nitric oxide (NO) synthase type 2 (NOS2) is considered to be the integral element of neuroinflammation. However, in spite of a robust up-regulation of chemokines and cytokines, we observed no up-regulation of the Nos2 gene expression in the brains of mice challenged with PIC (32). Also, we did not observe upregulation of the gene encoding the CX3CL1 chemokine, aka, fractalkine (23). Both NO and CX3CL1 can mediate brain tissue damage. NO is a highly reactive free radical that has diverse harmful effects on neuronal function, causing neuronal cell injury and death (83). CX3CL1 is a potent proinflammatory chemokine that promotes the proliferation of microglia and facilitates their activation into the pro-inflammatory and cytotoxic M1 phenotype (84). Consequently, PIC challenge models a “physiological” cerebral response expected during peripherally restricted infections that elicits protective sickness behavior in the absence (or suppression) of detrimental neuroinflammatory bystander effects.
5. Neuropathological models
Although sickness behavior per se represents a physiological adaptation that facilitates the healing process, in neurodegenerative disorders and in aged brains, the transient stimulation of cerebral immunity may enhance preexisting inflammatory levels, and consequently, promote further tissue destruction. In this context, even an innocuous virus may exacerbate neuropathological events. Such infectious triggers may underlie neuropathological deteriorations that are otherwise classified as idiopathic. In fact, peripheral viral infections are emerging as comorbid factors in neurodegeneration. For example, peripheral infections exacerbate cognitive dysfunction in Alzheimer's disease (AD) (85-89) and age-related dementia (90). The effects of peripheral viral infection on neuropathological conditions using the PIC model have been addressed in several preclinical studies as presented below.
5.1. Alzheimer's disease
The induction of cognitive impairments has been reported in animals peripherally challenged with PIC. For instance, PIC challenge disruptes hippocampus-dependent memory consolidation as seen from a worsening of performance on the contextual fear conditioning (CFC) test in mice (91). In addition, the cerebral response to PIC challenge has been shown to be exacerbated in the aging as compared to young brain (92). Together, these results indicate a possible link between a viral challenge and AD. Indeed, a subsequent study (28) revealed that the administration of seven daily i.p. injections of PIC elicited over a two-fold increase in the levels of the amyloid-beta peptide (Aβ1–42), a pathological signature of AD, in the hippocampus. Aβ production was concomitant with a profound cognitive dysfunction assessed by the CFC test. A linear regression analysis revealed that the increase in hippocampal Aβ greatly contributed to the deficient memory consolidation following PIC challenge. These data buttress the contention that viral infections exacerbate cognitive deficits in AD.
5.2. Prion disease
Field and collaborators reported that PIC challenge exacerbated neurodegeneration in the ME7 prion-diseased mice (93). A single intraperitoneal injection of PIC impaired motor co-ordination and muscle strength in ME7 but not in control mice. Three consecutive injections of PIC to ME7 mice at 2 week-intervals dramatically accelerated the neurological decline in a cumulative manner. At the molecular level, PIC challenge upregulated the expression of IFNα, IFNβ and two proinflammatory cytokines, i.e., IL-1β and IL-6, in the hippocampus and hypothalamus of diseased mice. Also, several IFN-responsive genes featured significant upregulation, indicating cerebral generation of type I IFNs. Histological examination revealed microglial activation with increased number of IL-1β positive microglia in the periventricular and dentate gyrus regions. There was also an increased COX2 staining of the endothelial cells. Moreover, PIC challenge activated proapoptotic pathways as seen from upregulated levels of mRNA encoding the dsRNA-dependent protein kinase (PKR) and Fas, as well as from an increase in TUNEL positive cells. Altogether, these results indicate the role of type I IFN signaling pathways in mediating the comorbid effects of PIC challenge on prion-related neurodegeneration.
5.3. Seizures
Intraperitoneal PIC injection also increases the vulnerability of the brain to excitotoxic insults as seen from hypersusceptibility to kainic acid (KA)-induced seizures (26, 94). This hypersusceptibility manifests as a robust increase in both the severity and protracted duration of status epilepticus as compared to saline-injected mice. The hypersusceptible phenotype lasts for three days after PIC challenge. This finding supports a causative role of peripheral viral infections in the increased seizure propensity inferred from epidemiological studies (95). A subsequent study of the hippocampus, the initial onset region for KA-induced seizures (96), revealed a robustly upregulated expression of the complement genes that was commensurate with seizure hypersusceptibility in PIC-challenged mice (33). The gene encoding complement factor B (CfB), the initiator of the alternative complement pathway featured the highest upregulation. Cerebral complement is a major modulator of neuronal activity through synaptic modification (97-99) and intracerebral injection of complement components has been shown to have a proconvulsant effect (100). Consequently, it is highly feasible that activation of the alternative complement pathway represents a mechanistic link between peripheral PIC challenge and hippocampal hyperexcitability.
In addition, the involvement of inflammatory cytokines in the induction of hyperexcitability leading to seizures is evident. For instance, IL-1β has been implicated as a major proictogenic cytokine (101, 102). IL-6 has a biphasic modality as either its over- or under-expression induces seizure hypersusceptibility (47, 48). Also, IFNβ was recently shown to mediate spontaneous interictal activity in acute hippocampal slices obtained from mice during the cytokine surge induced by peripheral PIC challenge (61).
Of note, neuronal hyperexcitability is a major contributing factor in the progression of neurodegeneration. For example, hippocampal over-activation correlates with cognitive impairment in the aging brain (103) and determines development of AD (104). As shown above, peripheral antiviral response robustly increases excitability of hippocampal circuits. Therefore, the comorbid effects of peripheral infections on neurodegeneration (85, 87-90, 105) are likely to involve hyperexcitability as a mechanistic link.
5.4. Schizophrenia and Autism
Epidemiological evidence has demonstrated a positive correlation between maternal viral infections and an increased risk of neurodevelopmental abnormalities in offspring that result in long-lasting behavioral dysfunctions in adulthood (106-108). Prenatal immune activation with PIC reproduces several features of schizophrenia and autism, and has been widely used to study these neuropsychiatric disorders (108, 109).
5.4.1. Prenatal exposure
The activation of maternal immunity with PIC has been shown to induce a range of psychopathological and neuropathological features characteristic for schizophrenia and autism. For example, PIC-challenge of pregnant dams reduces locomotor development, sensorimotor gaiting (110-114) and socialization (115) in the offspring. Other behavioral dysfunctions include cognitive deficits (110, 113, 116), depression-like behavior (116) and a reduction of EEG coherence between the dorsal hippocampus and medial prefrontal cortex (117). Other features pertaining to the neuropathology of schizophrenia and autism have also been observed in offspring exposed to maternal PIC-challenge. These include hippocampal hyperexcitability, seizure hypersusceptibility (115) and hypersensitivity to methamphetamine (110). Similar neurodevelopmental impairments were recently observed in nonhuman primates. Thus, rhesus monkeys born to females challenged with PIC during pregnancy displayed a host of behavioral abnormalities that include abnormal repetitive behavior, altered communication, atypical social interactions (118) and abnormal gaze patterns when viewing faces of unfamiliar conspecifics (119).
Morphologically, maternal PIC challenge dysregulates mouse brain development as seen from reduced proliferation of neural stem cells in the cortex (111), olfactory bulb (120) and dentate gyrus (116). Similar deficiencies in neuronal development have been observed in rats (114, 121). In rhesus monkeys, alterations of dendritic morphology of pyramidal neurons in the prefrontal cortex were recently reported (122). In addition to neuronal abnormalities, mouse offspring of PIC-challenged dams exhibited a transient decrease in axonal diameter and hypomyelination during the juvenile period (123), while microstructural alterations of the white matter within the fronto-striatal-limbic circuits persisted into adulthood (124). These findings corroborate clinical data showing a strong association of myelination defects with both schizophrenia (125) and autism (124).
At the molecular level, offspring born to PIC-challenged dams were found to feature downregulated expression of the GluN1 subunit of NMDA receptors (121), and upregulation of the metabotropic glutamate receptor 5 (mGluR5) (126), synaptobrevin (121) and choline acetyltransferase (127). The offspring also had increased tonic extracellular glutamate in the prefrontal cortex (128), impaired arginine metabolism in the hippocampus and prefrontal cortex (129) and reduced cerebral glutathione content (110).
5.4.2. Early postnatal exposure
Although important developmental events within the rodent brain occur in the early postnatal life, relatively few studies have addressed the effects of PIC challenge at this critical period. For example, repeated intraperitoneal injections of PIC during postnatal day 2-6 (P2-6) were shown to increase anxiety-like behavior, induce sensorimotor gating deficits, as well as to impair memory and social behavior in adult mice (130). PIC challenge also exacerbated behavioral deficits in transgenic mice deficient in the disrupted-in-schizophrenia 1 (DISC1) gene (131). In rats, a single intraperitoneal injection of PIC at P14 led to robust anxiety-like behaviors assessed two month later (132). These are significant findings because schizophrenia and autism feature an anxiety component. In addition, PIC challenge to P14 rat pups was shown to alter neuroimmune responses to PIC challenge in adult rats as seen from a mitigated febrile response concurrent with an augmented corticosteroid response (133). Altogether, these studies demonstrate that the window of behavioral vulnerability to the acute antiviral response in the periphery extends well into the postnatal period.
5.5. Neuroprotection
Besides the deleterious effects of PIC challenge discussed above, the resultant inflammatory milieu may be beneficial in certain circumstances. For example, intraperitoneal administration of low doses of PIC was shown to provide protection (tolerance) from a subsequent ischemic challenge (134). The administration of PIC three days before middle cerebral artery occlusion (MCAO) profoundly reduced the infarct volume and attenuated neurological and motor deficits. Subsequent studies from the same laboratory have demonstrated that the protection is mediated by type I IFNs generated in response to PIC challenge, and that the primary mechanism entails the attenuation of ischemia-induced BBB damage (135). Importantly, post-ischemic administration of PIC also has neuroprotective effect (136). Thus, peripheral PIC challenge three hours after MCAO reduced cerebral infarct volume and edema, and improved neurological scores. Neuronal cell death and mitochondrial damage in the ischemic tissue of the PIC-challenged mice was also mitigated. These protective effects are likely mediated by decreased expression of the pro-apoptotic Bax protein and increased expression of anti-apoptotic Bcl2, Hsp27 and Hsp70 proteins. Moreover, the protection was protracted and lasted for 14 days after the onset of ischemia. The therapeutic effects of PIC challenge seem to be related to cerebral IFNβ production and to the downregulation of TLR4-mediated cascades. The neuroprotective property of PIC is intriguing in view of the overwhelming deleterious effects discussed previously. This dual activity may be related to PIC dosing. For example, the ischemic neuroprotection is only observed at PIC doses of less than 4 mg/kg (134). It seems plausible that the low doses of PIC generate neuroprotective levels of type I IFNs, while at higher doses the neuroprotective effect of IFNs is obliterated by the generation of proinflammatory mediators, e.g., TNFα and IL-1β.
6. Conclusions
The studies reviewed above demonstrate that the PIC model provides a powerful heuristic tool for studies of the immune-to-brain communication pathways at the basic neurobiological level, as well as for the delineation of mechanisms underlying comorbid effects of peripheral viral infections on neurodegeneration. The clinical significance of such studies is underscored by the fact that most neurodegenerative conditions affect the aging brain, and that viral infections become more frequent than bacterial infections in the aging population. On the other hand, maternal PIC challenge dysregulates cerebral development of the offspring. This paradigm dovetails with the role of prenatal viral infections in the etiology of neuropsychiatric disorders. Consequently, the PIC model is well suited for the exploration of novel preventive strategies for neurodegenerative as well as neurodevelopmental disorders. Finally, there is compelling evidence that peripheral challenge with low doses of PIC may provide neuroprotection against ischemic damage. Peripherally administered PIC has been tested in clinical trials primarily as an adjuvant to anti-cancer vaccines, and has been proven to be safe even in long-term treatments. Therefore, studies employing the PIC model may lead to the development of efficient therapeutic interventions for stroke and possibly for other neuropathological conditions using PIC as a viable target drug.
Acknowledgments
This work was partly supported by a research grant from the National Institutes of Health/National Institute of General Medical Sciences, U54GM104942. The content is solely the responsibility of the author and does not necessarily represent the official views of the NIH. The author would like to thank Mr. Brent Lally for proofreading this manuscript.
References
- 1.Dantzer R. Cytokine, sickness behavior, and depression. Neurol Clin. 2006;24:441–460. doi: 10.1016/j.ncl.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dantzer R, Kelley KW. Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun. 2007;21:153–160. doi: 10.1016/j.bbi.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quan N, Banks WA. Brain-immune communication pathways. Brain Behav Immun. 2007;21:727–735. doi: 10.1016/j.bbi.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs BL, Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 5.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berke IC, Li Y, Modis Y. Structural basis of innate immune recognition of viral RNA. Cell Microbiol. 2013;15:386–394. doi: 10.1111/cmi.12061. [DOI] [PubMed] [Google Scholar]
- 7.Guha-Thakurta N, Majde JA. Early induction of proinflammatory cytokine and type I interferon mRNAs following Newcastle disease virus, poly [rI:rC], or low-dose LPS challenge of the mouse. J Interferon Cytokine Res. 1997;17:197–204. doi: 10.1089/jir.1997.17.197. [DOI] [PubMed] [Google Scholar]
- 8.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 9.Traynor TR, Majde JA, Bohnet SG, Krueger JM. Intratracheal double-stranded RNA plus interferon-gamma: a model for analysis of the acute phase response to respiratory viral infections. Life Sci. 2004;74:2563–2576. doi: 10.1016/j.lfs.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Fortier ME, Kent S, Ashdown H, Poole S, Boksa P, Luheshi GN. The viral mimic, polyinosinic:polycytidylic acid, induces fever in rats via an interleukin-1-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2004;287:R759–R766. doi: 10.1152/ajpregu.00293.2004. [DOI] [PubMed] [Google Scholar]
- 11.Katafuchi T, Kondo T, Take S, Yoshimura M. Enhanced expression of brain interferon-alpha and serotonin transporter in immunologically induced fatigue in rats. Eur J Neurosci. 2005;22:2817–2826. doi: 10.1111/j.1460-9568.2005.04478.x. [DOI] [PubMed] [Google Scholar]
- 12.Fang J, Bredow S, Taishi P, Majde JA, Krueger JM. Synthetic influenza viral double-stranded RNA induces an acute-phase response in rabbits. J Med Virol. 1999;57:198–203. [PubMed] [Google Scholar]
- 13.Kimura-Takeuchi M, Majde JA, Toth LA, Krueger JM. Influenza virus-induced changes in rabbit sleep and acute phase responses. Am J Physiol. 1992;263:R1115–R1121. doi: 10.1152/ajpregu.1992.263.5.R1115. [DOI] [PubMed] [Google Scholar]
- 14.Carter WA, De CE. Viral infection and host defense. Science. 1974;186:1172–1178. doi: 10.1126/science.186.4170.1172. [DOI] [PubMed] [Google Scholar]
- 15.Dunn AJ, Vickers SL. Neurochemical and neuroendocrine responses to Newcastle disease virus administration in mice. Brain Res. 1994;645:103–112. doi: 10.1016/0006-8993(94)91643-8. [DOI] [PubMed] [Google Scholar]
- 16.Majde JA. Viral double-stranded RNA, cytokines, and the flu. J Interferon Cytokine Res. 2000;20:259–272. doi: 10.1089/107999000312397. [DOI] [PubMed] [Google Scholar]
- 17.Loftis JM, Huckans M, Ruimy S, Hinrichs DJ, Hauser P. Depressive symptoms in patients with chronic hepatitis C are correlated with elevated plasma levels of interleukin-1beta and tumor necrosis factor-alpha. Neurosci Lett. 2008;430:264–268. doi: 10.1016/j.neulet.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huckans M, Seelye A, Parcel T, Mull L, Woodhouse J, Bjornson D, Fuller BE, Loftis JM, Morasco BJ, Sasaki AW, Storzbach D, Hauser P. The cognitive effects of hepatitis C in the presence and absence of a history of substance use disorder. J Int Neuropsychol Soc. 2009;15:69–82. doi: 10.1017/S1355617708090085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelligan JA, Loftis JM, Matthews AM, Zucker BL, Linke AM, Hauser P. Depression comorbidity and antidepressant use in veterans with chronic hepatitis C: results from a retrospective chart review. J Clin Psychiatry. 2008;69:810–816. doi: 10.4088/jcp.v69n0514. [DOI] [PubMed] [Google Scholar]
- 20.Lenczowski MJ, Van Dam AM, Poole S, Larrick JW, Tilders FJ. Role of circulating endotoxin and interleukin-6 in the ACTH and corticosterone response to intraperitoneal LPS. Am J Physiol. 1997;273:R1870–R1877. doi: 10.1152/ajpregu.1997.273.6.R1870. [DOI] [PubMed] [Google Scholar]
- 21.Romanovsky AA, Ivanov AI, Lenczowski MJ, Kulchitsky VA, Van Dam AM, Poole S, Homer LD, Tilders FJ. Lipopolysaccharide transport from the peritoneal cavity to the blood: is it controlled by the vagus nerve? Auton Neurosci. 2000;85:133–140. doi: 10.1016/S1566-0702(00)00232-0. [DOI] [PubMed] [Google Scholar]
- 22.Krasowska-Zoladek A, Banaszewska M, Kraszpulski M, Konat GW. Kinetics of inflammatory response of astrocytes induced by TLR 3 and TLR4 ligation. J Neurosci Res. 2007;85:205–212. doi: 10.1002/jnr.21088. [DOI] [PubMed] [Google Scholar]
- 23.Fil D, Borysiewicz E, Konat GW. A broad upregulation of cerebral chemokine genes by peripherally-generated inflammatory mediators. Metab Brain Dis. 2011;26:49–59. doi: 10.1007/s11011-010-9231-9. [DOI] [PubMed] [Google Scholar]
- 24.Hopwood N, Maswanganyi T, Harden LM. Comparison of anorexia, lethargy, and fever induced by bacterial and viral mimetics in rats. Can J Physiol Pharmacol. 2009;87:211–220. doi: 10.1139/y09-003. [DOI] [PubMed] [Google Scholar]
- 25.Thomson CA, McColl A, Cavanagh J, Graham GJ. Peripheral inflammation is associated with remote global gene expression changes in the brain. J Neuroinflammation. 2014;11:73. doi: 10.1186/1742-2094-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michalovicz LT, Konat GW. Peripherally restricted acute phase response to a viral mimic alters hippocampal gene expression. Metab Brain Dis. 2014;29:75–86. doi: 10.1007/s11011-013-9471-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behavior and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain Behav Immun. 2007;21:490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Weintraub MK, Kranjac D, Eimerbrink MJ, Pearson SJ, Vinson BT, Patel J, Summers WM, Parnell TB, Boehm GW, Chumley MJ. Peripheral administration of poly I:C leads to increased hippocampal amyloid-beta and cognitive deficits in a non-transgenic mouse. Behav Brain Res. 2014;266:183–187. doi: 10.1016/j.bbr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 29.Milton NG, Hillhouse EW, Milton AS. Activation of the hypothalamo-pituitary-adrenocortical axis in the conscious rabbit by the pyrogen polyinosinic:polycytidylic acid is dependent on corticotrophin-releasing factor-41. J Endocrinol. 1992;135:69–75. doi: 10.1677/joe.0.1350069. [DOI] [PubMed] [Google Scholar]
- 30.Deacon RM. Burrowing in rodents: a sensitive method for detecting behavioral dysfunction. Nat Protoc. 2006;1:118–121. doi: 10.1038/nprot.2006.19. [DOI] [PubMed] [Google Scholar]
- 31.Konat GW, Borysiewicz E, Fil D, James I. Peripheral challenge with double- stranded RNA elicits global up-regulation of cytokine gene expression in the brain. J Neurosci Res. 2009;87:1381–1388. doi: 10.1002/jnr.21958. [DOI] [PubMed] [Google Scholar]
- 32.Konat GW, Borysiewicz E. Cerebellar expression of inflammatory genes triggered by peripheral challenge with dsRNA. J Neurochem. 2009;108(Suppl. 1):133. [Google Scholar]
- 33.Michalovicz LT, Lally BE, Konat GW. Peripheral challenge with a viral mimic upregulates expression of the complement genes in the hippocampus. J Neuroimmunol. 2015;285:137–142. doi: 10.1016/j.jneuroim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plata-Salaman CR. Meal patterns in response to the intracerebroventricular administration of interleukin-1 beta in rats. Physiol Behav. 1994;55:727–733. doi: 10.1016/0031-9384(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 35.Plata-Salaman CR, Borkoski JP. Chemokines/intercrines and central regulation of feeding. Am J Physiol. 1994;266:R1711–R1715. doi: 10.1152/ajpregu.1994.266.5.R1711. [DOI] [PubMed] [Google Scholar]
- 36.MacManus A, Ramsden M, Murray M, Henderson Z, Pearson HA, Campbell VA. Enhancement of (45)Ca(2+) influx and voltage-dependent Ca(2+) channel activity by beta-amyloid-(1-40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1beta. J Biol Chem. 2000;275:4713–4718. doi: 10.1074/jbc.275.7.4713. [DOI] [PubMed] [Google Scholar]
- 37.Murray CA, McGahon B, McBennett S, Lynch MA. Interleukin-1 beta inhibits glutamate release in hippocampus of young, but not aged, rats. Neurobiol Aging. 1997;18:343–348. doi: 10.1016/s0197-4580(97)80317-x. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham AJ, Murray CA, O'Neill LA, Lynch MA, O'Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 39.Schneider H, Pitossi F, Balschun D, Wagner A, del RA, Besedovsky HO. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A. 1998;95:7778–7783. doi: 10.1073/pnas.95.13.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di LM, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang S, Liu ZW, Wen L, Qiao HF, Zhou WX, Zhang YX. Interleukin-1beta enhances NMDA receptor-mediated current but inhibits excitatory synaptic transmission. Brain Res. 2005;1034:172–179. doi: 10.1016/j.brainres.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 42.Viviani B, Gardoni F, Marinovich M. Cytokines and neuronal ion channels in health and disease. Int Rev Neurobiol. 2007;82:247–263. doi: 10.1016/S0074-7742(07)82013-7. [DOI] [PubMed] [Google Scholar]
- 43.Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–234. doi: 10.1016/0006-8993(93)90959-q. [DOI] [PubMed] [Google Scholar]
- 44.Wang S, Cheng Q, Malik S, Yang J. Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther. 2000;292:497–504. [PubMed] [Google Scholar]
- 45.Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7:153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- 46.Xiaoqin Z, Zhengli L, Changgeng Z, Xiaojing W, Li L. Changes in behavior and amino acid neurotransmitters in the brain of rats with seizure induced by IL-1beta or IL-6. J Huazhong Univ Sci Technolog Med Sci. 2005;25:236–239. doi: 10.1007/BF02828129. [DOI] [PubMed] [Google Scholar]
- 47.Samland H, Huitron-Resendiz S, Masliah E, Criado J, Henriksen SJ, Campbell IL. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J Neurosci Res. 2003;73:176–187. doi: 10.1002/jnr.10635. [DOI] [PubMed] [Google Scholar]
- 48.De Sarro G, Russo E, Ferreri G, Giuseppe B, Flocco MA, DiPaola ED, De Sarro A. Seizure susceptibility to various convulsant stimuli of knockout interleukin-6 mice. Pharmacol Biochem Behav. 2004;77:761–766. doi: 10.1016/j.pbb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 49.Balschun D, Wetzel W, del RA, Pitossi F, Schneider H, Zuschratter W, Besedovsky HO. Interleukin-6: a cytokine to forget. FASEB J. 2004;18:1788–1790. doi: 10.1096/fj.04-1625fje. [DOI] [PubMed] [Google Scholar]
- 50.Wei H, Chadman KK, McCloskey DP, Sheikh AM, Malik M, Brown WT, Li X. Brain IL-6 elevation causes neuronal circuitry imbalances and mediates autism-like behaviors. Biochim Biophys Acta. 2012;1822:831–842. doi: 10.1016/j.bbadis.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 51.Song C, Merali Z, Anisman H. Variations of nucleus accumbens dopamine and serotonin following systemic interleukin-1, interleukin-2 or interleukin-6 treatment. Neuroscience. 1999;88:823–836. doi: 10.1016/s0306-4522(98)00271-1. [DOI] [PubMed] [Google Scholar]
- 52.Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von ZM, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 53.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K. Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J Neurosci Res. 1997;50:990–999. doi: 10.1002/(SICI)1097-4547(19971215)50:6<990::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 56.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De CE, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 57.Koller H, Allert N, Oel D, Stoll G, Siebler M. TNF alpha induces a protein kinase C-dependent reduction in astroglial K+ conductance. Neuroreport. 1998;9:1375–1378. doi: 10.1097/00001756-199805110-00023. [DOI] [PubMed] [Google Scholar]
- 58.Murray C, Griffin EW, O'Loughlin E, Lyons A, Sherwin E, Ahmed S, Stevenson NJ, Harkin A, Cunningham C. Interdependent and independent roles of type I interferons and IL-6 in innate immune, neuroinflammatory and sickness behaviour responses to systemic poly I:C. Brain Behav Immun. 2015;48:274–286. doi: 10.1016/j.bbi.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hadjilambreva G, Mix E, Rolfs A, Muller J, Strauss U. Neuromodulation by a cytokine: interferon-beta differentially augments neocortical neuronal activity and excitability. J Neurophysiol. 2005;93:843–852. doi: 10.1152/jn.01224.2003. [DOI] [PubMed] [Google Scholar]
- 60.Muller M, Fontana A, Zbinden G, Gahwiler BH. Effects of interferons and hydrogen peroxide on CA3 pyramidal cells in rat hippocampal slice cultures. Brain Res. 1993;619:157–162. doi: 10.1016/0006-8993(93)91607-t. [DOI] [PubMed] [Google Scholar]
- 61.Costello DA, Lynch MA. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-beta. Hippocampus. 2013;23:696–707. doi: 10.1002/hipo.22129. [DOI] [PubMed] [Google Scholar]
- 62.Di Filippo M, Tozzi A, Arcangeli S, de IA, Durante V, Di GM, Gardoni F, Calabresi P. Interferon-beta1a modulates glutamate neurotransmission in the CNS through CaMKII and GluN2A-containing NMDA receptors. Neuropharmacology. 2016;100:98–105. doi: 10.1016/j.neuropharm.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 63.Vlkolinsky R, Siggins GR, Campbell IL, Krucker T. Acute exposure to CXC chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J Neuroimmunol. 2004;150:37–47. doi: 10.1016/j.jneuroim.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 64.Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 2004;156:74–87. doi: 10.1016/j.jneuroim.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207:92–100. doi: 10.1016/j.jneuroim.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lax P, Limatola C, Fucile S, Trettel F, Di BS, Renzi M, Ragozzino D, Eusebi F. Chemokine receptor CXCR2 regulates the functional properties of AMPA-type glutamate receptor GluR1 in HEK cells. J Neuroimmunol. 2002;129:66–73. doi: 10.1016/s0165-5728(02)00178-9. [DOI] [PubMed] [Google Scholar]
- 67.Giovannelli A, Limatola C, Ragozzino D, Mileo AM, Ruggieri A, Ciotti MT, Mercanti D, Santoni A, Eusebi F. CXC chemokines interleukin-8 (IL-8) and growth-related gene product alpha (GROalpha) modulate Purkinje neuron activity in mouse cerebellum. J Neuroimmunol. 1998;92:122–132. doi: 10.1016/s0165-5728(98)00192-1. [DOI] [PubMed] [Google Scholar]
- 68.Ragozzino D, Giovannelli A, Mileo AM, Limatola C, Santoni A, Eusebi F. Modulation of the neurotransmitter release in rat cerebellar neurons by GRO beta. Neuroreport. 1998;9:3601–3606. doi: 10.1097/00001756-199811160-00011. [DOI] [PubMed] [Google Scholar]
- 69.Puma C, Danik M, Quirion R, Ramon F, Williams S. The chemokine interleukin-8 acutely reduces Ca(2+) currents in identified cholinergic septal neurons expressing CXCR1 and CXCR2 receptor mRNAs. J Neurochem. 2001;78:960–971. doi: 10.1046/j.1471-4159.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 70.Gosselin RD, Varela C, Banisadr G, Mechighel P, Rostene W, Kitabgi P, Melik-Parsadaniantz S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem. 2005;95:1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x. [DOI] [PubMed] [Google Scholar]
- 71.Guyon A, Skrzydelski D, De GI, Rovere C, Conductier G, Trocello JM, Dauge V, Kitabgi P, Rostene W, Nahon JL, Melik PS. Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience. 2009;162:1072–1080. doi: 10.1016/j.neuroscience.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 72.Kuijpers M, van Gassen KL, de Graan PN, Gruol D. Chronic exposure to the chemokine CCL3 enhances neuronal network activity in rat hippocampal cultures. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Minano FJ, Fernandez-Alonso A, Myers RD, Sancibrian M. Hypothalamic interaction between macrophage inflammatory protein-1 alpha (MIP-1 alpha) and MIP-1 beta in rats: a new level for fever control? J Physiol. 1996;491(Pt 1):209–217. doi: 10.1113/jphysiol.1996.sp021208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tavares E, Minano FJ. Differential sensitivities of pyrogenic chemokine fevers to CC chemokine receptor 5 antibodies. Fundam Clin Pharmacol. 2004;18:163–169. doi: 10.1111/j.1472-8206.2003.00227.x. [DOI] [PubMed] [Google Scholar]
- 75.Machado RR, Soares DM, Proudfoot AE, Souza GE. CCR1 and CCR5 chemokine receptors are involved in fever induced by LPS (E. coli) and RANTES in rats. Brain Res. 2007;1161:21–31. doi: 10.1016/j.brainres.2007.05.054. [DOI] [PubMed] [Google Scholar]
- 76.Myers RD, Paez X, Roscoe AK, Sherry B, Cerami A. Fever and feeding: differential actions of macrophage inflammatory protein-1 (MIP-1), MIP-1 alpha and MIP-1 beta on rat hypothalamus. Neurochem Res. 1993;18:667–673. doi: 10.1007/BF00966780. [DOI] [PubMed] [Google Scholar]
- 77.Minano FJ, Myers RD. Anorexia and adipsia: dissociation from fever after MIP-1 injection in ventromedial hypothalamus and preoptic area of rats. Brain Res Bull. 1991;27:273–278. doi: 10.1016/0361-9230(91)90081-t. [DOI] [PubMed] [Google Scholar]
- 78.Adler MW, Geller EB, Chen X, Rogers TJ. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2005;7:E865–E870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melik-Parsadaniantz S, Rostene W. Chemokines and neuromodulation. J Neuroimmunol. 2008;198:62–68. doi: 10.1016/j.jneuroim.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 80.Voss T, Barth SW, Rummel C, Gerstberger R, Hubschle T, Roth J. STAT3 and COX-2 activation in the guinea-pig brain during fever induced by the Toll-like receptor-3 agonist polyinosinic:polycytidylic acid. Cell Tissue Res. 2007;328:549–561. doi: 10.1007/s00441-007-0386-6. [DOI] [PubMed] [Google Scholar]
- 81.Hein AM, O'Banion MK. Neuroinflammation and memory: the role of prostaglandins. Mol Neurobiol. 2009;40:15–32. doi: 10.1007/s12035-009-8066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- 83.Dawson VL, Dawson TM. Nitric oxide in neuronal degeneration. Proc Soc Exp Biol Med. 1996;211:33–40. doi: 10.3181/00379727-211-43950e. [DOI] [PubMed] [Google Scholar]
- 84.Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, Shi FD. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation. 2014;11:26. doi: 10.1186/1742-2094-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.George J, Bleasdale S, Singleton SJ. Causes and prognosis of delirium in elderly patients admitted to a district general hospital. Age Ageing. 1997;26:423–427. doi: 10.1093/ageing/26.6.423. [DOI] [PubMed] [Google Scholar]
- 86.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Holmes C. Review: systemic inflammation and Alzheimer's disease. Neuropathol Appl Neurobiol. 2013;39:51–68. doi: 10.1111/j.1365-2990.2012.01307.x. [DOI] [PubMed] [Google Scholar]
- 88.Murray AM, Levkoff SE, Wetle TT, Beckett L, Cleary PD, Schor JD, Lipsitz LA, Rowe JW, Evans DA. Acute delirium and functional decline in the hospitalized elderly patient. J Gerontol. 1993;48:M181–M186. doi: 10.1093/geronj/48.5.m181. [DOI] [PubMed] [Google Scholar]
- 89.Nee LE, Lippa CF. Alzheimer's disease in 22 twin pairs--13-year follow-up: hormonal, infectious and traumatic factors. Dement Geriatr Cogn Disord. 1999;10:148–151. doi: 10.1159/000017115. [DOI] [PubMed] [Google Scholar]
- 90.Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, Elkind MS. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology. 2013;80:1209–1215. doi: 10.1212/WNL.0b013e3182896e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kranjac D, McLinden KA, Koster KM, Kaldenbach DL, Chumley MJ, Boehm GW. Peripheral administration of poly I:C disrupts contextual fear memory consolidation and BDNF expression in mice. Behav Brain Res. 2012;228:452–457. doi: 10.1016/j.bbr.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 92.McLinden KA, Kranjac D, Deodati LE, Kahn M, Chumley MJ, Boehm GW. Age exacerbates sickness behavior following exposure to a viral mimetic. Physiol Behav. 2012;105:1219–1225. doi: 10.1016/j.physbeh.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 93.Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirschman LT, Borysiewicz E, Fil D, Konat GW. Peripheral immune challenge with dsRNA enhances kainic acid-induced status epilepticus. Metab Brain Dis. 2011;26:91–93. doi: 10.1007/s11011-011-9236-z. [DOI] [PubMed] [Google Scholar]
- 95.Tellez-Zenteno JF, Matijevic S, Wiebe S. Somatic comorbidity of epilepsy in the general population in Canada. Epilepsia. 2005;46:1955–1962. doi: 10.1111/j.1528-1167.2005.00344.x. [DOI] [PubMed] [Google Scholar]
- 96.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 97.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33:13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 100.Xiong ZQ, Qian W, Suzuki K, McNamara JO. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J Neurosci. 2003;23:955–960. doi: 10.1523/JNEUROSCI.23-03-00955.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797–803. doi: 10.1016/j.bbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 102.Vezzani A, Ravizza T, Balosso S, Aronica E. Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia. 2008;49(Suppl 2):24–32. doi: 10.1111/j.1528-1167.2008.01490.x. [DOI] [PubMed] [Google Scholar]
- 103.Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miller SL, Fenstermacher E, Bates J, Blacker D, Sperling RA, Dickerson BC. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J Neurol Neurosurg Psychiatry. 2008;79:630–635. doi: 10.1136/jnnp.2007.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 107.Meyer U, Feldon J. To poly(I:C) or not to poly(I:C): Advancing preclinical schizophrenia research through the use of prenatal immune activation models. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 108.Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Meyer U, Feldon J. Prenatal exposure to infection: a primary mechanism forabnormal dopaminergic development in schizophrenia. Psychopharmacology (Berl) 2009;206:587–602. doi: 10.1007/s00213-009-1504-9. [DOI] [PubMed] [Google Scholar]
- 110.Makinodan M, Yamauchi T, Tatsumi K, Okuda H, Noriyama Y, Sadamatsu M, Kishimoto T, Wanaka A. Yi-gan san restores behavioral alterations and a decrease of brain glutathione level in a mouse model of schizophrenia. J Brain Dis. 2009;1:1–6. doi: 10.4137/jcnsd.s2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.De Miranda J, Yaddanapudi K, Hornig M, Villar G, Serge R, Lipkin WI. Induction of Toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. MBio. 2010;1:1–10. doi: 10.1128/mBio.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yee N, Ribic A, de Roo CC, Fuchs E. Differential effects of maternal immune activation and juvenile stress on anxiety-like behaviour and physiology in adult rats: no evidence for the “double-hit hypothesis”. Behav Brain Res. 2011;224:180–188. doi: 10.1016/j.bbr.2011.05.040. [DOI] [PubMed] [Google Scholar]
- 113.Song X, Li W, Yang Y, Zhao J, Jiang C, Li W, Lv L. The nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate reduces polyinosinic-polycytidilic acid-induced immune response in pregnant rats and the behavioral defects of their adult offspring. Behav Brain Funct. 2011;7:50. doi: 10.1186/1744-9081-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mattei D, Djodari-Irani A, Hadar R, Pelz A, de Cossio LF, Goetz T, Matyash M, Kettenmann H, Winter C, Wolf SA. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun. 2014;38:175–184. doi: 10.1016/j.bbi.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 115.Pineda E, Shin D, You SJ, Auvin S, Sankar R, Mazarati A. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann Neurol. 2013;74:11–19. doi: 10.1002/ana.23898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Khan D, Fernando P, Cicvaric A, Berger A, Pollak A, Monje FJ, Pollak DD. Long-term effects of maternal immune activation on depression-like behavior in the mouse. Transl Psychiatry. 2014;4:e363. doi: 10.1038/tp.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dickerson DD, Overeem KA, Wolff AR, Williams JM, Abraham WC, Bilkey DK. Association of aberrant neural synchrony and altered GAD67 expression following exposure to maternal immune activation, a risk factor for schizophrenia. Transl Psychiatry. 2014;4:e418. doi: 10.1038/tp.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bauman MD, Iosif AM, Smith SE, Bregere C, Amaral DG, Patterson PH. Activation of the maternal immune system during pregnancy alters behavioral development of rhesus monkey offspring. Biol Psychiatry. 2014;75:332–341. doi: 10.1016/j.biopsych.2013.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Machado CJ, Whitaker AM, Smith SE, Patterson PH, Bauman MD. Maternal immune activation in nonhuman primates alters social attention in juvenile offspring. Biol Psychiatry. 2015;77:823–832. doi: 10.1016/j.biopsych.2014.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu YH, Lai WS, Tsay HJ, Wang TW, Yu JY. Effects of maternal immune activation on adult neurogenesis in the subventricular zone-olfactory bulb pathway and olfactory discrimination. Schizophr Res. 2013;151:1–11. doi: 10.1016/j.schres.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 121.Forrest CM, Khalil OS, Pisar M, Smith RA, Darlington LG, Stone TW. Prenatal activation of Toll-like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Mol Brain. 2012;5:22. doi: 10.1186/1756-6606-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weir RK, Forghany R, Smith SE, Patterson PH, McAllister AK, Schumann CM, Bauman MD. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun. 2015;48:139–146. doi: 10.1016/j.bbi.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Makinodan M, Tatsumi K, Manabe T, Yamauchi T, Makinodan E, Matsuyoshi H, Shimoda S, Noriyama Y, Kishimoto T, Wanaka A. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J Neurosci Res. 2008;86:2190–2200. doi: 10.1002/jnr.21673. [DOI] [PubMed] [Google Scholar]
- 124.Li Q, Cheung C, Wei R, Cheung V, Hui ES, You Y, Wong P, Chua SE, McAlonan GM, Wu EX. Voxel-based analysis of postnatal white matter microstructure in mice exposed to immune challenge in early or late pregnancy. Neuroimage. 2010;52:1–8. doi: 10.1016/j.neuroimage.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 125.Rowland LM, Spieker EA, Francis A, Barker PB, Carpenter WT, Buchanan RW. White matter alterations in deficit schizophrenia. Neuropsychopharmacology. 2009;34:1514–1522. doi: 10.1038/npp.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arsenault D, St-Amour I, Cisbani G, Rousseau LS, Cicchetti F. The different effects of LPS and poly I:C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav Immun. 2014;38:77–90. doi: 10.1016/j.bbi.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 127.Pratt L, Ni L, Ponzio NM, Jonakait GM. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res. 2013;74:393–401. doi: 10.1038/pr.2013.126. [DOI] [PubMed] [Google Scholar]
- 128.Roenker NL, Gudelsky G, Ahlbrand R, Bronson SL, Kern JR, Waterman H, Richtand NM. Effect of paliperidone and risperidone on extracellular glutamate in the prefrontal cortex of rats exposed to prenatal immune activation or MK-801. Neurosci Lett. 2011;500:167–171. doi: 10.1016/j.neulet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jing Y, Zhang H, Wolff AR, Bilkey DK, Liu P. Altered arginine metabolism in the hippocampus and prefrontal cortex of maternal immune activation rat offspring. Schizophr Res. 2013;148:151–156. doi: 10.1016/j.schres.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 130.Ibi D, Nagai T, Kitahara Y, Mizoguchi H, Koike H, Shiraki A, Takuma K, Kamei H, Noda Y, Nitta A, Nabeshima T, Yoneda Y, Yamada K. Neonatal polyI:C treatment in mice results in schizophrenia-like behavioral and neurochemical abnormalities in adulthood. Neurosci Res. 2009;64:297–305. doi: 10.1016/j.neures.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 131.Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206:32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Konat GW, Lally BE, Toth AA, Salm AK. Peripheral immune challenge with viral mimic during early postnatal period robustly enhances anxiety-like behavior in young adult rats. Metab Brain Dis. 2011;26:237–240. doi: 10.1007/s11011-011-9244-z. [DOI] [PubMed] [Google Scholar]
- 133.Ellis S, Mouihate A, Pittman QJ. Neonatal programming of the rat neuroimmune response: stimulus specific changes elicited by bacterial and viral mimetics. J Physiol. 2006;571:695–701. doi: 10.1113/jphysiol.2005.102939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Packard AE, Hedges JC, Bahjat FR, Stevens SL, Conlin MJ, Salazar AM, Stenzel-Poore MP. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2012;32:242–247. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gesuete R, Packard AE, Vartanian KB, Conrad VK, Stevens SL, Bahjat FR, Yang T, Stenzel-Poore MP. Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J Neurochem. 2012;123(Suppl 2):75–85. doi: 10.1111/j.1471-4159.2012.07946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang PF, Fang H, Chen J, Lin S, Liu Y, Xiong XY, Wang YC, Xiong RP, Lv FL, Wang J, Yang QW. Polyinosinic-polycytidylic acid has therapeutic effects against cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via TLR3. J Immunol. 2014;192:4783–4794. doi: 10.4049/jimmunol.1303108. [DOI] [PMC free article] [PubMed] [Google Scholar]