

Abstract
Purpose
Our objective was to compare observed and expected genotype proportions from newborn screening surveys of structural hemoglobin variants.
Methods
We conducted a systematic review of newborn screening surveys of hemoglobins S and C in Africa and the Middle-East. We compared observed frequencies to those expected assuming Hardy-Weinberg equilibrium (HWE). Significant deviations were identified by an exact test. The fixation index FIS was calculated to assess excess homozygosity. We compared newborn estimates corrected and uncorrected for HWE deviations using demographic data.
Results
Sixty samples reported genotype counts for hemoglobin variants in Africa and the Middle-East. Observed and expected counts matched in 27%. The observed number of sickle-cell anemia (SCA) individuals was higher than expected in 42 samples, reaching significance (p<0.05) in 24. High FIS were common across the study regions. The estimated total number of newborns with SCA, corrected based on FIS, were 33,261 annual births instead of 24,958 for the 38 samples across sub-Saharan Africa and 1,109 annual births instead of 578 for 12 samples from the Middle East.
Conclusion
Differences between observed and expected genotype frequencies are common in surveys of hemoglobin variants in the study regions. Further research is required to identify and quantify factors responsible for such deviations. Estimates based on HWE might substantially underestimate the annual number of SCA affected newborns (up to one third in sub-Saharan Africa and one half in the Middle East).
Keywords: sickle hemoglobin, hemoglobin C, newborn screening, Hardy-Weinberg equilibrium, deviation
INTRODUCTION
Hemoglobin S (HbS) is a structural variant of normal adult hemoglobin (HbA) caused by an amino acid substitution at position 6 of the β-globin gene (HBB c.20A>T; p.Glu6-Val).1 Individuals who have inherited HbS are usually asymptomatic when heterozygous (AS), while homozygous individuals with HbS (SS) suffer from sickle cell anemia (SCA), a disease associated with severe clinical complications (including recurring pain, vaso-occlusive crises, and inflammation, all of which can lead to organ damage) and high mortality rates in low-income, high-burden countries.2 Compound heterozygosity with other β-globin polymorphisms, the most common of which are hemoglobin C (HbC) and β-thalassemia, can also cause sickle cell disease (SCD); individuals with Sβ0-thalassemia suffer from a form of SCD that is clinically indistinguishable from SS. HbS is most prevalent in sub-Saharan Africa and parts of the Mediterranean, the Middle East and India, because of natural selection for heterozygous individuals through a survival advantage against Plasmodium falciparum malaria.3 Sickle hemoglobin is often considered to be the most common pathological hemoglobin variant worldwide. Globally, it has been conservatively estimated that 305,800 (confidence interval [CI]: 238,400–398,800) babies were born with SS in 2010, in addition to infants with other variants that cause sickle cell disease.4 Due to population growth, this number increases every year and could reach more than 400,000 by 2050.4
HbC is another structural variant of HbA caused by a different amino acid substitution at the same position of the β-globin gene (HBB c.19G>A; p.Glu6Lys).5 Heterozygous individuals (AC) are asymptomatic while homozygosity (CC) causes mild hemolytic anemia due to the reduced solubility of the red blood cells that can lead to crystal formation. HbC is mainly of clinical significance when inherited in combination with HbS (SC), causing chronic hemolytic anemia and intermittent sickle cell crises, slightly less severe or frequent than in SS individuals, and when co-inherited with β-thalassemia (hemoglobin C-β thalassemia), causing moderate hemolytic anemia with splenomegaly. HbC provides near full protection against complicated P. falciparum malaria in homozygous (CC) individuals and intermediate protection in heterozygous (AC) individuals.6 This variant has been under positive selection across West Africa, particularly in Burkina Faso, Ghana and Togo, where it reaches frequencies of up to 15%.7
The Hardy-Weinberg (HW) principle defines the relationship between allele frequencies and genotype counts in successive generations and predicts that in a random mating population of infinite size, allele and genotype frequencies should remain constant from one generation to the next in the absence of any disturbing factors.8,9 The HW non-evolutionary model is commonly referred to as the Hardy-Weinberg equilibrium (HWE) and represents one of the most basic hypotheses in population genetics and evolutionary biology.10,11 The estimate of 305,800 infants born with SS disease in 2010 is derived from a model that assumes HWE and projects the frequency of SS based on the frequency of the S allele in population studies.4
Multiple factors can result in deviations from HWE in population samples. The two primary factors traditionally assumed to account for significant deviations from HWE are inbreeding or consanguinity due to mating among close relatives (endogamy), and population admixture or stratification due to short- and/or long-distance migration (gene flow).10 Additional factors that can lead to deviation from HWE include the occurrence of new mutations, genetic drift in small populations, natural selection, cryptic relatedness in isolated populations,11 as well as several methodological artifacts, including selection bias,12 genotyping errors,13 and non-randomly missing genotypes.14 The most important of these factors, in addition to the two traditional factors of inbreeding and population stratification, appears to be genotyping errors and selection bias.15 In countries where consanguinity is uncommon, deviation from HWE is used for quality control in gene association analyses.16
Several factors can cause deviations from HWE at the same time in a given population, either with cumulative effects or with opposite evolutionary trends. For example, one factor (e.g. inbreeding or improved fitness of homozygotes) may tend to increase the allele frequency of the gene studied while another tends to reduce it (e.g. termination of affected pregnancies or elimination of malaria selective pressure, either naturally or through human control interventions). Furthermore, some of these disturbing factors have an immediate effect on allele or genotype frequencies (e.g. migrations) while others will cause slow changes over multiple generations (e.g. natural selection). Importantly, neglecting factors causing deviations from HWE can lead to an incorrect interpretation of the data from screening studies and to an under- or over-estimation of the number of individuals affected at local, national, regional or global scales.
In theory, data from universal newborn screening studies offer the unique advantage that they represent the best measure of true genotype frequencies. At ages beyond infancy, excess mortality is likely to reduce the frequency of individuals homozygous for deleterious mutations such as HbS. Furthermore, in areas endemic for malaria, survival among heterozygous individuals (with HbAS) is greater than that among normal individuals (with HbAA) due to their protection against severe malaria. Two main national, regional and global estimates of newborns affected by sickle cell anemia have been published in the last decade. In 2008, Modell and Darlison published conservative newborn estimates for all common hemoglobinopathies using the Hardy-Weinberg equation corrected by national population coefficients of consanguinity obtained from Bittles’s database and Murdock’s ethnographic atlas.17 In 2013, we published newborn frequency estimates for HbSS, HbAS, HbCC and HbAC based on a Bayesian geostatistical framework and on HWE that accounted for spatial heterogeneities in the frequency of hemoglobin variants and in the distribution of human populations within countries.4,7 Although we considered consanguinity as a potential confounder in the previous studies, it was not incorporated in our modelling framework due to the unavailability of a coefficient of consanguinity for many countries and to the lack of data on subnational variations for this covariate for all countries. We therefore assumed that the use of such a covariate in a framework designed to account for spatial heterogeneities would potentially introduce more biases than corrections. Our published estimates of HbSS and HbCC annual births, calculated using allele frequencies assuming HWE, likely represent underestimates of the prevalence of newborns affected by these genotypes in regions in which consanguinity is common. Because our modelling frameworks were independently developed for HbS and HbC, estimates of HbSC annual births were not calculated. Despite not correcting for consanguinity, our global HbSS and HbCC estimates were generally higher than those of Modell & Darlison, even when correcting for demographic changes. Those findings suggested that the global health burden of hemoglobin variants - HbS in particular - might be substantially higher than was believed at the time.
In the present study, we aim to assess the frequency and magnitude of deviations from HWE for HbS and HbC in Africa and the Middle East by reviewing existing data from newborn screening surveys conducted in these regions and discuss the impact of such deviations on estimates of affected newborns at various geographical scales.
MATERIAL AND METHODS
Newborn screening data
We conducted a systematic search of the published literature using PubMed, Web of Science and Scopus using the following search string: “sickle AND newborn AND screening AND (Africa OR Bahrain OR Iran OR Iraq OR Israel OR Jordan OR Kuwait OR Lebanon OR Oman OR Palestine OR Qatar OR “Saudi Arabia” OR Syria OR “United Arab Emirates” OR Yemen OR India)”. While the use of the term “Africa” seemed conservative, individual names of countries were used for the Middle East as the inclusion of this more generic term missed several key references. Searches, last updated on 26th January 2015, returned 159, 121 and 127 references, respectively in each of the above resources. After duplicate removal, 247 references remained, of which 30 contained relevant newborn screening data (see Supplementary Table S1). Six studies which did not appear in our systematic search but which had already been identified during preliminary manual searches,18 were also included.
The inclusion criteria for studies were: i) that subjects were tested at birth or within the first 28 days of life, either randomly or as part of a universal screening program and ii) that they reported detailed genotype counts for HbS. No minimum sample size cut-off value was used (accepting that results from small studies need to be considered with caution because of a likelihood of large differences in ratios due to chance). The diagnostic methods used in each screening study were also recorded (Supplementary Table S2). Because our search criteria revealed only two relatively small newborn screening studies from India,19,20 we excluded these studies and restricted our focus to Africa and the Middle East. Where more than one publication reported overlapping data, only the most recent or comprehensive report was included. Updated data for the Kumasi survey from Ghana were presented during a CDC webinar in March 2014 and were therefore used in place of the numbers published in 2008.
Deviation from HWE
HWE equations have been described in detail in several classic textbooks of population genetics12,21 and are only briefly presented in the Supplementary Information. Various tests have been designed to measure deviation from HWE. Although such a test is relatively straightforward for two alleles, it is more complex for multiple alleles (n≥3).22 Tests of HWE are often performed using Pearson’s χ2 goodness-of-fit test. It is now well established that this asymptotic method is unreliable for small samples and is associated with substantial errors in large samples with a low-prevalence of mutant alleles. As a result, exact tests are usually preferable.15,22 Here, we use the likelihood ratio full-enumeration exact test, as implemented in the “HWxtest” package in R.23 In this test, all possible tables with the same allele numbers as in the observed counts are examined. While this approach is computationally intensive, it does result in a robust P value. We use 0.05 as the standard cut-off for statistical significance (Supplementary Table S3).
Fixation index
The fixation index FIS (often referred to as Wright’s inbreeding coefficient) is used with genomic data on the distribution of alleles in population samples to identify gaps between the relative frequencies of homozygous or compound heterozygous individuals and those predicted on the basis of heterozygous individuals.24 Although typically interpreted as a measure of consanguinity at the population level, it can also indicate potential quality problems in a genetic study due to issues such as selection bias or genotyping errors. A high FIS index can be interpreted as a measure of consanguinity or inbreeding only in a population with direct evidence of high levels of consanguinity as well as reliable data on admixture and population structure.
The following equation, defining the proportionate reduction in heterozygosity relative to HWE, was used to calculate FIS in each population survey included in this study (Supplementary Table S3):,25
| Equation 1 |
where pi is the proportion of allele i, and Ptj is the observed frequency of genotype AiAj.
Newborn estimates at population level
In order to assess the influence of deviations from HWE on population-level estimates of newborns with Hb variants, we calculated the predicted number of HbSS newborns based on expected (HWE) and observed (HWE corrected using FIS) allele frequencies for study samples or subsamples stratified based on locality, citizenship, or place of origin. Due to high heterogeneities in FIS within countries, national estimates would be misleading even for those few studies with samples from multiple localities within a country.
As in previous studies,726 demographic data included local population and national crude birth rate (CBR). Population data were extracted from one of the following sources, investigated according to the sequence: UN Demographic Yearbook 2013 (Table 8), the CIA’s World Factbook, national census data, the study publication itself or Wikipedia. CBR data were extracted from the UN World Population Prospects 2012 Revision. HWE uncorrected (Equation 2) and corrected (Equation 3) newborn estimates were calculated as follows:
| Equation 2a |
| Equation 2b |
| Equation 3a |
| Equation 3b |
where Ni is the local population; CBRi is the crude birth rate; pi is the allele frequency of HbS; qi, the frequency of HbC, is equal to 1-pi; and is the fixation index at place i.
RESULTS
Newborn screening surveys
We identified 36 published surveys of newborns tested for structural hemoglobin variants (HbS and HbC) in the study regions matching our inclusion criteria. These surveys included 60 samples or subsamples; 40, 5, and 15 of which were from surveys conducted in sub-Saharan Africa, North Africa and the Middle East, respectively. Sample sizes ranged between 30 newborns in a survey conducted in Tanzania to more than half a million newborns tested over a decade in the United Arab Emirates. 57% of samples or subsamples had a size smaller than 1,000 newborns (Figure 1).
Figure 1.
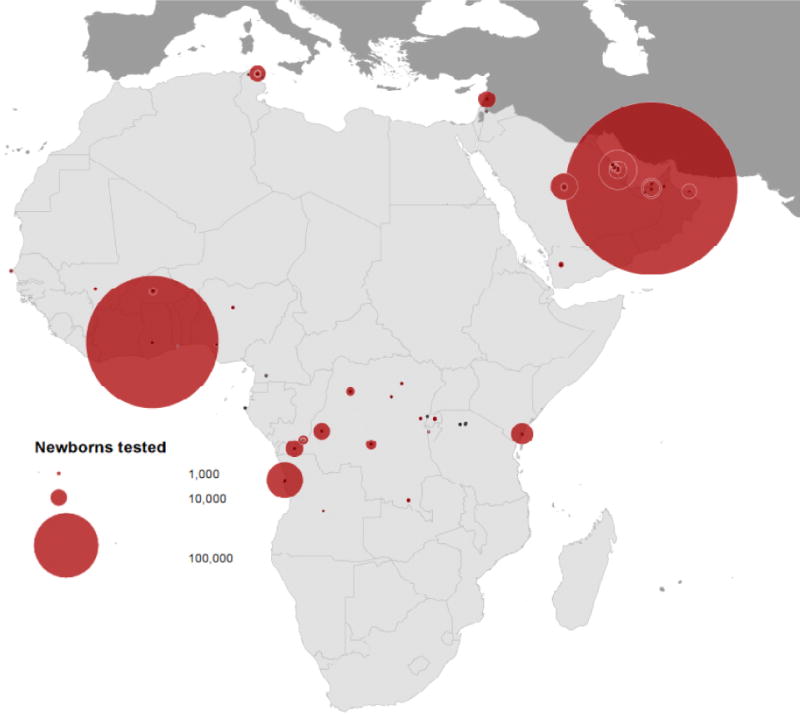
Distribution and size of newborn screening surveys of HbS and HbC in Africa and the Middle East.
HbS allele frequencies within individual study samples ranged between 1.4% (Rwanda) and 14.6% (infants of DRC-origin living in Burundi & DRC) in sub-Saharan Africa, between 0.3% and 2.8% in Tunisia, and between 0.5% (UAE) and 13.2% (Saudi) in the Middle East (Supplementary Table S2). HbC was observed in 20 population samples out of 38 that tested for HbC, covering an area extending from Senegal to Oman. Reported HbC frequencies in those studies that detected HbC ranged between 0.1% (Rwanda) and 10.4% (Burkina Faso) (Supplementary Table S2).
Deviation from HWE
While observed and expected genotype counts matched exactly in 16 study samples (27%), the observed number of HbSS individuals was higher than expected in 42 of them. The observed number of HbCC individuals was higher than expected in 29% (n=8) of study samples in which HbC was found (Figure 2). A statistically significant difference (p<0.05) between observed and expected HbS counts was found in 24 of 60 samples or subsamples: 1 in Burkina Faso, 6 out of 15 in the DRC, 1 in Gabon, 1 in Ghana, 1 out of 2 in Nigeria, 1 out of 4 in Senegal, 1 out of 5 in Tunisia, 1 out of 3 in Bahrain, 1 in Lebanon, 1 in Oman, 5 out of 5 in Saudi Arabia, and 3 out of 3 in the UAE (Supplementary Table S3). Among 29 samples or subsamples with >1000 newborns tested, 20 (69%) revealed differences between observed and expected frequencies of homozygotes that were statistically significant.
Figure 2.
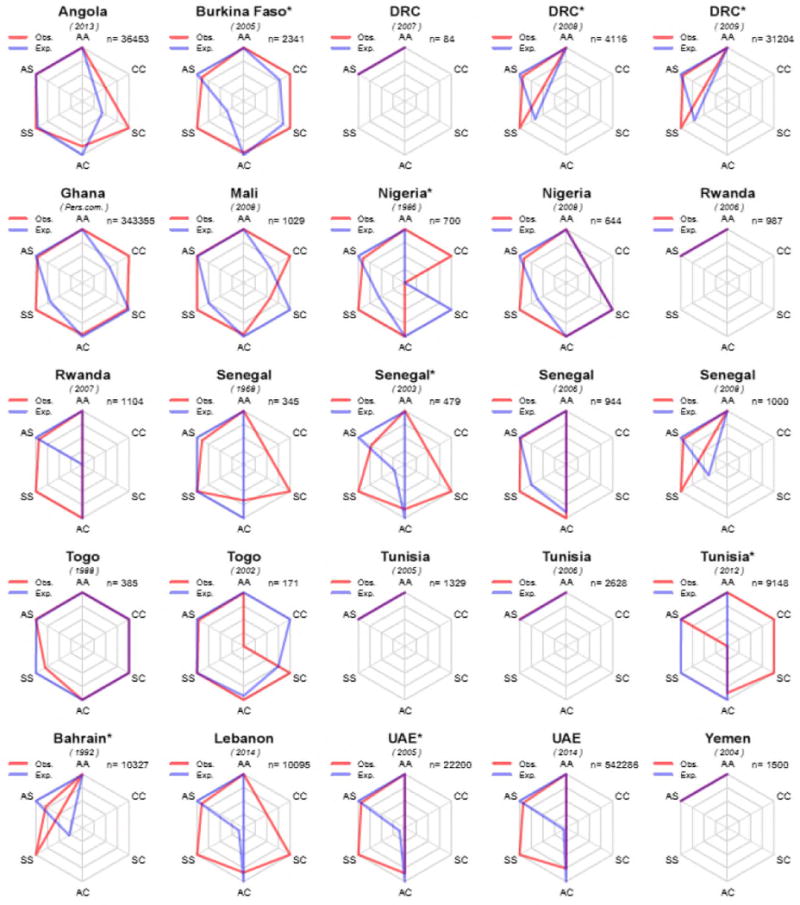
Radar charts showing differences between observed (in red) and expected (in blue) genotype counts in newborn screening surveys of HbS and HbC in Africa and the Middle East. Surveys for which only data on HbS were presented are not shown. To maximise the visualisation of the differences, each axis is scaled based on the maximum of the observed or expected counts for each survey independently. n = number of newborns tested. A * indicates a population sample for which deviation from Hardy-Weinberg Equilibrium was found to be statistically significant using the likelihood ratio full-enumeration exact test.
FIS coefficient
A positive FIS reflects an excess of homozygotes, while a negative value indicates an excess of heterozygotes. The highest FISs in sub-Saharan Africa were found in study samples from Senegal (25.7%) and Gabon (28.9%). In the Middle East, FIS reached values up to 18.8% in Saudi Arabia and 21.2% in Bahrain (Supplementary Table S3). For most countries, substantial differences in FIS were observed between various national population samples (Supplementary Table S3). For example, FIS ranged from −11.1% to 2.4% in Burundi, −5.7% to 6.4% in Tanzania -and from −0.9% to 21.2% in Bahrain.
Impact on estimates of prevalence of affected newborns
As most FIS were positive for the samples surveyed, estimates of the prevalence of HbSS in newborns corrected for the fixation index (i.e. based on Equation 3) were higher than the uncorrected estimates calculated using the simple HWE equation. In three large (n>30,000) studies from sub-Saharan Africa, the corrected to uncorrected HbSS prevalence estimate ratios were approximately 1.4 in the DRC and Ghana and 1.0 in Angola (Table 1). In the Middle East, the ratios were considerably higher in screening studies conducted in Lebanon (11.4) and the United Arab Emirates (up to 17.7) while in Bahrain and Saudi Arabia, ratios have fallen from 3.4 or greater during the 1980s to 1.0–1.5 in studies conducted more recently.
Table 1.
Demographic data and newborn estimates for HbSS at population level for each survey location based on Hardy-Weinberg equilibrium (uncorrected, q2) and accounting for excess homozygosity (corrected, q2+Fpq).
| ID | First Author | Year | Country | N | Survey location | Population | Source | CBR (2012) | HbS allele frequency | FIS (%) | HbSS newborn estimate | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HWE (1) |
HWE+FIS (2) |
Ratio (2)/(1) |
|||||||||||
| Sub-Saharan Africa | |||||||||||||
| 1 | McGann | 2013 | Angola | 36,453 | Luanda | 1,822,407 | UN | 44.3 | 0.120 | 0.62 | 1,169 | 1,222 | 1.0 |
| 2 | Kafando | 2005 | Burkina Faso | 2,341 | Ouagadougou | 1,475,223 | UN | 41.1 | 0.048 | 5.45 | 141 | 293 | 2.1 |
| 3 | Moreno | 1994 | Burundi | 382 | Bujumbura | 497,169 | UN | 44.8 | 0.071 | 0.37 | 111 | 117 | 1.1 |
| 4 | Mutesa | 2007 | Burundi | 637 | Bujumbura | 497,169 | UN | 44.8 | 0.024 | 4.42 | 12 | 35 | 2.9 |
| 5 | Le Hesran | 1999 | Cameroon | 156 | Ebolowa | 79,500 | Wikipedia | 37.5 | 0.135 | 6.46 | 54 | 76 | 1.4 |
| 6 | Diallo | 2008 | Congo | 1,186 | Brazzaville | 1,611,000 | CIA | 42.9 | 0.102 | −3.16 | 725 | 524 | 0.7 |
| 7 | Van Baelen | 1969 | DRC | 500 | Kinshasa | 8,798,000 | CIA | 42.9 | 0.146 | 3.76 | 8,045 | 9,813 | 1.2 |
| 8 | Lallemant | 1986 | DRC | 146 | Linzolo | NA | / | 42.9 | 0.099 | −3.37 | / | / | / |
| 9 | Mutesa | 2007 | DRC | 84 | Goma | 1,000,000 | Wikipedia | 42.9 | 0.024 | −2.86 | 24 | / | / |
| 10 | Tshilolo | 2008 | DRC | 4,116 | Kinshasa | 8,798,000 | CIA | 42.9 | 0.125 | 7.01 | 5,909 | 8,803 | 1.5 |
| 11 | Tshilolo | 2009 | DRC | 70 | Other Africans | 19,737 | PCT | 42.9 | 0.043 | −4.48 | 2 | 0 | / |
| 12 | Tshilolo | 2009 | DRC | 579 | Orientale | 163,250 | PCT | 42.9 | 0.097 | 1.15 | 66 | 73 | 1.1 |
| 13 | Tshilolo | 2009 | DRC | 717 | Kivu | 202,159 | PCT | 42.9 | 0.079 | 2.80 | 55 | 73 | 1.3 |
| 14 | Tshilolo | 2009 | DRC | 559 | Angola | 157,611 | PCT | 42.9 | 0.114 | 2.95 | 89 | 109 | 1.2 |
| 15 | Tshilolo | 2009 | DRC | 932 | Katanga | 262,778 | PCT | 42.9 | 0.082 | 6.88 | 75 | 133 | 1.8 |
| 16 | Tshilolo | 2009 | DRC | 1,016 | Not declared | 286,462 | PCT | 42.9 | 0.106 | 6.89 | 138 | 218 | 1.6 |
| 17 | Tshilolo | 2009 | DRC | 3,069 | Equateur | 865,308 | PCT | 42.9 | 0.102 | 6.65 | 390 | 617 | 1.6 |
| 18 | Tshilolo | 2009 | DRC | 4,217 | Kasai | 1,188,988 | PCT | 42.9 | 0.098 | 4.00 | 486 | 665 | 1.4 |
| 19 | Tshilolo | 2009 | DRC | 10,445 | Bas Congo | 2,944,979 | PCT | 42.9 | 0.097 | 4.02 | 1,188 | 1,633 | 1.4 |
| 20 | Tshilolo | 2009 | DRC | 9,600 | Bandundu | 2,706,730 | PCT | 42.9 | 0.100 | 4.65 | 1,170 | 1,657 | 1.4 |
| TshiloloΣ | 2009 | DRC | 31,204 | Kinshasa | 8,798,000 | CIA | 42.9 | 0.098 | 4.58 | 3,644 | 5,177 | 1.4 | |
| 21 | Agasa | 2010 | DRC | 520 | Kisangani | 820,000 | CIA | 42.9 | 0.126 | −5.68 | 558 | 338 | 0.6 |
| 22 | Richard-Lenoble | 1980 | Gabon | 96 | Port Gentil | 142,280 | Wikipedia | 32.1 | 0.063 | 28.89 | 18 | 95 | 5.3 |
| 23 | Ohene-Frempong | Pers.com. | Ghana | 343,355 | Kumasi | 1,922,130 | UN | 31.1 | 0.080 | 3.92 | 383 | 556 | 1.5 |
| 24 | Williams | Pers.com. | Kenya | 15,444 | Kilifi | 1,109,735 | UN | 35.1 | 0.084 | 1.40 | 273 | 315 | 1.2 |
| 25 | Diallo | 2008 | Mali | 1,025 | Bamako | 2,037,000 | CIA | 47.3 | 0.055 | 2.55 | 296 | 424 | 1.4 |
| 26 | Kulkarni | 1986 | Nigeria | 700 | Kaduna | 1,524,000 | CIA | 41.5 | 0.127 | 10.00 | 1,022 | 1,724 | 1.7 |
| 27 | Odunvbun | 2008 | Nigeria | 644 | Benin City | 1,147,188 | UN | 41.5 | 0.132 | 8.71 | 829 | 1,305 | 1.6 |
| 28 | Munyanganizi | 2006 | Rwanda | 987 | Kigali & Butare | 740,383 | UN | 35.4 | 0.014 | −1.39 | 5 | 0 | / |
| 29 | Mutesa | 2007 | Rwanda | 1,104 | Kigali & Butare | 740,383 | UN | 35.4 | 0.014 | 4.95 | 5 | 22 | 4.4 |
| 30 | Oudart | 1968 | Senegal | 345 | Dakar | 1,056,009 | UN | 38.1 | 0.042 | 7.40 | 71 | 191 | 2.7 |
| 31 | Mbodji | 2003 | Senegal | 479 | Dakar | 1,056,009 | Census | 38.1 | 0.059 | 25.69 | 142 | 721 | 5.1 |
| 32 | Tchamago | 2006 | Senegal | 944 | Dakar (CHAN & HPD) | 1,056,009 | UN | 38.1 | 0.052 | 5.87 | 108 | 225 | 2.1 |
| 33 | Diallo | 2008 | Senegal | 1,000 | Dakar | 1,056,009 | UN | 38.1 | 0.048 | 6.06 | 91 | 201 | 2.2 |
| 34 | Aebischer | 1990 | Tanzania | 92 | Nassa | 4,000 | Study | 39.3 | 0.054 | −5.75 | 0 | 0 | / |
| 35 | Aebischer | 1990 | Tanzania | 101 | Magu | 20,000 | Study | 39.3 | 0.074 | 6.38 | 4 | 8 | 2.0 |
| 36 | Aebischer | 1990 | Tanzania | 30 | Mwanza | 150,000 | Study | 39.3 | 0.017 | −1.69 | 2 | 0 | / |
| AebischerΣ | 1990 | Tanzania | 223 | Nassa + Magu + Mwanza | 174,000 | Sum | 39.3 | 0.058 | 1.98 | 23 | 31 | 1.3 | |
| 37 | North | 1988 | Togo | 385 | Lomé | 1,524,000 | CIA | 36.7 | 0.110 | −0.99 | 682 | 627 | 0.9 |
| 38 | Segbena | 2002 | Togo | 171 | Lomé | 1,524,000 | CIA | 36.7 | 0.105 | −3.26 | 620 | 448 | 0.7 |
| North Africa | |||||||||||||
| 39 | Diallo | 2008 | Tunisia | 1,329 | Tunis | 702,330 | UN | 17.3 | 0.008 | −0.76 | 1 | 0 | / |
| 40 | Fattoum | 2006 | Tunisia | 2,029 | Tunis | 702,330 | UN | 17.3 | 0.011 | −1.22 | 1 | 0 | / |
| 41 | Fattoum | 2006 | Tunisia | 335 | Nabeul | 56,387 | Wikipedia | 17.3 | 0.003 | −0.35 | 0 | 0 | / |
| 42 | Fattoum | 2006 | Tunisia | 264 | Béja | 106,441 | Wikipedia | 17.3 | 0.028 | −2.92 | 1 | 0 | / |
| FattoumΣ | 2006 | Tunisia | 2,628 | Tunis + Nabeul + Béja | 865,158 | Sum | 17.3 | 0.012 | −1.25 | 2 | 0 | / | |
| 43 | Hajer | 2012 | Tunisia | 9,148 | Tunis | 702,330 | UN | 17.3 | 0.010 | 1.32 | 1 | 3 | 3.0 |
| Middle East | |||||||||||||
| 44 | Mohammed | 1992 | Bahrain | 10,327 | Manama | 176,909 | UN | 15.5 | 0.077 | 21.21 | 16 | 58 | 3.6 |
| 45 | Al Arrayed | 2005 | Bahrain | 2,000 | Manama | 176,909 | UN | 15.5 | 0.090 | 1.02 | 22 | 25 | 1.1 |
| 46 | Al Arrayed | 2012 | Bahrain | 39,016 | Manama | 176,909 | UN | 15.5 | 0.073 | −0.94 | 15 | 13 | 0.9 |
| 47 | Talafih | 1996 | Jordan | 181 | Irbid | 313,820 | UN | 27.3 | 0.025 | −2.55 | 5 | 0 | / |
| 48 | Khoriaty | 2014 | Lebanon | 10,095 | Countrywide | 5,882,562 | CIA | 13.5 | 0.009 | 9.93 | 7 | 80 | 11.4 |
| 49 | Alkindi | 2010 | Oman | 5,694 | Muscat | 24,893 | UN | 21.4 | 0.029 | 7.03 | 0 | 1 | / |
| 50 | Al-Awamy | 1984 | Saudi Arabia | 1,697 | Al Khobar | 165,799 | UN | 19.6 | 0.028 | 18.78 | 3 | 19 | 6.3 |
| 51 | Al-Awamy | 1984 | Saudi Arabia | 2,800 | Dammam | 744,321 | UN | 19.6 | 0.039 | 10.27 | 22 | 78 | 3.5 |
| 52 | Al-Awamy | 1984 | Saudi Arabia | 1,124 | Qatif | 871,000 | Wikipedia | 19.6 | 0.109 | 7.80 | 204 | 334 | 1.6 |
| Al-AwamyΣ | 1984 | Saudi Arabia | 5,621 | Al Khobar + Dammam + Qatif | 1,781,120 | Sum | 19.6 | 0.050 | 12.46 | 86 | 292 | 3.4 | |
| 53 | Nasserullah | 2003 | Saudi Arabia | 2,154 | Qatif (Non-Saudi) | 78,133 | PCT | 19.6 | 0.013 | 6.07 | 0 | 1 | / |
| 54 | Nasserullah | 2003 | Saudi Arabia | 21,858 | Qatif (Saudi) | 792,867 | PCT | 19.6 | 0.132 | 7.40 | 269 | 401 | 1.5 |
| NasserullahΣ | 2003 | Saudi Arabia | 24,012 | Qatif (Saudi + Non-Saudi) | 871,000 | Wikipedia | 19.6 | 0.121 | 8.39 | 250 | 402 | 1.6 | |
| 55 | Al Hosani | 2005 | UAE | 9,165 | UAE (citizens) | 1,400,000 | Wikipedia | 14.8 | 0.008 | 7.14 | 1 | 13 | 13.0 |
| 56 | Al Hosani | 2005 | UAE | 13,035 | UAE (non-citizens) | 7,800,000 | Wikipedia | 14.8 | 0.004 | 6.33 | 2 | 32 | 16.0 |
| Al HosaniΣ | 2005 | UAE | 22,200 | UAE | 9,200,000 | Wikipedia | 14.8 | 0.006 | 6.84 | 5 | 58 | 11.6 | |
| 57 | Al Hosani | 2014 | UAE | 542,286 | UAE | 9,200,000 | Wikipedia | 14.8 | 0.005 | 8.13 | 3 | 53 | 17.7 |
| 58 | Al-Nood | 2004 | Yemen | 1,500 | Sana’a | 1,978,286 | UN | 31.4 | 0.011 | −1.11 | 8 | 0 | / |
N: number of newborns tested; Source: UN = United Nations Statistics Division (http://unstats.un.org/unsd/demographic/products/dyb/City_Page.htm), CIA = The World Factbook (https://www.cia.gov/library/publications/the-world-factbook/fields/2219.html), PCT = percentage, equivalent to the proportions of sub-populations in the newborn screening study, Sum = sum of sub-populations sampled in the newborn screening study. NA = not available. FIS = fixation index as a measure of homozygosity excess.
Across all surveys, the estimated total number of HbSS newborns, corrected based on FIS, were 33,261 instead of 24,958 in sub-Saharan Africa and 1,109 instead of 578 in the Middle East. Corrected and uncorrected estimates of annual births with HbCC and HbSC are presented in Supplementary Table S4 for the 20 samples for which the frequency of HbC was reported and not null. Although most of these estimates need to be considered with caution due to the relatively small number of births affected by HbCC or HbSC (compared to HbSS), they suggest that uncorrected estimates based on HbC alleles assuming HWE could substantially underestimate the number of HbCC births and slightly overestimate the number of HbSC births.
DISCUSSION
This study, which to our knowledge includes all relevant published studies across the study regions, reveals a striking lack of newborn screening surveys in the areas most affected by sickle cell disorders. Universal newborn screening programs for sickle cell disorders have long been implemented in the United States of America and the United Kingdom, where it has led to substantial improvements in childhood mortality.27,28 More generally, universal newborn screening can contribute to the reduction of health disparities.29 Ghana is the only African country which has so far implemented a large scale newborn screening program, with data so far published from Kumasi and a neighboring community. Local screening efforts have begun in a number of African countries, but those often lack financial and political support to be scaled up to subnational or national levels.30 This is reflected by the rather small sample sizes of most newborn screening studies conducted in sub-Saharan African countries, with 30 of 40 samples or sub-samples (75%) having tested less than 1,000 newborns and only three studies reporting results for more than 30,000. With the ongoing increase of the health burden of hemoglobinopathies, the implementation of large-scale newborn screening programs could contribute to saving millions of lives in the coming decades.4
In countries for which several newborn screening studies have been published, the present study highlights heterogeneities in the allele frequencies of hemoglobin variants and the magnitude of deviation from HW expectations. Although the former can to some extent be taken into account using geostatistical methods26, to date the latter has been under-appreciated in the calculation of national, regional and global burden estimates.
Differences between observed and expected frequencies of genetic disorders based on allele frequencies within a study sample are common in newborn screening surveys of structural hemoglobin variants in Africa and the Middle East. Various factors can explain an excess of homozygosity, including consanguinity and population structure, but their respective effects are difficult to dissect. These results give insight into the relative magnitudes by which current national and regional estimates based on extrapolations from the frequencies of HbS alleles might represent underestimates of the birth prevalence of sickle cell disease. The magnitude of underestimation may be as much as one-third in African populations and almost one-half in Middle Eastern surveys. Regrettably, we did not find substantial data from screening studies conducted in India and could not, therefore, extrapolate this conclusion to that country.
Surprisingly high FIS’s found in small study samples from Gabon and Senegal suggest either quality control issues or chance variation. The threats to quality and potential bias in estimates of FIS include small sample size, selection bias, and mistaken genotyping. The Gabon study illustrates the first problem, with fewer than 100 newborns tested, a sample size that is inadequate for reliable estimates to be made. Despite a larger sample size (n=479), the high FIS coefficient found in one sample of Senegalese newborns should be interpreted with caution because the reported frequency of SS (1.9%) is several times higher than reported in all other studies from Senegal (0.3–0.5%). This finding could potentially be the result of referral or selection bias or due to diagnostic errors.
An apparent excess of homozygotes is often attributed to consanguinity, which is assumed to be high in populations living in the Sahel, other parts of West Africa, the Middle East and Central/South Asia.31 Nevertheless, data availability on consanguinity are very patchy.31 Existing national estimates of consanguinity may reflect the behavior of specific localized ethnic groups studied, rather than that of the overall population of a country. In much of sub-Saharan Africa, however, consanguinity is reported to be widespread, and may be found in similar frequencies among different ethnic groups in a country.32 Moreover, estimates of the frequency of consanguineous marriage may differ widely for a single ethnic group. For example, two published estimates of consanguinity among the Yoruba living in south-western Nigeria were 51% in a rural sample in the 1970s and 6% in an urban sample in the 1990s.33
Consanguinity is generally thought to be common in many parts of The Middle East.31 For example, well over one-half of marriages among Saudi nationals in Saudi Arabia are consanguineous, with one-third of all marriages between first cousins.34 As illustrated by Figure 2, we found a marked excess of SS homozygotes in most of the earlier surveys conducted in Bahrain, Oman, Saudi Arabia, and the United Arab Emirates (UAE).
In Bahrain, data have shown a reduction over time of the excess homozygosity, an observation that may be accounted for by more recent introduction of wide-spread carrier testing. The excess of SS homozygotes observed in 1984–1985 had essentially disappeared in screening data collected in 2002 and 2008–2010. The introduction of prenatal carrier screening beginning in 1993, student screening beginning in 1998, voluntary premarital screening and counselling of couples identified as carriers (mandatory for Bahraini citizens beginning in 2004), appear to have driven this trend. Between 1984–1985 and 2010 the prevalence of HbSS in Bahraini newborns decreased from 2.1% to 0.4%, while that of HbAS increased from 11.2% to 14.7%. The public health implication is that the direct influence of parental consanguinity on high rates of sickle cell disease relative to the prevalence of the sickle cell allele can be attenuated if carrier testing leads to fewer carrier couples marrying and having children together. In various Mediterranean countries programs combining premarital screening, genetic counseling, and prenatal diagnosis for carrier couples offered during early pregnancy long ago resulted in 90% or more decreases in live births with thalassemia. More recently, a similar program in parts of coastal Turkey resulted in similar decreases in births for both thalassemia and sickle cell disease.35 In the Middle East, attitude towards prenatal diagnosis and abortion are greatly influenced by religious values. Education about a fatwa which allows the abortion of a diseased fetus within the first 120 days of a pregnancy can promote the acceptance of genetic counselling in Islamic societies.36
Ethnographic or genealogical approaches to assessing consanguinity often yield results that differ markedly from genome-based measures of supposed interbreeding.33 Differences in the presumed frequency of inbreeding suggested by the FIS coefficient often reflect the influences of genetic isolation and genetic drift, including past population bottlenecks and founder effects rather than consanguineous marriage.37 Differences in the FIS coefficient may also reflect differences in study quality, including mistaken attribution of genotype due to testing errors and selection bias. A study that analyzed data from 26 populations around the world with both FIS coefficients and estimates of consanguinity found that the latter explained a little more than one-tenth of variation in the former (r = 0.349, p = 0.040).37
One of the highest ethnographic estimates of consanguineous marriage in Nigeria (56–61%) comes from rural Hausa communities in the northern part of the country.38 Although FIS coefficients are moderately correlated with ethnographic estimates of marital consanguinity,37 it has not been established that the same association holds true among studies in sub-Saharan Africa.
An excess of homozygotes or deficit of heterozygotes can also be explained by population genetic structure or stratification, also referred to as the Wahlund Effect.39 Population stratification can result from random mating within sub-populations of distinct ancestral origins. In population stratification, each sub-population may be characterized by HWE but an aggregate sample from the stratified population can show deviation from HWE and apparent inbreeding or assortative mating. Population stratification is particularly likely to occur in countries or regions in which hemoglobinopathies were historically rare but now have distinct high frequency sub-populations due to historical or recent population migrations.40 In particular, population stratification is present in heterogeneous populations in North America and Western Europe in which most cases of sickle cell disease occur among specific sub-populations with a migration history or ancestry from areas in which the HbS allele has been present for millennia. In African and Middle Eastern countries with heterogeneous allele frequencies of hemoglobin variants, population stratification can be expected in large cities to which people migrate. This is illustrated by Tshilolo’s 2009 study of newborns in Kinshasa in which HbS allele frequencies varied between 4.3% and 11.4% in different sub-populations, and the FIS ranged from −4.5 to 6.9. While screening studies usually provide little information about the actual structure of local populations, the inclusion in future analyses of data from national censuses and/or genome-wide association studies41 could potentially contribute to a better understanding of the factors underlying HWE deviations observed and to improving current and future newborn estimates.
Finally, the sensitivity and specificity of the diagnostic methods used, as well as the main objectives of screening surveys represent another potential source of deviation. Most newborn screening programs attempt to minimize the chances of false positives and false negatives for individuals with sickle cell anemia, leading to strict diagnosis confirmation and minimal errors, while the limited clinical implications of a misdiagnosis of a heterozygote carrier as a wild-type homozygote and vice versa might not justify the expenses of further confirmation, leading to more frequent misclassification of AS and AA than SS. While we recorded the diagnostic method used in each survey, further work is needed to identify the precise role of this parameter on deviations from HWE.
In conclusion, taking potential deviation from HWE into account in assessments of the prevalence of hemoglobinopathies could have several benefits. First, it would help refine existing estimates of numbers of SCA-affected newborns at national, regional and global levels. Due mostly to the limited availability of data on consanguinity, the most recent published global estimates of the prevalence of HbSS4 presumed HWE. That could have led to a substantial underestimation of the number of HbSS annual births in some countries, as suggested by the present study. Second, the refined model presented here allows for improved estimation of numbers of SCD-affected newborns, including both HbSS and HbSC, based on allele frequencies, although other SCD variants are still excluded. Third, the prevention of these disorders could be improved by the adoption of appropriate policy measures tailored to the genetic, social and cultural factors responsible for HWE deviation. Finally, further analyses into the respective contributions of various factors responsible for deviations from HWE might reveal interesting insights into the evolutionary dynamics of these genes in different populations across the globe.
Supplementary Material
Acknowledgments
The authors thank Krista Crider for comments on an earlier version of the manuscript, and Danielle Lena from the Association Méditerranée Hémoglobine (Med-Hem) for sharing additional data about the surveys described in Diallo, 2008. FBP has been partly supported by an advanced grant (Diversity) from the European Research Council. TNW is funded by a senior clinical fellowship from the Wellcome Trust (#091758). This report was submitted with permission of the Director of the Kenya Medical Research Institute. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the United States Centers for Disease Control and Prevention.
Footnotes
Conflict of Interest: The authors have no conflict of interest to declare.
SUPPLEMENTARY INFORMATION
Supplementary Material Hardy-Weinberg Equilibrium
References
- 1.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376(9757):2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- 2.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6 Suppl 4):S398–405. doi: 10.1016/j.amepre.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. doi: 10.1038/ncomms1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itano HA, Neel JV. A new inherited abnormality of human hemoglobin. Proc Natl Acad Sci U S A. 1950;36(11):613–617. doi: 10.1073/pnas.36.11.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Modiano D, Luoni G, Sirima BS, et al. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature. 2001;414(6861):305–308. doi: 10.1038/35104556. [DOI] [PubMed] [Google Scholar]
- 7.Piel FB, Howes RE, Patil AP, et al. The distribution of haemoglobin C and its prevalence in newborns in Africa. Sci Rep. 2013;3:1671. doi: 10.1038/srep01671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J. The Hardy-Weinberg principle and its applications in modern population genetics. Front Biol. 2010;5(4):348–353. [Google Scholar]
- 9.Crow JF. Eighty years ago: the beginnings of population genetics. Genetics. 1988;119(3):473–476. doi: 10.1093/genetics/119.3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson A, Hill WG. Deviations from Hardy-Weinberg proportions: sampling variances and use in estimation of inbreeding coefficients. Genetics. 1984;107(4):703–718. doi: 10.1093/genetics/107.4.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi Y, Wijsman EM, Weir BS. Case-control association testing in the presence of unknown relationships. Genet Epidemiol. 2009;33(8):668–678. doi: 10.1002/gepi.20418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillespie JH. Population genetics: a concise guide. 2nd. Baltimore, Md; London: Johns Hopkins University Press; 2004. [Google Scholar]
- 13.Hosking L, Lumsden S, Lewis K, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12(5):395–399. doi: 10.1038/sj.ejhg.5201164. [DOI] [PubMed] [Google Scholar]
- 14.Graffelman J, Sanchez M, Cook S, Moreno V. Statistical inference for Hardy-Weinberg proportions in the presence of missing genotype information. PLoS One. 2013;8(12):e83316. doi: 10.1371/journal.pone.0083316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Shete S. Testing departure from Hardy-Weinberg proportions. Methods Mol Biol. 2012;850:77–102. doi: 10.1007/978-1-61779-555-8_6. [DOI] [PubMed] [Google Scholar]
- 16.Deng HW, Chen WM, Recker RR. Population admixture: detection by Hardy-Weinberg test and its quantitative effects on linkage-disequilibrium methods for localizing genes underlying complex traits. Genetics. 2001;157(2):885–897. doi: 10.1093/genetics/157.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi: 10.2471/BLT.06.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grosse SD, Williams TN, Amendah DD, Piel FB. Second National Conference on Blood Disorders in Public Health. Westin Atlanta Perimeter North; Atlanta, GA: 2012. Estimates of the birth prevalence of sickle cell disease in African populations: implications for newborn screening programs in Africa and the US. [Google Scholar]
- 19.Panigrahi S, Patra PK, Khodiar PK. Neonatal screening of sickle cell anemia: a preliminary report. Indian J Pediatr. 2012;79(6):747–750. doi: 10.1007/s12098-011-0682-8. [DOI] [PubMed] [Google Scholar]
- 20.Italia Y, Krishnamurti L, Mehta V, et al. Feasibility of a newborn screening and follow-up programme for sickle cell disease among South Gujarat (India) tribal populations. J Med Screen. 2014 doi: 10.1177/0969141314557372. [DOI] [PubMed] [Google Scholar]
- 21.Hartl DL, Clark AG. Principles of population genetics. 4th. Sunderland, Mass: Sinauer Associates; 2007. [Google Scholar]
- 22.Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet. 2005;76(5):887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engels WR. Exact tests for Hardy-Weinberg proportions. Genetics. 2009;183(4):1431–1441. doi: 10.1534/genetics.109.108977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Excoffier L. Handbook of Statistical Genetics. John Wiley & Sons, Ltd; 2004. Analysis of population subdivision. [Google Scholar]
- 25.Wang J. A new likelihood estimator and its comparison with moment estimators of individual genome-wide diversity. Heredity. 2011;107(5):433–443. doi: 10.1038/hdy.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):142–151. doi: 10.1016/S0140-6736(12)61229-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N Engl J Med. 1986;314(25):1593–1599. doi: 10.1056/NEJM198606193142501. [DOI] [PubMed] [Google Scholar]
- 28.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92(7):905–912. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]
- 29.Brosco JP, Grosse SD, Ross LF. Universal state newborn screening programs can reduce health disparities. JAMA Pediatr. 2015;169(1):7–8. doi: 10.1001/jamapediatrics.2014.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. British Journal of Haematology. 2011;154(6):736–744. doi: 10.1111/j.1365-2141.2011.08742.x. [DOI] [PubMed] [Google Scholar]
- 31.Bittles AH, Black ML. Consanguinity, human evolution, and complex diseases. P Natl Acad Sci. 2010;107(suppl 1):1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tshilolo L, Aissi LM, Lukusa D, et al. Neonatal screening for sickle cell anaemia in the Democratic Republic of the Congo: experience from a pioneer project on 31 204 newborns. J Clin Pathol. 2009;62(1):35–38. doi: 10.1136/jcp.2008.058958. [DOI] [PubMed] [Google Scholar]
- 33.Leutenegger AL, Sahbatou M, Gazal S, Cann H, Genin E. Consanguinity around the world: what do the genomic data of the HGDP-CEPH diversity panel tell us? Eur J Hum Genet. 2011;19(5):583–587. doi: 10.1038/ejhg.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Mouzan MI, Al-Salloum AA, Al-Herbish AS, Qurachi MM, Al-Omar AA. Regional variations in the prevalence of consanguinity in Saudi Arabia. Saudi Med J. 2007;28(12):1881–1884. [PubMed] [Google Scholar]
- 35.Canatan D. Thalassemias and hemoglobinopathies in Turkey. Hemoglobin. 2014;38(5):305–307. doi: 10.3109/03630269.2014.938163. [DOI] [PubMed] [Google Scholar]
- 36.Alkuraya FS, Kilani RA. Attitude of Saudi families affected with hemoglobinopathies towards prenatal screening and abortion and the influence of religious ruling (Fatwa) Prenatal Diagnosis. 2001;21(6):448–451. doi: 10.1002/pd.76. [DOI] [PubMed] [Google Scholar]
- 37.Pemberton TJ, Rosenberg NA. Population-genetic influences on genomic estimates of the inbreeding coefficient: a global perspective. Hum Hered. 2014;77(1–4):37–48. doi: 10.1159/000362878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manvell A. Sahelian action spaces: an examination of livelihood configurations in a rural Hausa community. J Int Dev. 2006;18(6):803–818. [Google Scholar]
- 39.Sinnock P. The Wahlund effect for the two-locus model. Am Nat. 1975;109(969):565–570. [Google Scholar]
- 40.Piel FB, Tatem AJ, Huang Z, Gupta S, Williams TN, Weatherall DJ. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health. 2014;2(2):e80–89. doi: 10.1016/S2214-109X(13)70150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hellenthal G, Busby GBJ, Band G, et al. A genetic atlas of human admixture history. Science. 2014;343(6172):747–751. doi: 10.1126/science.1243518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.