

Abstract
G protein-coupled receptors (GPCRs) are essential membrane proteins that facilitate cell-to-cell communication and co-ordinate physiological processes. At least 30 human GPCRs contain a Type I PSD-95/DLG/Zo-1 (PDZ) ligand in their distal C-terminal domain; this four amino acid motif of X-[S/T]-X-[ϕ] sequence facilitates interactions with PDZ domain-containing proteins. Because PDZ protein interactions have profound effects on GPCR ligand pharmacology, cellular localization, signal-transduction effector coupling and duration of activity, we analyzed the importance of Type I PDZ ligands for the function of 23 full-length and PDZ-ligand truncated (ΔPDZ) human GPCRs in cultured human cells. SNAP-epitope tag polyacrylamide gel electrophoresis revealed most Type I PDZ GPCRs exist as both monomers and multimers; removal of the PDZ ligand played minimal role in multimer formation. Additionally, SNAP-cell surface staining indicated removal of the PDZ ligand had minimal effects on plasma membrane localization for most GPCRs examined. Label-free dynamic mass redistribution functional responses, however, revealed diverging effects of the PDZ ligand. While no clear trend was observed across all GPCRs tested or even within receptor families, a subset of GPCRs displayed diminished agonist efficacy in the absence of a PDZ ligand (i.e. HT2RB, ADRB1), whereas others demonstrated enhanced agonist efficacies (i.e. LPAR2, SSTR5). These results demonstrate the utility of label-free functional assays to tease apart the contributions of conserved protein interaction domains for GPCR signal-transduction coupling in cultured cells.
Keywords: G protein-coupled receptor, Label-free signaling, PDZ domain, Pharmacology
Graphical Abstract
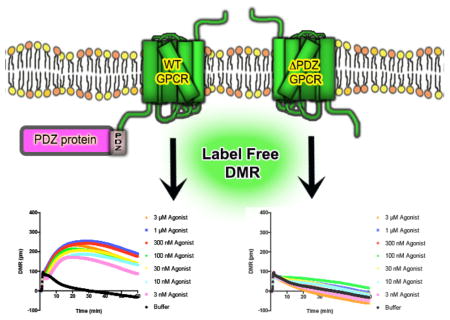
1. Introduction
G-protein coupled receptors (GPCRs) are essential for cell-to-cell communication and regulation of physiological events. To perform their designated functions, GPCRs must specifically interact with key proteins, the most thoroughly characterized being the heterotrimeric G-proteins (Gα, β and γ), which transmit the energy of agonist-GPCR binding to cellular response [1]. Interestingly, proteomic (i.e. affinity purification/mass spectrometry) and yeast-based (i.e. 2-hybrid) screening approaches developed over the last decade permitted high-throughput, unbiased identification of numerous novel GPCR-interacting proteins [2,3]. Indeed, GPCRs are expressed as intricate macromolecular complexes in cell membranes, with the GPCR acting as the central hub of signaling networks; a dynamic scaffold that temporally and spatially directs cellular traffic.
With this next era of GPCR molecular pharmacology comes the promise of innovative approaches to drug discovery. Targeting interaction interfaces between GPCRs and associated proteins may permit molecular tweaking of distinct GPCR signaling events, simultaneously inhibiting signaling events that are toxic whilst enhancing those that are beneficial. This endeavor is in its infancy, requiring thorough identification of GPCR interacting proteins with cell-type accuracy, and identifying divergent downstream signaling cascades linked to individual GPCR interaction modules.
Thus far, assessing how interacting proteins contribute to GPCR function has been limited to reductionist outputs: second messenger formation (i.e. cAMP/cGMP, Ca2+, ERK1/2), enzyme activity (i.e. phospholipase C, protein kinase A/C), bioluminescence/fluorescence energy transfer (BRET/FRET), cellular localization with high resolution microscopy and arrestin-association. Although informative, these assays are narrow in scope, each unable to identify unknown components of GPCR signaling networks. Label-free dynamic mass redistribution (DMR) technology represents an innovative approach to analyze complex GPCR signaling networks [4–6]. This assay involves passing polarized light through the glass bottom of a biosensor microtiter plate seeded with cells, then measuring shifts in the wavelength of reflected light over time. The shifts in wavelength are due to changes in intracellular mass near the membrane in response to exogenous stimulation, such as an agonist. As small as 1 picomter (pm) changes in wavelength can be reliably detected, and the direction of the overall change in cellular mass is indicated by whether the response is positive or negative. A positive response indicates an overall increase of cellular mass towards the cell membrane, whereas a negative response is indicative of cellular mass moving away from the membrane [7]. Similar to classic organ-tissue bath assays, which in effect are a summation of all the signaling events linking GPCR-stimulation to a contraction/relaxation event, DMR responses represent holistic changes in cellular mass and permit divergent GPCR signaling cascades to be analyzed without the need of a cell reporter. This is particularly useful for directly comparing GPCRs that couple to varying G proteins such as Gαs (i.e. β-adrenergic receptors), Gαi (i.e. β2-adrenergic receptors) or Gαq (i.e. β1-adrenergic receptors) [6].
Remarkably, at least 30 human GPCRs contain putative Type I PSD-95/DLG/Zo-1 (PDZ) ligands on their distal C-terminus with amino acid sequence X-[S/T]-X-[ϕ] [8]. This small protein-protein interaction domain permits GPCRs to associate with one or more of the ~180 PDZ domain-containing proteins encoded in the human genome. Once bound, PDZ-proteins may modulate GPCR pharmacodynamic properties via scaffolding effector proteins in close proximity, organizing GPCR complexes as discrete microdomains in cells, or linking GPCRs to non-canonical signaling events [2,3]. We previously demonstrated the Type I PDZ α1D-adrenergic receptor (AR) forms a macromolecular complex with PDZ-proteins scribble (SCRIB) and multiple isoforms of syntrophin (SNTA, SNTB1, and SNTB2), which impart functionality and distinct cellular localization to the receptor [9–13]. The specific contributions of each PDZ protein for ADRA1D function and agonist efficacy in human cells was determined by DMR technology. SCRIB and syntrophins bind C-terminal ADRA1D PDZ-ligands on discrete protomers within ADRA1D homodimers/oligomers, and in doing so, differentially regulate agonist efficacy [13].
Given the importance of PDZ-protein interactions for GPCR function, we subjected 23 human Type I PDZ GPCRs to label-free DMR assays in human cells with and without PDZ ligand truncation. Although no definitive role of the PDZ ligand was identified across all GPCRs tested or even within GPCR families, we identified 9 GPCRs for which the PDZ ligand differentially regulated receptor activation.
2. Materials and Methods
2.1. Plasmids, chemicals and antibodies
Human GPCR cDNAs were purchased from the Missouri S&T cDNA resource center (cdna.org) or cloned from human brain cDNA library (kindly provided by Prof. Ning Zheng, HHMI, University of Washington Department of Pharmacology). cDNAs were subcloned into pSNAPf/pCLIPf (New England Biolabs) using In-Fusion HD cloning technology (Clontech).
GPCR agonists (−)-isoproterenol hydrochloride (I6504), 5-hydroxytryptamine hydrochloride (H9523), clonidine hydrochloride (C7897), somatostatin (S9129), interleukin-8 (CXCL8, SRP3098), histamine dihydrochloride (H7250), UTP (U6875) and benzeneacetamide (C4494) were purchased from Sigma; [Ala17]-melanin concentrating hormone (3434), sphingosine-1-phosphate (1370) and galanin 1–30 (1179) from Tocris Bioscience.
SNAP-surface 782 substrate from New England Biolabs (S9142S). Topro-3 iodide (T3605) is from Life Technologies. Anti-HA mouse mAb (6E2, #2367) from Cell Signaling. IRdye 680 goat antimouse IgG and IRdye 800cw goat antirabbit IgG from Li-Cor.
2.2. Cell Culture and Reagents
Human embryonic kidney (HEK) 293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 2 mM L-glutamine. Cells were transfected with 1mg/ml polyethylenimine (PEI) and used 24–48 hr post transfection.
2.3. SNAP Cell Surface Assays
HEK293T cells were seeded in 6 well plates at 8 x 105 cells/well. Cells were transfected with SNAP-tagged cDNA constructs/PEI and replated in 96 well black optical bottom cell culture plates. Cell density was ~ 90% confluency prior to assay commencement. SNAP-Surface 782 substrate was diluted in DMEM to designated concentrations and incubated at 37°C/5% CO2 for 30 min. Cells were washed, fixed with 4% paraformaldehyde, then incubated with 1:10000 nuclear stain TOPRO-3 to normalize for cell number. Plates were analyzed with the LI-COR Odyssey Scanner (Li-Cor Biotechnology) and signal intensity quantified.
2.4. SNAP-PAGE
HEK293T cells were transfected with SNAP-tagged proteins. 48 hours after transfection, cells were lysed with 50 mM Tris-HCl, 150 mM NaCl, 1% NP40, and 0.1% Tween 20 buffer. Final concentration of 0.5 μM BG-782 substrate and 1 mM DTT were added to lysates for substrate binding reaction and samples were incubated for 30 minutes @37°C in the dark. Samples were then run on SDS-PAGE without boiling and gels were imaged using LI-COR Odyssey Scanner.
2.5. Label-free Dynamic Mass Redistribution (DMR) Assays
HEK293T cells were seeded at ~500k/well in Corning Epic sensor microplates and cultured for 24 hr. Cells were washed 3x with HBSS buffer and transferred to Corning Epic BT reader @37°C. Baseline DMR measurements were recorded for 1 hr. Compounds were added with the Sorenson Biosciences 96-well Benchtop Pipettor and agonist DMR responses were recorded for 1 hr. Data were exported to Microsoft Excel using Epic Analyzer Software. DMR background responses (buffer triggered responses) were subtracted from all datasets, as they are thought to occur as a result of (1) buffer bulk refractive index difference between the assay buffer and compound solution, (2) temperature mismatch or (3) a mechanical issue resulting from movement of the Epic 384 well plate between the Epic BT apparatus and the Sorenson Pipettor, as previously described in [7]. Error bars are displayed in agonist-concentration response curves, and not Epic DMR traces, to improve figure clarity. For Schild Plot analysis, data were analyzed using linear regression analysis using the method first described in [14].
2.6. Data Analysis
Data were analyzed with GraphPad Prism 6 software and expressed mean ± SEM. Differences in agonist-stimulated DMR responses were tested for significance using student’s t-test (p < 0.05).
3. Results
3.1. Importance of Type I PDZ ligands for GPCR protein expression
Before examining the potential effects of PDZ-ligands for GPCR DMR responses, we ensured that any observed effects were not a result of significant changes in GPCR expression. Thus, we first examined the impact of PDZ ligand truncation (ΔPDZ) on GPCR protein levels. To do so, full length and ΔPDZ GPCRs were subcloned into the pSNAP vector to add N-terminal human O6-alkylguanine-DNA alkyltransferase epitope tags. SNAP technology eliminates the need for traditional antibody-based Western blotting [15], which is highly problematic for transmembrane spanning GPCRs; commercially available GPCR antibodies are of questionable value, often detecting numerous non-specific artifacts [16].
SNAP-tagged GPCRs were transfected into HEK293T cells and imaged on a polyacrylamide gel in the 700–800 nm wavelength range by LI-COR (Fig. 1). We consistently observed a GPCR band at the predicted monomer size, but also as higher order bands, which may represent homo/heterodimers, and/or higher order oligomers. In many cases GPCRs displayed multiple bands of similar size to the full length receptor (i.e. CXCR1–3, HTR2A, SSTR1, SSTR2), possibly representing post-translation modifications (i.e. glycosylation). Smaller bands in the 20–36 kDa range were observed for CXCR2, CXCR3, and P2RY12.
Fig. 1. SNAP-PAGE of WT and ΔPDZ-GPCRs.

N-terminal SNAP-tagged GPCRs were transfected into HEK293T cells, lysed, incubated with BG 782 and run on PAGE. Full length (WT) and C-terminal Type I PDZ ligand truncated (ΔPDZ) GPCRs were analyzed.
While most WT and ΔPDZ GPCRs examined were expressed at similar densities, several exhibited differential expression. For example, ΔPDZ ADRA2B expression was diminished compared to WT ADRA2B at all observed fragments. Interestingly, increased levels of lower molecular weight fragments in the ΔPDZ versions of SSTR3 and GALR1 were observed, whereas decreased levels of a lower molecular weight fragment were observed for ΔPDZ CXCR3 as compared to the WT receptor. These observations suggest that in several instances, the PDZ ligand regulates expression levels and cleavage events involved in receptor processing.
3.2. Role of Type I PDZ ligands for GPCR membrane localization
We next employed SNAP cell surface expression assays to determine if PDZ-ligand truncation significantly impacted the ability of GPCRs to be trafficked to the plasma membrane. WT and ΔPDZ SNAP-GPCRs were transfected into HEK293T cells, seeded in 96-well glass bottom plates, fixed and incubated with increasing concentrations of the cell-impermeable SNAP-substrate BG782 (Fig. S1). As shown, no detectable SNAP-staining in untransfected (UT) or empty vector (SNAP) transfected HEK293T cells is present.
HTR2A demonstrated the greatest cell surface expression of all GPCRs tested and was used to normalize expression of all other GPCRs (Fig. S1, data summarized in Table 1). Only 4 receptors achieved > 50% HTR2A cell surface expression levels: P2RY1 (86.61 ± 3.83), LPAR2 (58.82 ± 4.74), SSTR3 (58.46 ± 6.51) and CXCR1 (56.27 ± 2.89). As previously reported by us and others [17–21], minimal ADRA1D cell surface expression was achieved in HEK293T cells, as it requires co-transfection of syntrophins and/or SCRIB, or truncation of the N-terminal domain. Other receptors that achieved minimal cell surface expression included MCHR2 (3.38 ± 7.79), C3AR1 (5.65 ± 0.96) and ADRA2B (7.26 ± 0.91).
Table 1. SNAP cell surface expression of WT and ΔPDZ GPCRs.
Cell surface expression of wild type (WT) and PDZ-ligand truncated (ΔPDZ) GPCRs was quantified in HEK293T cells using SNAP cell impermeable substrate BG782. Data are expressed as % of HTR2A CSE. Data were analyzed with GraphPad Prism and are expressed as mean ± SEM (n = 3–4).
| GPCR gene name | WT CSE (%WT HTR2A) | ΔPDZ CSE (%WT HTR2A) |
|---|---|---|
| ADRA1D | 1.39 ± 0.645 | 3.57± 0.99 |
| ADRA2B | 7.26 ± 0.91 | 6.87 ± 0.97 |
| ADRB1 | 22.72 ± 4.41 | 25.03 ± 2.76 |
| ADRB2 | 9.77 ± 0.97 | 11.21 ± 2.27 |
| HTR2A | 100 ± 1.85 | 75.44 ± 6.12 |
| HTR2B | 30.72 ± 6.73 | 34.86 ± 5.77 |
| HTR2C | 22.71 ± 4.40 | 25.02 ± 2.76 |
| SSTR1 | 22.88 ± 2.96 | 21.08 ± 4.05 |
| SSTR2 | 28.53 ± 7.94 | 29.01 ± 5.85 |
| SSTR3 | 58.46 ± 6.51 | 46.26 ± 12.16 |
| SSTR4 | 33.7 ± 3.50 | 32.80 ± 2.63 |
| SSTR5 | 19.98 ± 3.82 | 27.69 ± 8.92 |
| CXCR1 | 56.27 ± 2.89 | 53.88 ± 3.43 |
| CXCR2 | 23.28 ± 2.20 | 25.68 ± 4.90 |
| CXCR3 | 15.48 ± 6.10 | 14.7 ± 3.27 |
| CXCR5 | 20.1 ± 1.02 | 17.6 ± 4.41 |
| GALR1 | 29.11 ± 1.41 | 23.06 ± 2.22 |
| HRH3 | 37.99 ± 5.99 | 52.90 ± 6.03 |
| P2RY1 | 86.61 ± 3.83 | 78.81 ± 8.78 |
| P2RY12 | 17.88 ± 3.19 | 20.93 ± 2.08 |
| MCHR2 | 3.38 ± 7.79 | 0.532 ± 8.36 |
| C3AR1 | 5.65 ± 0.96 | 1.16 ± 0.39 |
| LPAR2 | 58.82 ± 4.74 | 68.05 ± 6.39 |
| S1PR2 | 11.49 ± 0.67 | 14.81 ± 7.79 |
Overall, deletion of the PDZ ligand produced subtle effects on cell surface expression. In certain cases, PDZ ligand truncation caused a >10% reduction in cell surface expression (HTR2A, SSTR3, P2YR1, MCHR2, C3AR1), or a >10% increase (LPAR2, SSTR5, HRH3). However, in no examples did removal of the PDZ ligand completely abolish GPCR cell surface expression, suggesting that this domain is not absolutely essential for plasma membrane localization for the GPCRs as examined by SNAP cell-surface assays.
3.3. DMR measures direct activation of GPCRs
Although others have demonstrated that observed DMR signals generated by the addition of GPCR agonists are induced by heterotrimeric G-protein signal transduction pathways [6], we wished to confirm that our DMR responses are also due to direct GPCR signaling. Thus, concentration-response curves (CRCs) were generated for the β-agonist isoproterenol in HEK293T cells expressing ADRB1, in the absence and presence of increasing amounts of the competitive β-adrenergic receptor antagonist propranolol (Fig. 2). Propranolol progressively inhibited 3 μM isoproterenol-stimulated DMR curves (Fig. 2a), and rightward-shifted isoproterenol DMR CRCs (Fig. 2b, Fig. S2). Schild plot analysis calculated the propranolol functional affinity constant for inhibiting isoproterenol DMR responses to be 4.57 nM (pA2 = −8.34 ± 0.49 M) (Fig. 2c), which is in the range of previously reported propranolol functional affinities [21,22]. The slope of the Schild plot was 1.04 ± 0.08, indicative of one-site competition mode of antagonist binding. Taken together, these observations validate that DMR responses are a direct result of GPCR stimulation.
Fig. 2. Propranolol functional affinity for antagonizing isoproterenol-stimulated DMR responses in HEK293T cells expressing ADRB1.

a, DMR responses stimulated by 3 μM isoproterenol in the absence and presence of increasing concentrations of the β-adrenergic receptor antagonist propranolol in β1-adrenergic receptor (ADRB1) transfected HEK293T cells. b, Isoproterenol-stimulated DMR concentration-response curves in the absence and presence of propranolol. c, Schild plot analysis of data in (B). Data are the mean ± SEM of n = 4.
3.4. DMR signatures of Type I PDZ GPCRs
Confident that PDZ ligand truncated GPCRs can still achieve significant levels of protein and plasma membrane expression and that DMR responses are the result of direct activation of GPCRs, we next subjected all WT and ΔPDZ GPCRs to label-free DMR. GPCR cDNAs were expressed in HEK293T cells and DMR signatures were recorded for 30–60 minutes following agonist stimulation. The time point at which the greatest DMR signal was achieved was determined and termed time-to-peak (TTP) response. TTP was used to create CRCs, from which agonist potencies (EC50) and maximal DMR responses (Max) were calculated (Table 2). A plethora of GPCR signatures were observed (Figs. S2–S6). With the exception of S1PR2, all maximum DMR values were positive, suggesting that activation of these receptors leads to an overall increase in cellular mass towards the membrane. The majority of WT GPCRs produced biphasic curves, with a rapid TTP occurring in the initial 5–10 minutes. After reaching TTP, certain GPCRs reached a steady state DMR which remained stable for the remainder of the experiment (HTR2A, HTR2C, MCHR2, ADRA2B, ADRB1). Other GPCRs displayed either a rapid, (CXCR1, CXCR2, P2YR1, P2YR2, HRH3) or prolonged (SSTR1, SSTR4, GALR1) return to baseline DMR. The greatest DMR responses were stimulated by the ADRB1 (255.12 ± 3.74) and ADRB2 (251.48 ± .15.45) subtypes; other GPCRs that produced robust DMR responses included HTR2A (118.1 ± 10.5), P2YR12 (170.04 ± 2.93) and P2YR1 (116.19 ± 2.22). In certain cases GPCRs produced very weak, if any, DMR responses (CXCR3, CXCR5, C3AR1). Agonist potencies ranged from the subnanomolar (Interleukin-8 for CXCR1 and CXCR2; Galanin 1–30 for GALR1) to micromolar concentrations (UTP for P2Y receptors).
Table 2. Pharmacological properties of agonists targeting WT and ΔPDZ GPCRs.
Agonist stimulated DMR concentration response curves were constructed for wild type (WT) and PDZ-ligand truncated (ΔPDZ) GPCRs, and used to calculate potencies (pEC50). Maximal agonist DMR responses (Max) and time when maximal response reached (Time To Peak, TTP) in seconds are shown. All data were analyzed with GraphPad Prism and are expressed as mean ± SEM (n = 2–4). Max DMR of ΔPDZ GPCRs significantly different than WT GPCR Max DMR are denoted with ** (student’s t-test, p < 0.05). ND = not determined.
| GPCR | Agonist | WT pEC50 |
Max (DMR) | TTP (sec) | ΔPDZ pEC50 |
Max (DMR) | TTP (sec) |
|---|---|---|---|---|---|---|---|
| ADRA2B | Clonidine | −7.31 ± 0.17 | 74.40 ± 2.54 | 11.10 | −7.15 ± 0.10 | 90.10 ± 6.31 | 9.51 |
| ADRB1 | Isoproterenol | −8.42 ± 0.15 | 255.12 ± 3.74 | 31.67 | −9.09 ± 0.12 | 48.17 ± 8.08** | 61.30 |
| ADRB2 | Isoproterenol | −8.01 ± 0.15 | 251.48 ± 15.45 | 28.37 | −7.65 ± 0.26 | 219.84 ± 5.73 | 30.37 |
| HTR2A | 5-hydroxytryptamine | −6.84 ± 0.07 | 118.10 ± 10.50 | 14.10 | −6.78 ± 0.13 | 106.10 ± 3.96 | 14.70 |
| HTR2B | 5-hydroxytryptamine | −7.02 ± 0.17 | 80.60 ± 12.92 | 60.0 | −7.34 ± 0.13 | 213.20 ± 15.52** | 60.0 |
| HTR2C | 5-hydroxytryptamine | −8.04 ± 0.25 | 90.30 ± 9.89 | 5.90 | −7.83 ± 0.14 | 106.61 ± 12.89 | 39.21 |
| SSTR1 | Somatostatin | −8.98 ± 0.21 | 35.01 ± 2.47 | 4.17 | −8.50 ± 0.44 | 32.48 ± 2.57 | 5.87 |
| SSTR2 | Somatostatin | −9.29 ± 0.16 | 41.06 ± 2.27 | 5.37 | −8.91 ± 0.15 | 75.62 ± 7.34** | 5.57 |
| SSTR3 | Somatostatin | −8.84 ± 0.16 | 60.58 ± 1.29 | 5.07 | −8.97 ± 0.11 | 62.07 ± 2.52 | 4.97 |
| SSTR4 | Somatostatin | −8.65 ± 0.30 | 38.76 ± 3.73 | 5.60 | −8.83 ± 0.22 | 54.74 ± 2.07** | 5.60 |
| SSTR5 | Somatostatin | −9.39 ± 0.17 | 31.09 ± 3.49 | 3.90 | −8.46 ± 0.16 | 155.86 ± 10.81** | 5.60 |
| CXCR1 | Interleukin-8 | −8.29 ± 0.07 | 69.69 ± 1.88 | 3.60 | −7.08 ± 0.45 | 9.48 ± 1.53** | 3.80 |
| CXCR2 | Interleukin-8 | −8.53 ± 0.11 | 34.89 ± 2.08 | 3.20 | −8.45 ± 0.13 | 37.50 ± 2.20 | 3.50 |
| CXCR3 | Interleukin-8 | ND | 10.17 ± 2.00 | 11.70 | ND | 18.3 ± 3.16 | 15.60 |
| CXCR5 | Interleukin-8 | −10.34 ± 0.16 | 23.22 ± 1.80 | 2.50 | −9.97 ± 0.12 | 40.70 ± 2.54** | 2.30 |
| GALR1 | Galanin 1–30 | −9.98 ± 0.12 | 67.70 ± 3.45 | 4.07 | −5.67 ± 0.09 | 93.60 ± 2.08** | 3.97 |
| HRH3 | Histamine | −7.26 ± 0.16 | 46.97 ± 3.71 | 7.30 | −7.74 ± 0.18 | 60.19 ± 5.07 | 6.30 |
| P2RY1 | UTP | −5.39 ± 0.15 | 116.19 ± 2.22 | 4.87 | −5.69 ± 0.19 | 118.68 ± 10.27 | 5.27 |
| P2RY12 | UTP | −5.47 ± 0.07 | 170.04 ± 2.93 | 5.40 | −5.70 ± 0.08 | 192.60 ± 6.35** | 5.20 |
| MCHR2 | Ala17-MCH | −6.27 ± 0.12 | 92.70 ± 4.57 | 60.0 | −8.70 ± 0.61 | 19.16 ± 7.57** | 60.0 |
| C3AR1 | Benzeneacetamide | ND | 36.90 ± 1.82 | 4.50 | ND | 38.77 ± 2.59 | 3.50 |
| LPAR2 | Lysophosphatidic acid | −9.89 ± 0.92 | 19.70 ± 8.56 | 10.55 | −8.17 ± 0.10 | 110.73 ± 2.72** | 27.07 |
| S1PR2 | Sphingosine-1-phosphate | −8.42 ± 0.36 | −40.54 ± 3.76 | 17.61 | −8.10 ± 0.85 | −10.16 ± 4.91** | 16.31 |
3.4.1. GPCR DMR responses affected by PDZ ligand truncations
We next focused on DMR responses in the absence of PDZ ligands. As expected multiple GPCRs required an intact PDZ ligand for DMR response. For example, the PDZ ligand is required for isoproterenol activation of ADRB1, which gradually reaches a steady state and remains relatively stable. Similarly, the PDZ ligand is also required for interleukin-8 (IL-8) activation of CXCR1. However, unlike the DMR response of ADRB1 activation, activation of CXCR1 is rapid and transient (Fig. 3). In addition to ADRB1 and CXCR1, deletion of the PDZ ligand in MCHR2 and S1PR2 also lead to decreased DMR response to agonist, suggesting the PDZ domain is required for DMR responses by these receptors (Fig. 4).
Fig. 3. Epic DMR responses diminished by removal of the GPCR C-terminal PDZ ligand.

Epic DMR responses in HEK293T cells expressing WT (a) or ΔPDZ (b) β1-adrenergic receptor (ADRB1); WT (c) or ΔPDZ (d) chemokine type I receptor (CXCR1). Data are the mean ± SEM (n = 4). ISO = isoproterenol; IL-8 = interleukin-8.
Fig. 4. Agonist efficacies abrogated by C-terminal PDZ ligand truncation.

Concentration-response curves for agonist-stimulated DMR responses in HEK293T cells expressing WT or ΔPDZ β1-adrenergic receptor (ADRB1) (a), chemokine subtype 1 receptor (CXCR1) (b), Melanin-Concentrating Hormone Receptor 2 (MCHR2) (c) or Sphingosine-1-Phosphate Receptor 2 (S1PR2) (d). ISO = isoproterenol; IL-8 = interleukin-8; 17ALA-MCH = [Ala17]-melanin concentrating hormone; S1P= sphingosine-1-phosphate. Data are the mean ± SEM (n = 4).
In addition to the above examples, where the PDZ ligand was required for DMR responses, we also observed multiple receptors in which deletion of the PDZ ligand resulted in an enhanced DMR response. For example, agonist stimulation of WT HTR2B produced a modest DMR response (Fig. 5a). However, the maximal response more than doubled when the PDZ ligand was deleted (Fig. 5b). Similarly, the maximal response more than tripled when the PDZ ligand was deleted from SSTR5 (Fig. 5c, 5d). In addition to HTR2B and SSTR5, deletion of the PDZ ligand enhanced the maximal DMR response in SSTR2, LPAR2, and GALR1, suggesting the PDZ ligand inhibits signaling by these receptors (Fig. 6).
Fig. 5. Epic DMR responses enhanced by removal of the GPCR C-terminal PDZ ligand.

Epic DMR responses in HEK293T cells expressing WT (a) or ΔPDZ (b) 5-hydroxytryptamine type 2B receptor (HTR2B); WT (c) or ΔPDZ (d) somatostatin receptor 5 (SSTR5). 5-HT = 5-hydroxytryptamine; SST = somatostatin. Data are the mean ± SEM (n = 4).
Fig. 6. Agonist efficacies enhanced by C-terminal PDZ ligand truncation.

Concentration-response curves for agonist-stimulated DMR responses in HEK293T cells expressing WT or ΔPDZ 5-hydroxytryptamine type 2B receptor (HTR2B) (a), somatostatin receptor 5 (SSTR5) (b), somatostatin receptor 2 (SSTR2) (c), or lysophosphatidic acid receptor 2 (LPAR2) (d) GPCRs. 5-HT = 5-hydroxytryptamine; SST = somatostatin; LPA = lysophosphatidic acid. Data are the mean ± SEM (n = 4).
Interestingly, no definitive trends were observed across the entire set of GPCRs tested or within GPCR families when the PDZ ligand was deleted. For example, within the 5-HT receptor family, deletion of the PDZ ligand in HTR2B resulted in an increased DMR response upon the addition of 5-HT, whereas deletion of the PDZ ligand in HTR2A or HTR2C had no effect compared to the WT receptors (Fig. S3). This was also apparent for the somatostatin family of receptors, although the shape of the DMR signature was vastly different. PDZ ligand removal did not affect SSTR1, SSTR3, or SSTR4 DMR responses. In striking contrast, PDZ ligand removal enhanced the efficacy of both SSTR2 and SSTR5 somatostatin-stimulated DMR responses (Fig. S4). Combined, our DMR results highlight the diversity and importance of PDZ ligands for GPCR functional responses, and represents an alternative method to functionally classify GPCRs.
Discussion
With the recent advances in methodologies to provide high resolution GPCR crystal structures, and biophysical approaches to study GPCR signaling at the level of protein-protein interaction with BRET-FRET, it is increasingly clear that GPCR cell signaling is an intricately complex, tightly orchestrated physiological event. Given GPCRs can selectively associate with numerous accessory proteins, which in turn, impart specific functional properties to a GPCR, novel functional assays are required that provide a readout that encompasses all downstream signaling events with temporal accuracy [24,25].
Our study demonstrates the utility in using label-free DMR assays to analyze complex GPCR signaling events in real time, and to assess the contribution of conserved protein interaction domains for GPCR signaling. In this case, we examined the Type I PDZ ligand, which is found on no less than 30 human GPCRs of broad physiological significance, each of which is coupled to myriad molecular signaling events and regulates a wide variety of physiological processes. This subfamily of GPCRs activates divergent cellular responses, thus making it difficult to compare the importance of Type I PDZ ligands for GPCR activity with common end-point assays. For example, intracellular Ca2+ or IP3 generation is typically used to examine Gαq-coupled receptors, whereas alterations in cytosolic cAMP levels are used to quantify drug efficacy when examining Gαs- and Gαi-coupled receptors. As a result, a multitude of GPCR reporter systems have been created to circumvent this issue [26], yet none share the major advantage of the label-free DMR assays, in that a single functional readout (DMR) can be obtained for any GPCR examined, regardless of the signal-transduction cascade to which it is coupled.
The major finding of this study was that agonist-stimulated DMR responses of at least 9 GPCRs were significantly modified by truncation of the PDZ ligand. Interestingly, no definitive trend was identified based on known characteristics of each GPCR (i.e. Gα subtype, agonist, PDZ ligand sequence), demonstrating the complexity and uniqueness of each GPCR signaling cascade. Nevertheless, these observations provide an invaluable resource for understanding individual GPCR responses.
Given that the PDZ ligand is a well-known protein-protein interaction domain, it is likely that PDZ-domain containing proteins have a direct role in binding and modifying the pharmacodynamic properties of the GPCR. We and others have identified multiple GPCR:PDZ protein interactions that regulate GPCR activity. For example, ADRB1 interacts with multiple PDZ proteins, including MAGI2 and PSD95, both of which have been shown to regulate ADRB1 internalization [27–29]. As shown in this study, the ADRB1 PDZ ligand is required for an agonist-induced DMR response, which may suggest that the response is partially comprised of receptor internalization or dependent upon it. Of note, three additional GPCRs that are poorly understood, namely CXCR1, MCHR2, and S1PR2, also require an intact PDZ ligand for DMR response. Thus, it is plausible that the PDZ ligands in these receptors also regulate receptor internalization, which is important for the function of these receptors. It would be of interest to delineate and compare CXCR1, MCHR2 and S1PR2 downstream functional responses with the more thoroughly characterized ADRB1 signal transduction cascade.
In addition to receptors that require an intact PDZ ligand for a DMR response, we also identified at least five GPCRs (HTR2B, SSTR5, SSTR2, LPAR2, and GALR1) where the PDZ domain inhibits agonist responses. For example, somatostatin-stimulated DMR responses are minimal in cells expressing WT SSTR5, whereas the response is increased at least 3-fold in the absence of the PDZ domain. In our previous proteomic analysis of SSTR5 [12], we were unable to identify PDZ proteins associated with unstimulated or steady-state levels of SSTR5, suggesting that PDZ proteins may be recruited to the receptor upon activation by agonist, which then act to inhibit downstream signaling events. Conversely, in the absence of a PDZ ligand, it is possible that SSTR5 is now able to internalize and proceed with downstream signaling events. In support of this, previous studies demonstrate the PDZ ligand of SSTR5 is not required for plasma membrane targeting, but multiple PDZ proteins, including SNX27, PIST/GOPC and SLC9A3R1/NHERF1, regulate its subcellular trafficking, endocytosis, and recycling to the membrane following agonist stimulation and washout [30, 31].
Finally, we did not detect any noticeable differences in DMR responses between WT and ΔPDZ for 15 GPCRs containing a Type I PDZ ligand. At least two possible explanations exist. First, it is entirely feasible that the PDZ ligand is dispensable for GPCR activation in specific receptors. Second, as we only analyzed these GPCRs in HEK293T cells, it is possible that the expression levels of putative PDZ proteins are insufficient to regulate GPCR activation. In our previous analysis of ADRA1D, we observed weak activation by phenylephrine for both WT and ΔPDZ ADRA1D. However, overexpression of SCRIB and or syntrophins enhanced the DMR response, nearly 5 fold higher with SCRIB alone [13]. Furthermore, others have shown that DMR response profiles are cell type dependent [6], adding another layer of complexity to GPCR activation and function.
While it is clear that DMR responses comprise G-protein dependent signaling [4–6], our results suggest that it is much more complex, with hints of receptor trafficking and recycling having significant contributions to agonist-induced responses, and possibly G protein-independent signaling events. Future studies that directly assess the relative contributions of each component of GPCR signaling to the DMR responses will provide robust datasets that enhance our general understanding of GPCR biology.
Conclusion
This study demonstrates the utility of label free DMR assays to study complex functional responses stimulated by modular GPCR oligomers in living cells, critical features in the future of drug discovery. Historically, most drugs targeting GPCRs bind to the orthosteric site, in that they compete with the endogenous ligand for binding. A major hurdle in this approach involves imparting ligand selectivity while ensuring adequate bioavailability of investigational small molecules/peptides; chemical similarities of ligand binding pockets in closely related GPCR subtypes makes achieving this task arduous and costly, and as a result, high risk. Thus, the next era of pharmacology may very well involve targeting discrete protein-protein interaction domains within a specific GPCR macromolecular complex. This innovative approach may enhance medicinal therapeutic index and improve clinical outcomes by honing in on discrete signaling events, while reducing off-target modulation of highly homologous GPCR subtypes. To achieve this goal, functional assays, like label-free DMR, that provide a real-time, holistic, consolidated vantage point of all GPCR signaling events with cell-type specificity will be required.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health RO1 GM100893. T.K. is a Mary Gates University of Washington Research Scholar. D.-A.H. and M.E. are Ronald E. McNair Scholars. N.D.C is supported by National Human Genome Research Institute T32 HG00035.
Abbreviations
- ADRA1D
α1D adrenergic receptor
- ADRA2B
α2B-adrenergic receptor
- ADRB1
β1-adrenergic receptor
- ADRB2
β2-adrenergic receptor
- C3AR1
Complement Component 3a Receptor 1
- CXCR1
Chemokine Receptor 1
- CXCR2
Chemokine Receptor 2
- CXCR3
Chemokine Receptor 3
- CXCR5
Chemokine Receptor 5
- GALR1
Galanin Receptor 1
- HRH3
Histamine Receptor H3
- HTR2A
5-Hydroxytryptamine (Serotonin) Receptor 2A
- HTR2B
5-Hydroxytryptamine (Serotonin) Receptor 2B
- HTR2C
5-Hydroxytryptamine (Serotonin) Receptor 2C
- LPAR2
Lysophosphatidic Acid Receptor 2
- MCHR2
Melanin-Concentrating Hormone Receptor 2
- P2RY1
Purinergic Receptor P2Y1
- P2RY12
Purinergic Receptor P2Y12
- S1PR2
Sphingosine-1-Phosphate Receptor 2
- SSTR1
Somatostatin Receptor 1
- SSTR2
Somatostatin Receptor 2
- SSTR3
Somatostatin Receptor 3
- SSTR4
Somatostatin Receptor 4
- SSTR5
Somatostatin Receptor 5
Footnotes
Competing Financial Interests Statement
The authors have no competing financial interests.
Author Contributions
N.D.C., K.-S.L., J.L.W.-M., N.S., A.W.-Y. and C.H. designed experiments.
N.D.C., K.-S.L., A.C., J.L.W.-M., T.K., J.-M.P., D.-A.H., M.E., A.S., A.E.C., A.W.-Y. and C.H. performed experiments.
N.D.C., N.S., A.W.-Y. and C.H. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Bio. 2009;10:819–830. doi: 10.1038/nrm2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol. 2012;165(6):1717–36. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang Y, et al. Resonant waveguide grating biosensor for living cell sensing. Biophys J. 2006;91:1925–1940. doi: 10.1529/biophysj.105.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007;55:314–322. doi: 10.1016/j.vascn.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Schroder R, et al. Deconvolution of complex G-protein coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nature Biotech. 2010;28:943–950. doi: 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- 7.Fang Y. Label-Free Biosensor Methods in Drug Discovery. In: Fang Y, editor. Methods in Pharmacology and Toxicology. Springer Science + Business Media; New York: 2015. pp. 17–33. [Google Scholar]
- 8.Marchese A, Paing MM, Temple BRS, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Ann Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Hague C, Hall RA, Minneman KP. Syntrophins regulate α1D-ARs through a PDZ domain-mediated interaction. J Biol Chem. 2006;281:12414–12420. doi: 10.1074/jbc.M508651200. [DOI] [PubMed] [Google Scholar]
- 10.Lyssand JS, et al. Blood pressure is regulated by an α1D-AR/dystrophin signalosome. J Biol Chem. 2008;283:18792–18800. doi: 10.1074/jbc.M801860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyssand JS, et al. β-dystrobrevin-1 recruits α-catulin to the α1D-AR/DAPC signalosome. Proc Natl Acad Sci USA. 2010;107:21854–21859. doi: 10.1073/pnas.1010819107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyssand JS, Lee KS, DeFino M, Adams ME, Hague C. Syntrophin isoforms play specific functional roles in the α1D-AR/DAPC signalosome. Biochem Biophys Res Commun. 2011;412(4):596–601. doi: 10.1016/j.bbrc.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camp ND, Lee KS, Wacker-Mhyre JL, Kountz TS, Park JM, Harris DA, Estrada ME, Stewart A, Wolf-Yadlin A, Hague C. Individual protomers of a G-protein coupled receptor dimer integrate distinct functional modules. Cell Disc. 2015;1:15011. doi: 10.1038/celldisc.2015.11. doi:10.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14(1):48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gong H, et al. Near-Infrared fluorescence imaging of mammalian cells and xenograft tumors with SNAP-tag. PLoS One. 2012;7(3):e34003. doi: 10.1371/journal.pone.0034003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen BC, Swigart PM, Simpson PC. Ten commercial antibodies for α1-adrenergic receptor subtypes are nonspecific. Naunyn Schmiedebergs Arch Pharmacol. 2009;379(4):409–12. doi: 10.1007/s00210-008-0368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalothorn D, et al. Differences in the cellular localization and agonist-mediated internalization properties of the α1-adrenoceptor subtypes. Mol Pharmacol. 2002;61(5):1008–16. doi: 10.1124/mol.61.5.1008. [DOI] [PubMed] [Google Scholar]
- 18.Pupo AS, Uberti MA, Minneman KP. N-terminal truncation of human α1D-adrenoceptors increases expression of binding sites but not protein. Eur J Pharmacol. 2003;462:1–8. doi: 10.1016/s0014-2999(03)01292-5. [DOI] [PubMed] [Google Scholar]
- 19.Hague C, et al. The N terminus of the human α1D-adrenergic receptor prevents cell surface expression. J Pharmacol Exp Ther. 2004;309:388–397. doi: 10.1124/jpet.103.060509. [DOI] [PubMed] [Google Scholar]
- 20.Petrovska R, Kapa I, Klovins J, Schloth HB, Uhlen S. Addition of a signal peptide sequence to the α1D-adrenoceptor gene increases the density of receptors, as determined by [3H]-prazosin binding in the membranes. Br J Pharmacol. 2005;144:651–659. doi: 10.1038/sj.bjp.0706087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Cazarin ML, et al. The α1D-adrenergic receptor is expressed intracellularly and coupled to increases in intracellular calcium and reactive oxygen species in human aortic smooth muscle cells. J Mol Signal. 2008;3(6):1–9. doi: 10.1186/1750-2187-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gille E, Lemoine J, Ehle B, Kaumann AJ. The affinity of (−)-propranolol for β1 and β1-autoreceptors of the human heart. Naunyn Schmiedebergs Arch Pharmacol. 1985;331(1):60–70. doi: 10.1007/BF00498852. [DOI] [PubMed] [Google Scholar]
- 23.Baker JG. The selectivity of β-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br J Pharmacol. 2005;144(3):317–22. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- 25.Lefkowitz RJ, Whalen EJ. β-arrestins: traffic cops of cell signaling. Curr Opin Cell Biol. 16(2):162–8. doi: 10.1016/j.ceb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012;33(3):372–384. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, et al. β1-adrenergic receptor association with the synaptic scaffolding protein membrane-associated guanylate kinase inverted-2 (MAGI-2): differential regulation of receptor internalization by MAGI-2 and PSD-95. J Biol Chem. 2001;276:41310–41317. doi: 10.1074/jbc.M107480200. [DOI] [PubMed] [Google Scholar]
- 28.Hu LA, et al. GIPC interacts with the β1-adrenergic receptor and regulates β1-adrenergic receptor-mediated ERK activation. J Biol Chem. 2003;278:26295–26301. doi: 10.1074/jbc.M212352200. [DOI] [PubMed] [Google Scholar]
- 29.He J, et al. Proteomic analysis of β1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2005;281:2820–2827. doi: 10.1074/jbc.M509503200. [DOI] [PubMed] [Google Scholar]
- 30.Wente W, Stroh T, Beaudet A, Richter D, Kreienkamp HJ. Interactions with PDZ domain proteins PIST/GOPC and PDZK1 regulate intracellular sorting of SSTR5. J Biol Chem. 2005;280:32419–32425. doi: 10.1074/jbc.M507198200. [DOI] [PubMed] [Google Scholar]
- 31.Bauch C, Koliwer J, Buck F, Honck H, Kreienkamp H. Subcellular sorting of the G-protein coupled mouse somatostatin receptor 5 by a network of PDZ-domain containing proteins. PLoS One. doi: 10.1371/journal.pone.0088529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.