

Abstract
Clostridium sordellii infections have been reported in women following natural childbirth and spontaneous or medically-induced abortion, injection drug users and patients with trauma. Death is rapid and mortality ranges from 70–100%. Clinical features include an extreme leukemoid reaction, the absence of fever, and only minimal pain or erythema at the infected site. In the current study, we developed a murine model of C. sordellii soft tissue infection to elucidate the pathogenic mechanisms. Mice received 0.5, 1.0 or 2.0 × 106 CFU C. sordellii (ATCC 9714 type strain) in the right thigh muscle. All doses caused fatal infection characterized by intense swelling of the infected limb but no erythema or visible perfusion deficits. Survival rates and time to death were inoculum dose-dependent. Mice developed a granulocytic leukocytosis with left shift, the onset of which directly correlated with disease severity. Histopathology of infected tissue showed widespread edema, moderate muscle damage and minimal neutrophil infiltration. Circulating levels of granulocyte colony-stimulating factor (G-CSF), soluble tumor necrosis factor receptor I (sTNF-RI) and interlukin-6 (IL-6) were significantly increased in infected animals, while TNF-α, and IL-1β levels were only mildly elevated, suggesting these host factors likely mediate the leukocytosis and innate immune dysfunction characteristic of this infection. Thus, this model mimics many of the salient features of this infection in humans and has allowed us to identify novel targets for intervention.
Keywords: C. sordellii, pathogenesis, myonecrosis, host immune response, leukemoid reaction
I. Introduction
Clostridium sordellii causes a rapidly progressive soft tissue infection [1–3]. Our recent review of 45 C. sordellii cases demonstrated an overall mortality of 70% [1]. Of the fatal infections, 74% occurred in women undergoing childbirth or spontaneous or induced abortions and mortality was 100%. C. sordellii infections associated with traumatic wounds, injecting drug use or surgery were fatal in 52% of cases [1]. Initial symptoms included nausea, dizziness, lethargy, and mild tenderness at sites of infection. Most patients (73%) were afebrile. Within hours of presentation, most developed a unique constellation of clinical features including refractory hypotension, severe tachycardia, profound capillary leak syndrome with hemoconcentration. Development of an extreme leukemoid reaction (LR: 75–200 ×103 cells/μL whole blood) with a left shift was the sole predictor of a fatal outcome [1]. Though the development of an extreme LR is pathognomonic for C. sordellii infections and portends fatal outcome, the mechanisms remain entirely unknown.
The virulence of C. sordellii is strain variable and reflects the profile of exotoxins produced by individual organisms. Pathogenic strains of C. sordellii produce up to seven known exotoxins [4]. Of these, the hemorrhagic toxin (HcsT) and lethal toxin (LcsT) are thought to mediate the pathogenesis of infection [4]. Other toxins include a cholesterol-dependent hemolysin, phospholipase C, neuraminidase, DNAse, hyaluronidase, and collagenase. However, the specific roles of these auxiliary exotoxins during infection remain to be clearly defined.
Animal models are extremely important to elucidating the mechanisms associated with disease pathogenesis, dynamics of the host response and the specific roles of bacterial toxins during infection. A murine intrauterine model of C. sordellii infection has been developed to investigate C. sordellii-associated endometritis [5]. In addition, experimental fatal toxic shock syndrome studies have utilized intraperitoneal injections of the lethal toxin of C. sordellii in mice and rats [6]. However, an in vivo model to investigate C. sordellii intramuscular infections (IM) has yet to be established, despite the fact that soft tissue infections represent nearly one-third of all reported cases in humans [1].
The goals of the present work were to establish the first murine model of C. sordellii muscle infection that mimics the salient features of this infection in humans and to use this model to investigate the mechanisms driving the extreme C. sordellii LR. Mice challenged intramuscularly with a clinically relevant strain of C. sordellii developed signs and symptoms comparable to those associated with human infection, including development of an LR, and had a rapidly lethal outcome. Reduced circulating TNFα and IL-1β levels and marked increases in sTNF-RI, G-CSF and IL-6 provide the first-ever clues as to the mechanisms responsible for the characteristic absence of fever and extreme leukemoid reaction observed in humans with C. sordellii infection. These findings demonstrate the relevance and utility of this model of C. sordellii infection in elucidating the mechanisms responsible for the local and systemic effects of infection with this important human pathogen and suggest new interventional targets.
II. Materials and Methods
Bacteria
C. sordellii type strain #9714 (American Type Culture Collection [ATCC], Manassas, VA) was used. This strain was originally isolated from an acute human soft tissue infection [4]. A Bactron II anaerobic chamber (Sheldon Manufacturing, Cornelius, OR) was utilized to maintain an anaerobic environment for C. sordellii growth and manipulation.
Bacterial cultivation and inoculum preparation
Stock cultures were streaked on Brucella blood agar plates (Anaerobe Systems, Morgan Hill, CA) and a single isolated colony was used to inoculate Brain Heart Infusion (BHI; Becton Dickinson, San Jose, CA) broth. Cultures were grown anaerobically overnight at 37°C. The following morning, 1% of the overnight culture was used to inoculate 250 mL of fresh, pre-reduced BHI liquid media. This culture was grown to an OD600 of approximately 1.7 (late-stationary phase culture). Organisms were collected by centrifugation (5000 × g, 15 min, 4°C), washed twice and resuspended in pre-chilled saline. A working stock of 2.0 × 107 colony forming units (CFU)/mL was prepared based on a previously determined relationship between CFU and OD600. The working stock was serially diluted 2-fold in saline to achieve desired concentrations. Bacterial concentrations of these preparations were confirmed by plating serially diluted samples in duplicate on BHI agar plates. Plates were incubated anaerobically at 37°C and colonies counted the following day.
Experimental C. sordellii infection
All animal experiments were approved by the Boise, ID Veterans Affairs Medical Center’s Institutional Animal Care and Use Committee and adhered to guidelines of the National Institutes of Health. Adult female C57BL/6 mice (20/group) were injected intramuscularly in the right upper thigh with 0.5, 1.0 or 2.0 ×106 CFU (low, medium and high doses, respectively) of C. sordellii ATCC 9714. These concentrations were selected based on similar studies previously performed by our group (data not published). Immediately following challenge, infected animals were randomly divided into ‘survival’ and ‘pathogenesis’ subgroups. The survival subgroup was closely monitored over 5 days and mortalities noted. In the pathogenesis subgroup, blood specimens were taken from 2 animals/time point and infected tissues were examined for histologic abnormalities as described below.
Histopathological analysis
Two mice per inoculum group from the ‘pathogenesis’ subgroups were randomly selected at 12, 21, 30, 39 and 48 hrs after infection and a single blood specimen was obtained by a retro-orbital draw from isoflurane-anesthetized animals to determine circulating white blood cell counts and differentials (see below). Animals were then sacrificed by cervical dislocation and the infected muscle tissue (and the contra-lateral non-infected control muscle) was surgically removed and fixed in 10% neutral buffered formalin. Formalin-fixed tissues were embedded in paraffin, stained with hematoxylin-eosin and examined and scored by a blinded pathologist for evidence of; 1) inflammation, 2) edema, 3) tissue necrosis, and 4) hemorrhage, on a scale of 0 – 3 as follows; 0: none, 1: mild, 2: moderate, and 3: marked.
White Blood Cell (WBC) Counts and Differentials
WBC counts were determined in two animals per time point by adding 10 μL of each whole blood specimen to 180 μL of Turks stain solution (1:20 dilution). Each sample was counted twice by hemocytometer, and the final WBC count is given as the average of these two counts. For WBC differential counts, cytospin samples were prepared. Here, 10 μL of each whole blood specimen was added to 0.5 mL of PharM Lyse (BD Biosciences, San Jose, CA) to lyse red blood cells. Samples were zap vortexed, incubated at room temperature for 15 min, and spun at 300 × g for 5 min. Overlaying supernatants were aspirated and the remaining WBC pellets resuspended in 500 μL of PBS + 7% culture grade bovine serum albumin (BSA). 450 μL of this material were then added to individual chambers of a Shandon Cytospin 2 centrifuge (Block Scientific, Inc, Bohemia, NY) and spun at 200 × g for 10 min. After air drying, slides were stained via an Aerospray 7120 hematology automatic slide stainer-cytocentrifuge (Wescore, Inc, Logan, UT) and sent to the University of Idaho Animal and Veterinary Science Department (Caldwell, ID) where WBC differentials were determined by a blinded observer with expertise in this area. Cells were counted using an Olympus CX31 microscope (1000× magnification). 100 cells were counted in two separate fields, and data are reported as the average of these 2 counts. Population ratios for polymorphonuclear leukocytes, lymphocytes, monocytes, eosinophils, band cells, and metamyelocyte/myelocyte cells were determined.
Murine Protein Cytokine Array
Relative serum levels of 30 cytokines and chemokines were measured by a custom slide-based mouse cytokine array (RayBiotech, Norcross, GA) according to the manufacturer’s instructions. Briefly, the arrays were blocked with the supplied blocking buffer for 30 min, then incubated with 100 μl of serum collected from C. sordellii-infected or no treatment control animals (diluted 1:10 with blocking buffer) at 25°C for 2 hrs. After washing, arrays were incubated with biotin-conjugated primary antibody and horseradish peroxidase-conjugated streptavidin. Arrays were sent to RayBiotech where a semi-quantitative analysis of the comparative intensity of the spots was performed. The relative intensities of each cytokine were normalized to control spots on the same array. The final cytokine values were calculated by subtracting control animal measurements from those obtained from infected animals. Cytokines/chemokins tested were: Cluster of Differentiation (CD) 30 ligand (CD30L), CD40, Fas Ligand, granulocyte colony-stimulation factor (G-CSF), granulocyte macrophage colony-stimulating factor (GM-CSF), interferon-gamma (IFN-γ), interleukin (IL)-1 alpha (IL-1α), interleukin-1 beta (IL-1β), IL-2, IL-3, IL-6, IL-9, IL-10, (monocyte chemotactic protein-1 (MCP-1, MCP-5, macrophage colony-stimulating factor (M-CSF), monokine induced by gamma interferon (MIG), macrophage inflammatory protein (MIP)-1 alpha (MIP-1α), macrophage inflammatory protein-1 gamma (MIP-1γ), MIP-2, macrophage inflammatory protein 3 alpha (MIP-3α), macrophage inflammatory protein 3 beta (MIP-3β), regulated on activation normal T-cell expressed and secreted (RANTES), stem cell factor (SCF), IL-12p70, tumor necrosis factor-alpha (TNF-α), tumor necrosis factor receptor I (sTNF-RI), vascular cell adhesion molecule (VCAM)-1, platelet selectin (P-selectin), and lymphocyte selectin (L-selectin). Serum concentrations of G-CSF and sTNF-R1 were quantitated by commercial ELISA purchased from R&D Systems (Minneapolis, MN) and RayBiotech respectively, using conditions suggested by the suppliers and the methods described for murine serum samples.
III. Results
Survival Studies
Survival of infected mice was inoculum dose-dependent. The minimum LD100 dose of C. sordellii ATCC 9714 was 1.0×106 CFU, and all mice injected with this dose died by 48 hrs (Fig. 1, medium dose). Mice infected with 2.0 × 106 CFU exhibited a more rapid demise, with all animals succumbing to infection by 24 hrs (Fig. 1, high dose). Challenge with <1×106 CFU was not uniformly fatal, as 30% of animals challenged with 0.5 × 106 CFU remained alive at 48 hrs (Fig. 1, low dose). With this model, all animals still alive at 48 hrs consistently survived the infection (not shown).
Figure 1. Survival curves of mice infected intramuscularly with C. sordellii ATCC 9714.
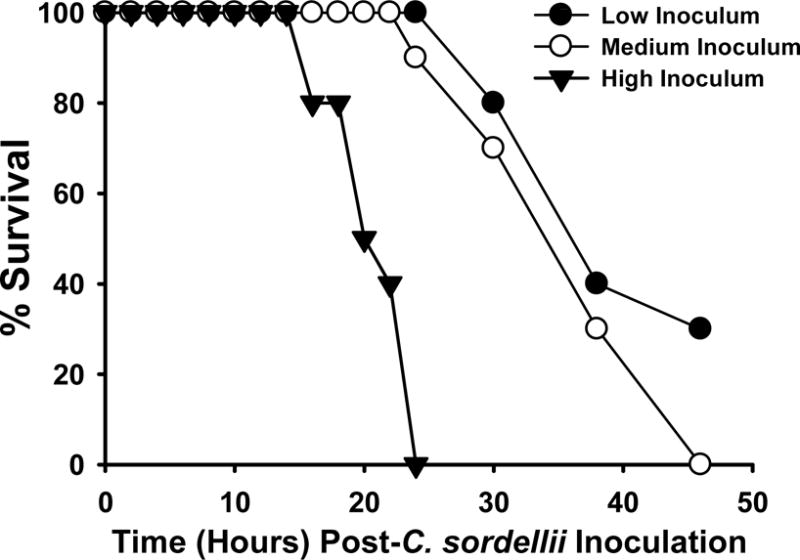
Groups of C57BL/6 mice (N = 10) were inoculated in the right thigh muscle with either 0.5 (low), 1.0 (medium) or 2.0 (high) × 106 colony forming units of washed, log phase C. sordellii ATCC 9714. Mice were observed for 5 days with mortalities recorded.
Clinical features and histopathology of C. sordellii infection
Irrespective of the inoculum size, all animals challenged with C. sordellii displayed progressive swelling of the infected leg (Fig. 2a,b) and loss of function over the observation period. However, the onset and severity of these signs was dose-dependent and correlated with mortality. Interestingly, none of the C. sordellii-infected animals displayed gross muscle or skin erythema or blackening of the infected foot, findings that are characteristic of experimental C. perfringens myonecrosis (Fig. 2c).
Figure 2. Comparative physical findings of mice infected intramuscularly with C. sordellii (A), or control (B) versus C. perfringens (C).

C57BL/6 mice were inoculated with 1.0×106 CFU of washed, log phase C. sordellii ATCC 9714 (A). C. sordellii infected animals developed severe edema at the infection site compared to non-infected mice (A vs B) but little erythema of the muscle or overlying skin (not shown) or blackening of the foot that are characteristic of experimental C. perfringens infection (A vs C).
On the histological level, non-infected muscle had normal myofibrillar structure, little to no edema and no infiltrating PMNL (Fig. 3a). In contrast, infected muscle demonstrated mild to moderate PMNL infiltration, edema, disruption of muscle fiber architecture and myofiber engulfment by surrounding PMNLs in the muscle and connective tissues of infected legs (Fig. 3b,c). The severity and consistency of these pathologies is reflected in the substantially elevated histopathological scores of 4 infected animals (Fig. 3d). Additionally, the severity of these pathologic findings was dependent upon the inoculum dose as well as duration of the infection (data not shown).
Figure 3. C. sordellii muscle infection causes marked histologic abnormalities.

Routine hematoxylin-eosin staining of muscle specimens obtained from 4 mice 39 hrs after infection with 1.0×106 CFU C. sordellii ATCC 9714; control mice received sterile saline injection. (A) H&E of normal muscle tissue at 20× magnification. H&E of infected muscle at (B) 20× and (C) 40× magnification shows widespread edema and areas of focal PMNL influx and tissue necrosis. (D) Muscle specimens from infected animals were scored by a blinded observer for evidence of inflammation, edema, tissue necrosis and hemorrhage on a scale of 0–3 for each abnormality (max score 12). No pathologies were observed in sham-infected control muscles (not shown).
WBC counts and differentials
Uninfected mice had an average circulating WBC count of 4.25×103/μL, which were comprised of 17.5% PMNL, 3.0% monocytes, and 79.5% lymphocytes. All animals infected with C. sordellii mounted an increase in circulating WBC count that was comparable across groups (7.8 – 8.4×103/μL; Fig. 4a). However, the time to the peak LR directly correlated with inoculum size and paralleled infection severity (Fig. 4a). Specifically, peak WBC counts were observed at 12, 21 and 30 hours post-infection in mice in the high, medium and low dose groups, respectively (Fig. 4a). The LR was attributable to expansion of the granulocyte population (Fig. 4b), including a marked left shift to immature forms (i.e., band cells, myelocytes, metamyeloctyes; Fig. 4c). Lymphocytes were notably reduced by 10 hrs in all challenge groups (Fig. 4d).
Figure 4. Intramuscular infection with varying doses of C. sordellii generates a leukemoid reaction.

C57BL/6 mice were infected intramuscularly with either, 0.5 (low), 1.0 (medium) or 2.0 (high) × 106 colony forming units of washed, log phase C. sordellii ATCC 9714. (A) Total WBC counts were obtained on blood specimens from 2 randomly chosen mice at that time point. Data are the mean maximal counts (± standard deviation) observed for each inoculum group. Sham infected mice had a constant WBC count of 4.2 × 103/μL whole blood. WBC differential ratios were determined by a blinded observer with expertise in this area. (B) Polymorphonuclear leukocytes, (C) Granulocyte precursors, and (D) Lymphocytes.
Circulating cytokine levels during C. sordellii infection
Relative levels of circulating cytokines were assessed by commercial microarray for mice infected with the medium dose inoculum (1×106 CFU) of C. sordellii (Table 1). At 10 hrs-post-infection, G-CSF was elevated 1500-fold in mice when compared to sham infected controls (Fig. 5a), and by 39 hrs, a > 6,000-fold increase in G-CSF was observed (Fig. 5a). Quantitative ELISA confirmed elevated G-CSF serum concentrations of 6,577 pg/mL and 158,484 pg/mL at 24 hrs and 48 hrs post-infection, respectively, compared to sham infected controls (14 pg/mL) (Fig. 5b).
Table 1. Changes in circulating cytokine profiles in animals following intramuscular infection with C. sordellii.
Following inoculation with the medium dose (1.0×106 CFU: LD100-48 hr) of C. sordellii, a custom slide-based mouse cytokine array was used to semi-quantitate the relative serum levels of 30 individual cytokines and chemokines. Designated values are given as the relative percentage above those obtained for sham infected animals at each time point. Relative serum levels were determined for two mice from sham and infected animals at the indicated time points.
| Cytokine/Chemokine | 12 hr | 21 hr | 39 hr |
|---|---|---|---|
| CD30 L | 82 | 210 | 27 |
| CD40 | 0 | 4 | −6 |
| Fas Ligand | 66 | 27 | 26 |
| G-CSF | 873 | 5,507 | 17,966 |
| GM-CSF | 21 | 6 | 13 |
| IFN-gamma | 15 | 1 | −9 |
| IL-1 alpha | 65 | 8 | 52 |
| IL-1 beta | 165 | 147 | 182 |
| IL-2 | 55 | 74 | 12 |
| IL-3 | 44 | 16 | 0 |
| IL-6 | 144 | 4,421 | 8,736 |
| IL-9 | 70 | 94 | 55 |
| IL-10 | 32 | 8 | 15 |
| MCP-1 | 47 | 44 | 52 |
| MCP-5 | 37 | 36 | 67 |
| M-CSF | 28 | −2 | 43 |
| MIG | 36 | −8 | 35 |
| MIP-1 alpha | 44 | 51 | 3 |
| MIP-1 gamma | 78 | 208 | 174 |
| MIP-2 | 4 | 41 | 213 |
| MIP-3 alpha | 66 | 120 | 183 |
| MIP-3 bate | 83 | 47 | 33 |
| RANTES | 49 | 28 | 19 |
| SCF | 14 | 4 | 41 |
| IL-12p70 | 21 | 15 | 15 |
| TNF alpha | 57 | 35 | 38 |
| sTNF RI | 1,233 | 1,505 | 4,949 |
| VCAM-1 | 24 | 18 | 12 |
| P-Selectin | 45 | 33 | 30 |
| L-Selectin | 12 | −15 | 25 |
Fig. 5. Granulocyte-colony stimulating factor response in mice infected intramuscularly with C. sordellii.
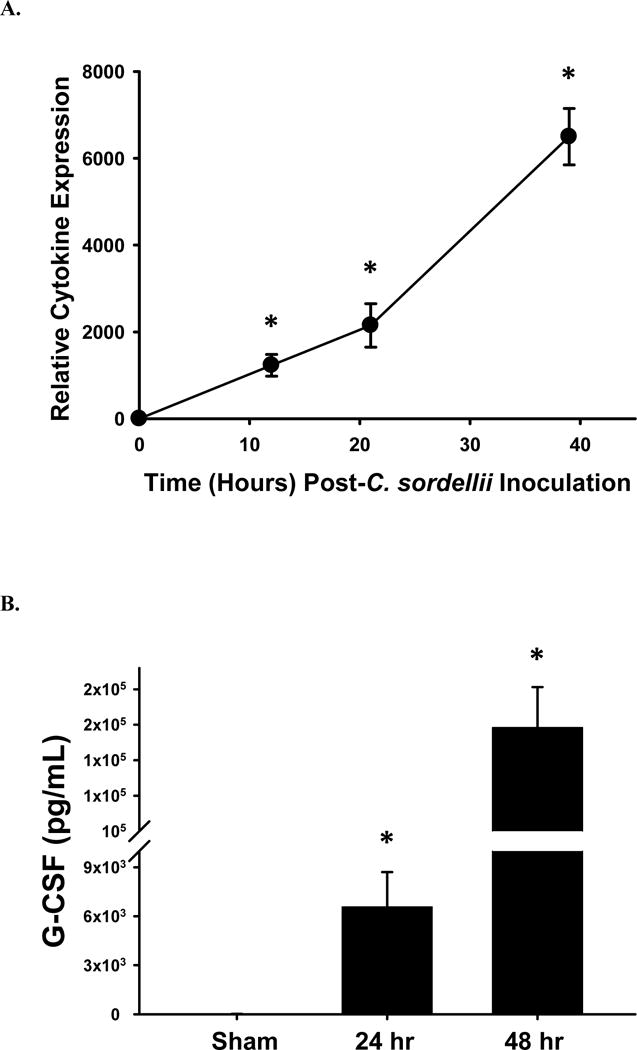
(A) Fold change differences in circulating G-CSF levels in mice infected with 1 × 106 CFU of C. sordellii ATCC 9714 as determined by murine cytokine array analysis. Serum was collected from two infected mice at each indicated time. (B) Serum concentrations of G-CSF as determined by ELISA. Data represents the average serum concentration from four mice infected with either nothing or 1 × 106 CFU of C. sordellii at 24 hrs and 48 hrs. *P< .05, compared with the no-toxin (NoTx) control (1-way analysis of variance with Tukey’s test).
Cytokine array analysis also demonstrated little to no increase in the pro-inflammatory mediators TNF-α and IL-1β over the course of infection (Fig. 6a). However, significant increases in IL-6 were observed during the fulminant phase of infection (21 and 39 hrs, 1,904- and 5,528-fold, respectively; Fig. 6a). Higher levels of soluble TNF receptor type 1 (sTNF-R1), a TNF-α antagonist and modulator of the innate immune response, were also seen by cytokine array at these stages of infection (data not shown) and were confirmed by quantitative ELISA (24 hr: 5,331 pg/mL; 48 hr: 18,511 pg/mL) versus sham infected controls (1,232 pg/mL) (Fig. 6b). No substantial differences in other cytokines/chemokines were observed (data not shown).
Fig. 6. Serum concentrations of innate immune modulators from mice infected intramuscularly with C. sordellii.

(A) Relative fold change differences in circulating TNF-α, IL-1β and IL-6 in mice infected with 1 × 106 CFU of C. sordellii ATCC 9714 as determined by murine cytokine array analysis. Serum was collected from two infected mice at each indicated time. (B) Serum concentrations of sTNF-R1 as determined by ELISA. Data represents the average serum concentration from four mice challenged with either nothing or 1 × 106 CFU of C. sordellii at 24 hrs and 48 hrs post-infection. *P< .05, compared with the no-toxin (NoTx) control (1-way analysis of variance with Tukey’s test).
IV. Discussion
C. sordellii infections in humans are characterized by an extreme leukemoid reaction (LR) which is the sole predictor of fatal outcome [1]. Leukemoid reactions of lesser intensity are also characteristic of C. difficile and C. novyi infections. Development of an LR in these infections also predicts poor outcomes. While the clostridial LR has been a clinically important observation for decades, the mechanisms responsible are not fully understood.
Each of these organisms produces large clostridial cytotoxins (LCCs) [7]. LCCs possess remarkable amino acid similarity between species, with C. sordellii lethal toxin (LT) and C. difficile Toxin B (TcdB) having the highest homology (90%). LCCs are glycosyltransferases that inactivate Rho GTPases controlling cell cycle, apoptosis, gene transcription and the structural functions of actin (reviewed in [7]). LCC-induced actin disruption causes loss of endothelial barrier integrity, leading to the systemic capillary leakage characteristic of C. sordellii and C. difficile infections [6, 8–10].
LCC production by those clostridial species that cause infections with associated LRs suggests their potential role in LR development. Indeed, Geny et al showed that mice challenged intraperitoneally with 15 ng of purified C. sordellii LT (i.e., the 24 hr minimum 100% lethal dose, MLD24) developed a significant 1.56-fold increase in WBC count (5.40 ± 2.5 vs 8.39 ± 2.8 cells/mm3) 6 hrs after toxin injection [6]. In our model of active C. sordellii soft tissue infection, mice exhibited a 2.1-fold increase in circulating WBCs, the onset of which directly correlated with disease severity. In humans, the C. sordellii LR is primarily attributable to expansion of the granulocytic population with a marked left shift to immature cells of this lineage [1] and our infection model accurately reproduces these findings. In contrast, in the LT intoxication model used by Geny et al [6], the increased WBC count included a nearly 3-fold increase in monocytes [6]. This suggests that our active infection model more closely mimics the human condition.
In the current study, mice infected with C. sordellii displayed a robust increase in circulating G-CSF. Significantly increased levels of G-CSF have also been described in the cecal and colonic contents of mice with C. difficile gastrointestinal infection [11]. Previous studies have demonstrated that administration of G-CSF in both mice and humans induces the mobilization of leukocytes, including hematopoietic stem cells, from the bone marrow into circulation; a process commonly used for the clinical transplantation of stem cells [12, 13]. Specifically, G-CSF stimulates the release of proteases (i.e., elastase, cathepsin G) from mature bone marrow PMNL resulting in the cleavage of VCAM-1 [12, 13] and CXCR4 [14–16] which are key adhesins that regulate leukocyte egress from the hematopoietic compartment. Thus, G-CSF likely mediates the common clostridial LR shared by C. sordellii, C. difficile and C. novyi.
However, the mechanisms driving the extreme C. sordellii LR are not completely understood. We have demonstrated that a C. sordellii-specific metalloproteinase cleaves both human VCAM-1 and CXCR4 (manuscript in progress). Such direct toxin-induced cleavage of these vital adhesins could augment that caused by G-CSF-induced elastase release. We have also shown that the C. sordellii neuraminidase augments the ability of GM-CSF to stimulate proliferation of a granulocytic precursor cell line in vitro [17]. Thus, we conclude that C. sordellii-specific exotoxins, including the neuraminidase and metalloproteinase, amplify the common LCC-driven, G-CSF-mediated release of bone marrow-derived leukocytes to drive C. sordellii LR to extreme levels.
Human C. sordellii infections are also rapidly progressive and our model mimics this finding. However, achieving 100% mortality required at least 106 CFU of C. sordellii. The requirement for large numbers of organisms to achieve 100% mortality has been observed in other murine models of soft tissue infections such as myonecrosis due to Streptococcus pyogenes [18, 19], Clostridium perfringens [20–22], or C. septicum [23] or fatal pneumonia with methicillin-resistant Staphylococcus aureus (MRSA) [24]. Clearly, many orders of magnitude fewer organisms are required to cause similar infections in humans. With C. sordellii specifically, the human lethal dose is presumed to be extremely low. This conclusion is based on the observation that even in fatal human infection, the number of C. sordellii can be so low that organisms are undetectable by culturing or Gram stain, and the diagnosis is made only by PCR methods or specific immunofluorescence staining on autopsy specimens [25]. Further, prospective studies of the vaginal and stool microbiota in human females demonstrate that C. sordellii is rarely found; when it is recovered, it is in low numbers (Dr. Sharon Hillier, Professor of Reproductive Infectious Diseases, University of Pittsburg School of Medicine, personal communication).
In humans, early clinical symptoms of C. sordellii infection include only mild tenderness with minimal erythema and a paucity of infiltrating inflammatory cells at the infection site – features that are reproduced by our model. Patients also fail to develop fever. Taken together, these findings suggest that a prominent step(s) in the pro-inflammatory innate immune response to C. sordellii is inhibited or absent. Consistent with this notion, C. sordellii-infected mice in our study displayed only minimal elevations in the pro-inflammatory cytokines TNF-α and IL-1β over the entire disease course. Yet we have shown that washed, heat-killed C. sordellii are efficiently recognized in vitro via Toll-like receptors 2 and 6 [26], and that such recognition activates the NF-kB signaling pathway, resulting in the production of TNF-α by isolated human monocytes [26]. Thus, products of active infection (i.e., bacterial toxins or host response modifiers) are required to down-regulate the pro-inflammatory innate immune response observed both in humans and in our experimental animal model.
Suppression of the TNF-α/IL-1β pro-inflammatory response may be a consequence of the large increase in circulating G-CSF and sTNF-RI seen in our model of C. sordellii infection. G-CSF has been reported to switch the responsiveness of circulating leukocytes from a pro-inflammatory to an anti-inflammatory state [27]. Specifically, administration of G-CSF to humans attenuates the production of pro-inflammatory cytokines, including TNF-α, and enhances release of anti-inflammatory regulatory molecules such as IL-1 receptor antagonist (IL-1ra) and soluble TNF-α receptors by PMNLs [28]. Similarly, increases in circulating IL-6 may also contribute to the weak inflammatory response during C. sordellii infection. While IL-6 is clearly associated with the acute innate response to infection, it also negatively modulates both local and systemic inflammatory responses by dampening TNF-α and IL-1β production and inhibiting tissue inflammatory infiltration [29]. Collectively, our data indicates that IL-6, G-CSF and sTNF-RI may act in concert to down modulate both the local and systemic innate immune responses observed during C. sordellii infection. It also implies that neutralization of G-CSF in humans may provide therapeutic benefit.
In summary, we have established a murine model of C. sordellii soft tissue infection that mimics many of the salient features of this infection in humans. Using this model, we have identified potential novel targets for intervention that could modulate the extreme LR, reverse the innate immune system dysfunction and improve outcomes in patients with these infections.
Highlights for Review.
The bacterial virulence factors responsible, the molecular mechanisms involved and the significance of the response to pathogenesis of C. sordellii infection are entirely unknown. With this study, wanted to develop and utilize an animal model of C. sordellii soft tissue infection to elucidate the mechanisms associated with the extreme leukemoid reaction and weak innate immune response; both hallmark symptoms typically used to identify this deadly infection.
The results from this study successfully demonstrated, for the first time 1) the development of an intramuscular model of C. sordellii infection; and 2) pathogenic mechanisms responsible for the display of distinct clinical symptoms displayed by patients suffering from C. sordellii infection. Specifically, we showed that increases in circulating G-CSF and sTNF-RI in conjunction with decreases in TNF-α and IL-1β production contribute to the leukemoid reaction and absent inflammatory response during infection. These findings may lead to novel forms of diagnosis, prevention and treatment for this infection, and insights gained may bridge large gaps in our knowledge of hematopoiesis and leukocyte mobilization during both normal cellular turnover and during infection.
The findings from this study promote advancements in basic science and infectious disease pathogenesis research. Leukemoid reactions are hallmark features of C. sordellii, C. difficile, and C. novyi infections and their development during infection portends a fatal outcome. Thus, the results of the gained from this investigation will have very defined, and potentially very broad, significance to human health.
Acknowledgments
This material is based upon work supported in part by the U.S. Department of Veterans Affairs, Office of Research and Development Biomedical Laboratory Research Program (MJA, AEB, DLS) and by the National Institutes of Health (Grant P20 RR016454/GM103408).
Financial support: This work was supported in part by the U. S. Department of Veterans Affairs, Office of Research and Development, Biomedical Laboratory Research Program (MJA, AEB, DLS) and by the National Institutes of Health (Grant P20 RR016454/GM103408).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: No authors involved with this manuscript have a commercial or other association that might pose a conflict of interest.
Presentation of information: No information provided herein has been presented at any national or international scientific forum.
Reference List
- 1.Aldape MJ, Bryant AE, Stevens DL. Clostridium sordellii infection: epidemiology, clinical findings, and current perspectives on diagnosis and treatment. Clin Infect Dis. 2006 Dec 1;43(11):1436–46. doi: 10.1086/508866. [DOI] [PubMed] [Google Scholar]
- 2.Sicard D. Deaths from Clostridium sordellii after medical abortion. N Engl J Med. 2006 Apr 13;354(15):1645–7. [PubMed] [Google Scholar]
- 3.Sinave C, Le TG, Blouin D, Leveille F, Deland E. Toxic shock syndrome due to Clostridium sordellii: a dramatic postpartum and postabortion disease. Clin Infect Dis. 2002 Dec 1;35(11):1441–3. doi: 10.1086/344464. [DOI] [PubMed] [Google Scholar]
- 4.Smith LDS. Clostridium sordellii. In: Smith LDS, editor. The pathogenic anaerobic bacteria. Springfield, IL: Charles C. Thomas Publishing; 1975. pp. 291–8. [Google Scholar]
- 5.Aronoff DM, Hao Y, Chung J, et al. Misoprostol impairs female reproductive tract innate immunity against Clostridium sordellii. J Immunol. 2008 Jun 15;180(12):8222–30. doi: 10.4049/jimmunol.180.12.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geny B, Khun H, Fitting C, et al. Clostridium sordellii lethal toxin kills mice by inducing a major increase in lung vascular permeability. Am J Pathol. 2007 Mar;170(3):1003–17. doi: 10.2353/ajpath.2007.060583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Just I, Gerhard R. Large clostridial cytotoxins. Rev Physiol Biochem Pharmacol. 2004;152:23–47. doi: 10.1007/s10254-004-0033-5. [DOI] [PubMed] [Google Scholar]
- 8.Adamson RH, Curry FE, Adamson G, et al. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol. 2002 Feb 15;539(Pt 1):295–308. doi: 10.1113/jphysiol.2001.013117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waschke J, Baumgartner W, Adamson RH, et al. Requirement of Rac activity for maintenance of capillary endothelial barrier properties. Am J Physiol Heart Circ Physiol. 2004 Jan;286(1):H394–H401. doi: 10.1152/ajpheart.00221.2003. [DOI] [PubMed] [Google Scholar]
- 10.Hippenstiel S, Tannert-Otto S, Vollrath N, et al. Glucosylation of small GTP-binding Rho proteins disrupts endothelial barrier function. Am J Physiol. 1997 Jan;272(1 Pt 1):L38–L43. doi: 10.1152/ajplung.1997.272.1.L38. [DOI] [PubMed] [Google Scholar]
- 11.Pawlowski SW, Calabrese G, Kolling GL, et al. Murine model of Clostridium difficile infection with aged gnotobiotic C57BL/6 mice and a BI/NAP1 strain. J Infect Dis. 2010 Dec 1;202(11):1708–12. doi: 10.1086/657086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiq S, Pamphilon D, Brunskill S, Doree C, Hyde C, Stanworth S. Bone marrow harvest versus peripheral stem cell collection for haemopoietic stem cell donation in healthy donors. Cochrane Database Syst Rev. 2009;1:CD006406. doi: 10.1002/14651858.CD006406.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlo-Stella C, Di NM, Magni M, et al. Defibrotide in combination with granulocyte colony-stimulating factor significantly enhances the mobilization of primitive and committed peripheral blood progenitor cells in mice. Cancer Res. 2002 Nov 1;62(21):6152–7. [PubMed] [Google Scholar]
- 14.Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003 Jan;111(2):187–96. doi: 10.1172/JCI15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002 Jul;3(7):687–94. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 16.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002 Oct;17(4):413–23. doi: 10.1016/s1074-7613(02)00424-7. [DOI] [PubMed] [Google Scholar]
- 17.Aldape MJ, Bryant AE, Ma Y, Stevens DL. The leukemoid reaction in Clostridium sordellii infection: neuraminidase induction of promyelocytic cell proliferation. J Infect Dis. 2007 Jun 15;195(12):1838–45. doi: 10.1086/518004. [DOI] [PubMed] [Google Scholar]
- 18.Stevens DL, Yan S, Bryant AE. Penicillin-binding protein expression at different growth stages determines penicillin efficacy in vitro and in vivo: an explanation for the inoculum effect. J Infect Dis. 1993 Jun;167(6):1401–5. doi: 10.1093/infdis/167.6.1401. [DOI] [PubMed] [Google Scholar]
- 19.Stevens DL, Gibbons AE, Bergstrom R, Winn V. The Eagle effect revisited: efficacy of clindamycin, erythromycin, and penicillin in the treatment of streptococcal myositis. J Infect Dis. 1988 Jul;158(1):23–8. doi: 10.1093/infdis/158.1.23. [DOI] [PubMed] [Google Scholar]
- 20.Stevens DL, Titball RW, Jepson M, Bayer CR, Hayes-Schroer SM, Bryant AE. Immunization with the C-Domain of alpha-Toxin prevents lethal infection, localizes tissue injury, and promotes host response to challenge with Clostridium perfringens. J Infect Dis. 2004 Aug 15;190(4):767–73. doi: 10.1086/422691. [DOI] [PubMed] [Google Scholar]
- 21.Stevens DL, Maier KA, Laine BM, Mitten JE. Comparison of clindamycin, rifampin, tetracycline, metronidazole, and penicillin for efficacy in prevention of experimental gas gangrene due to Clostridium perfringens. J Infect Dis. 1987 Feb;155(2):220–8. doi: 10.1093/infdis/155.2.220. [DOI] [PubMed] [Google Scholar]
- 22.Stevens DL, Maier KA, Mitten JE. Effect of antibiotics on toxin production and viability of Clostridium perfringens. Antimicrob Agents Chemother. 1987 Feb;31(2):213–8. doi: 10.1128/aac.31.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballard J, Bryant A, Stevens D, Tweten RK. Purification and characterization of the lethal toxin (alpha-toxin) of Clostridium septicum. Infect Immun. 1992 Mar;60(3):784–90. doi: 10.1128/iai.60.3.784-790.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis. 2011 Mar 15;203(6):880–8. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatnagar J, Deleon-Carnes M, Kellar KL, et al. Rapid, simultaneous detection of Clostridium sordellii and Clostridium perfringens in archived tissues by a novel PCR-based microsphere assay: diagnostic implications for pregnancy-associated toxic shock syndrome cases. Infect Dis Obstet Gynecol. 2012;2012:972845. doi: 10.1155/2012/972845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aldape MJ, Bryant AE, Katahira EJ, et al. Innate immune recognition of, and response to, Clostridium sordellii. Anaerobe. 2010 Apr;16(2):125–30. doi: 10.1016/j.anaerobe.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Hartung T, Docke WD, Gantner F, et al. Effect of granulocyte colony-stimulating factor treatment on ex vivo blood cytokine response in human volunteers. Blood. 1995 May 1;85(9):2482–9. [PubMed] [Google Scholar]
- 28.Hartung T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr Opin Hematol. 1998 May;5(3):221–5. doi: 10.1097/00062752-199805000-00013. [DOI] [PubMed] [Google Scholar]
- 29.Xing Z, Gauldie J, Cox G, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998 Jan 15;101(2):311–20. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]