

Abstract
Human immunodeficiency virus type 1 (HIV-1) envelope (Env) glycoprotein surface subunit gp120 and transmembrane subunit gp41 play important roles in HIV-1 entry, thus serving as key targets for the development of HIV-1 entry inhibitors. T20 peptide (enfuvirtide) is the first U.S. FDA-approved HIV entry inhibitor; however, its clinical application is limited by the lack of oral availability. Here, we have described the structure and function of the HIV-1 gp120 and gp41 subunits and reviewed advancements in the development of small-molecule HIV entry inhibitors specifically targeting these two Env glycoproteins. We then compared the advantages and disadvantages of different categories of HIV entry inhibitor candidates and further predicted the future trend of HIV entry inhibitor development.
Keywords: HIV, viral entry, entry inhibitor, gp120, gp41
1. INTRODUCTION
Three decades ago, HIV was identified as the pathogenic causative agent of AIDS. So far, more than 70 million people have been infected with HIV. About one half of them have died of AIDS, and 35 million people are living with HIV infection (www.unaids.org). Since the first anti-HIV drugs, azidothymidine (AZT) and zidovudine (ZDV), were approved for clinical use by the U.S. FDA in 1987 [1, 2], a series of antiretroviral therapies (ARTs), including nucleoside reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), and protease inhibitors, have been developed. The combinatorial use of 3 or 4 ARTs, the so-called “highly active antiretroviral therapy” (HAART), has resulted in significant reduction of the viral load and extension of the patients’ lives [3, 4]. However, an increasing number of patients cannot use these reverse transcriptase inhibitors (RTIs) and protease inhibitors because of the emergence of resistant HIV variants, calling for development of new anti-HIV drugs targeting different steps of the HIV life cycle, particularly HIV-1 entry into the target cell.
Two decades ago, Jiang and coworkers at the New York Blood Center [5–7], Wild and coworkers at the Duke University [8–10], and Lu and coworkers at the Massachusetts Institute of Technology [11–13] discovered that synthetic peptides derived from the C-terminal heptad repeat (CHR) region of HIV-1 were highly potent HIV fusion inhibitors. Identification of these anti-HIV peptides opened a new avenue for the development of viral entry inhibitors against HIV and other enveloped viruses with class I membrane fusion proteins, such as paramyxoviruses, including respiratory syncytial virus (RSV), human parainfluenza virus type 3, measles virus, Sendai virus, Menangle virus, mumps virus or Newcastle disease virus [14–19], simian immunodeficiency virus (SIV) [20], feline immunodeficiency virus (FIV) [21], influenza virus [22], Dengue virus [23], and those causing emerging infectious diseases, such as Ebola virus [24, 25], Nipah virus and Hendra virus [26, 27], as well as severe acute respiratory syndrome coronavirus (SARS-CoV) [28–32].
One decade ago, T20 (generic name: enfuvirtide; brand name: Fuzeon) was approved by the U.S. FDA as the first HIV entry inhibitor-based antiviral drug for use with other anti-HIV medicines to treat adults and children ages 6 – 16 years old with HIV infection [33–35](http://www.fuzeon.com). However, its clinical application is limited by the twice daily dosage of 90 mg per dose, leading to high cost and serious local injection reactions. Therefore, it is essential to develop small-molecule HIV entry inhibitors with oral availability.
Since both viral Env (gp120 and gp41) and cellular receptors (CD4 and CCR5/CXCR4) are involved in HIV-1 fusion and entry, they can all serve as attractive targets for developing HIV-1 entry inhibitors. These HIV-1 entry inhibitors can be classified as those targeting gp120 and gp41, and HIV-1 coreceptor antagonists that bind to CCR5 or CXCR4. This review will be focused on HIV-1 entry inhibitors specifically targeting gp120 and blocking gp120-CD4 interaction and those specifically targeting gp41 and blocking gp41-mediated membrane fusion. This article will not discuss nonspecific HIV attachment inhibitors, including polyanionic compounds, such as sodium cellulose sulfate (Ushercell®), Carrageenan (Carraguard®), PRO 2000, cellulose acetate phthalate and Aurintricarboxylic acid, or the carbohydrate-binding agents, such as Griffithsin, Cyanovirin-N, Microvirin, Pradimicin A and Alcian Blue. These anti-HIV agents have been thoroughly reviewed by Flores and Quesada [36]. The compounds targeting the CCR5-binding region in gp120 and the CCR5 antagonists are not discussed in this review since they have been extensively reviewed by others [37–39].
2. STRCUTURE AND FUNCTION OF THE HIV-1 ENV GP120 AND GP41
Like other enveloped viruses, human immunodeficiency virus type 1 (HIV-1), the causative pathogen of acquired immunodeficiency syndrome (AIDS) [40–42], enters into and infects host cells through an envelope glycoprotein (Env)-mediated virus-cell membrane fusion. HIV-1 Env is synthesized as a polyprotein precursor gp160, which is cleaved by a cellular protease into a surface subunit gp120 and a transmembrane subunit gp41, corresponding to residues 1–511 and 512–856 of the HXB2 gp160, respectively.
HIV-1 gp120 contains five conserved constant regions, C1–C5, and five variable loop regions, V1–V5 [43]. The crystal structure reveals that the unliganded gp120 exists in a state of conformational equilibrium, with its core exhibiting a strong intrinsic propensity to snap into the CD4-bound conformation. This propensity is modulated by gp41 interaction and the V1/V2 and V3 variable loops [44]. The core structure of gp120 in complex with the two membrane-distal domains of CD4 and the Fab fraction of the X5 antibody was resolved by Kwong’s group in 2005 (Fig. 1a)[45]. The structure contains an intact V3 loop, an immunodominant part of gp120, which plays an important role in coreceptor binding. The crystal structure of gp120 core showing the gp120 N- and C-terminus with intact gp41-interactive region was also resolved recently (Fig. 1a)[46]. It displays three topologically separate and structurally plastic layers, which are required for buffering movements of gp120 from gp41.
Fig. 1. A model of HIV-1 entry into the target cell and the crystal structures of HIV-1 gp120 and gp41 core.
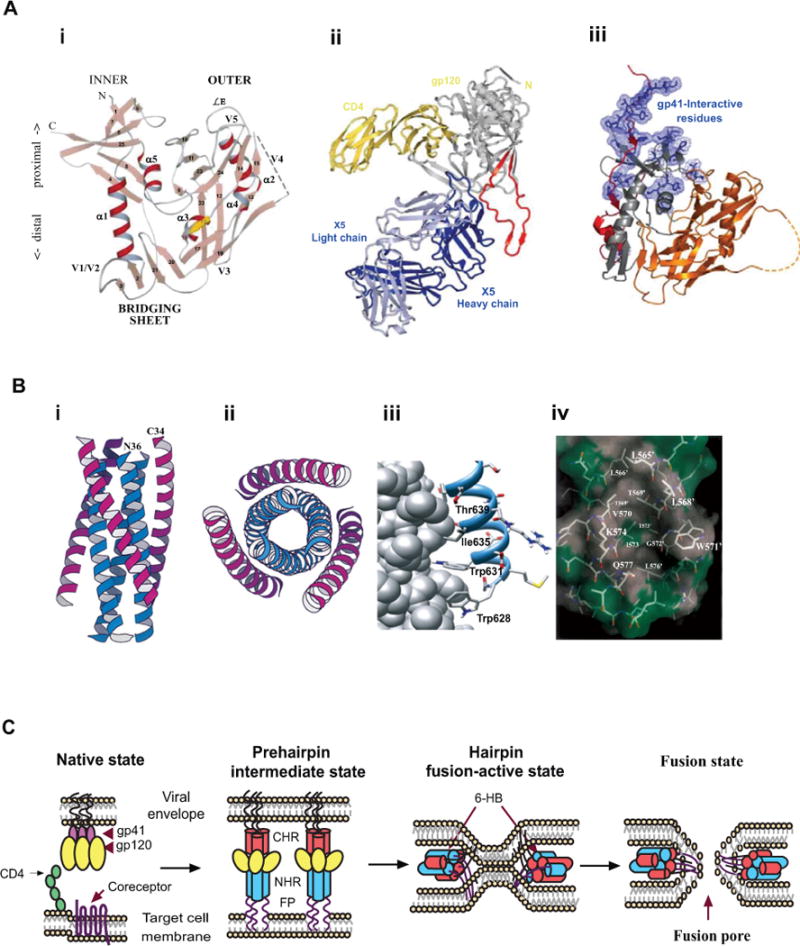
A) A model of HIV-1 fusion and entry. (i) the crystal structures of gp120 (adapted from [135] with permission); (ii) gp120 core in complex with two membrane-distal domains of CD4 and the Fab fraction of an antibody X5 (adapted from [45] with permission); (iii) the HIV-1 gp120 core in complex with CD4, and the gp41-interactive residues identified by mutation analysis (adapted from [46] with permission). B) Crystal structure of gp41 core (adapted from ref [13] with permission). (i) side-view of the 6-HB core formed by N36 and C34 peptides; (ii) top-view of the 6-HB core; (iii) three conserved hydrophobic residues (W628, W631, and I635) in the gp41 CHR region binding to the hydrophobic pocket; and (iv) the deep pocket of gp41 NHR and its interaction with the WWI motif in CHR. C) Model of HIV-1 gp41-mediated membrane fusion. HIV-1 entry into the target cell is initiated by gp120 binding to CD4 and then to CCR5 or CXCR4, followed by conformational change of gp41. The fusion peptide (FP) at the N-terminus of gp41 inserts into the target cell membrane and the NHR and CHR regions in gp41 interact with each other to form 6-HB, which brings the HIV-1 envelope and the target cell membrane into close proximity for fusion.
HIV-1 gp41 consists of an ectodomain (residues 511–684), a transmembrane domain (TM: residues 685–705) and a cytoplasmic domain (CP: residues 706–856). The ectodomain contains three important functional regions: the fusion peptide (FP: residues 512–527), the N-terminal heptad repeat (NHR: residues 536–590), and the C-terminal heptad repeat (CHR: residues 628–673). Formation of a six-helical bundle (6-HB) between NHR and CHR is an important feature of gp41 ectodomain in virus-cell membrane fusion. In 1997, three groups independently resolved the core structure of gp41 6-HB [13, 47, 48], in which three NHRs form an inner core and then associate with three CHRs in an antiparallel manner (Fig. 1b)[13]. Crystallographic analysis demonstrated that each of the grooves on the NHR trimer contains a deep hydrophobic pocket (~16-Å long, ~7-Å wide, and 5–6 Å deep), which can accommodate three conserved hydrophobic residues (W628, W631, and I635) in the CHR domain [13] and play important roles in gp41-mediated membrane fusion [49, 50] and stabilization of gp416-HB formation [51]. This pocket is an attractive target for the development of small-molecule HIV fusion inhibitors (Fig. 1b)[13, 50].
HIV-1 entry into the host cell is initiated by binding of gp120 to CD4 receptor, which causes gp120 to expose its coreceptor binding sites. Next, gp120 binds to a chemokine coreceptor CCR5 or CXCR4, depending on the subtype of HIV-1 isotopes, which brings the HIV-1 virions closer to the target cells. The binding of receptor and coreceptor causes large conformational changes in the gp120/gp41 complex, resulting in the release of the metastable gp41 subunit and, finally, triggering HIV-1-cell membrane fusion. The gp41 first inserts into target cell membrane via its FP and adopts an extended pre-hairpin intermediate (PHI) conformation. Its C-terminus anchors the viral membrane through the transmembrane domain (TD), while the N-terminus anchors the target cell membrane via FP, thus bridging virus and cell membranes (Fig. 1c). The PHI undergoes continuous conformational changes, and the NHR and CHR fold towards each other to form a 6-HB, where three NHRs form the inner core and three CHRs pack in an antiparallel manner into their respective NHRs. The energy release from 6-HB formation brings the virus and cell membrane into close apposition which finally results in the formation of a fusion pore, enabling HIV-1 to release its genomic material into the host cells to finish infection (Fig. 1c).
3. SMALL-MOLECULE HIV ENTRY INHIBITORS SPECFICALLY TARGETING GP120
3.1. BMS-378806, BMS-488043 and their analogs
Developed by Bristol-Myers Squibb Pharmaceutical Research Institute, BMS-378806, a 4-methoxy-7-azaindole derivative, 1-[(2R)-4-benzoyl-2-methylpiperazin-1-yl]-2-(4-methoxy-1H-pyrrolo[2,3-b]pyridin-3-yl)ethane-1,2-dione (Fig. 2a), is a small-molecule HIV entry inhibitor (MW = 406.5) targeting gp120 [52]. It specifically inhibited infection by a panel of R5, X4, and R5/X4 HIV-1 laboratory and clinical isolates of the B subtype with median EC50 of 0.04 μM. It showed relatively lower activity against clinical isolates of C subtype and very poor to virtually no activity against subtypes A, D, E, F, G and O. BMS-378806 had no inhibitory effect on infection by HIV-2, SIV and a panel of other viruses [53], indicating its high specificity.
Fig. 2. HIV entry inhibitors specifically targeting gp120.
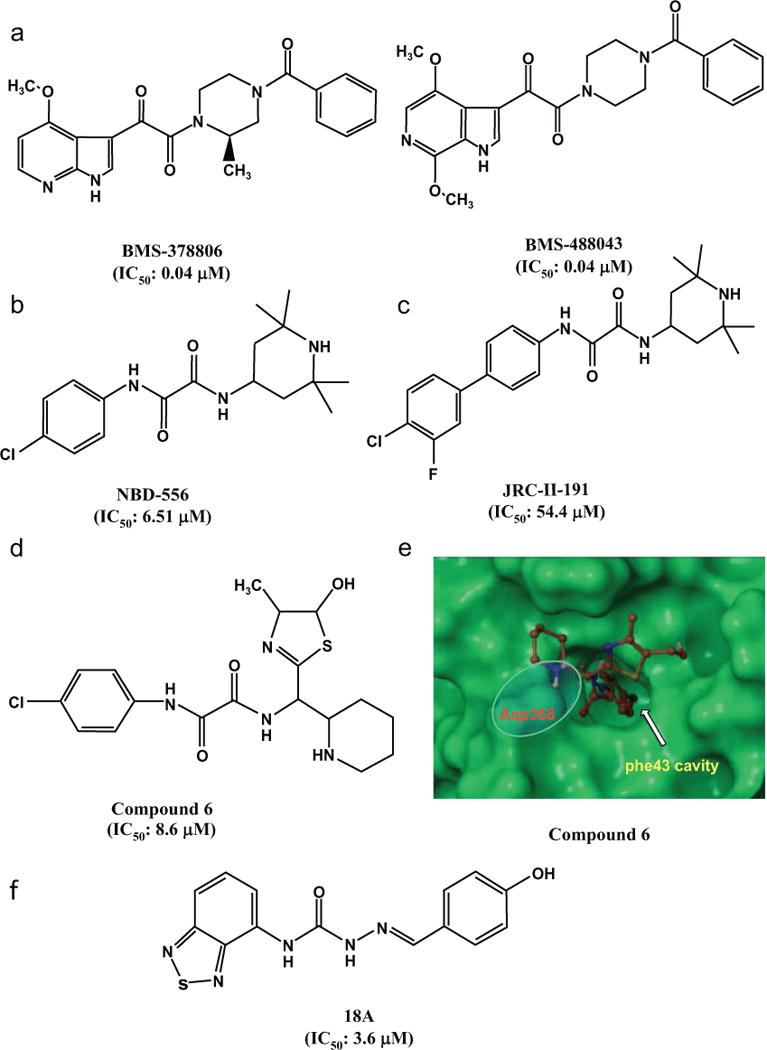
A) Chemical structures of BMS-378806 and its derivatives. B) Chemical structure of NBD-556. C) Chemical structures of JRC-II-191. D) Chemical structures of compound 6 (NBD-09027). E) GLIDE-based docking of compound 6 in the Phe43 cavity. The 4-chlorophenyl moiety of compound 6 is located deep inside the cavity, and the protonated “N” of piperidine ring is within salt-bridge (H-bond interaction) distance from Asp368 (adapted from [75] with permission).
In order to identify the molecular target of this attachment inhibitor and find out its potential mechanism, extensive in vitro experiments were performed to identify resistant mutants. Although a couple of mutations were located in the gp41 region (I595F and K655E), most of the mutations (V68A, D185N, R350K, M426L, M434I/V, M475I and S440R) were located in the gp120 region. More significantly, M434I and M475I, which play the most critical role in resistance development, are located at the CD4 binding site in gp120. The location of the mutations led researchers to believe that the putative binding site of BMS-378806 is the CD4 binding site, the Phe43 cavity in gp120 [54]. However, Si et al. suggested that BMS-378806 functions as a post-CD4 inhibitor [55]. Subsequently, the BMS group convincingly has shown that this inhibitor binds to gp120 and induces conformational change in gp120 that prevents CD4 binding [56].
BMS-378806 has a number of favorable pharmacological properties, including low protein binding, minimal human serum effect on anti-HIV-1 potency, and good oral bioavailability and safety profile in animal studies. However, the inhibitor showed poor pharmacokinetic properties, such as short half-life (t1/2), and, subsequently, its development was discontinued during Phase I clinical trials because it failed to achieve target exposure [53, 57]. Also developed by Bristol-Myers Squibb, BMS-488043, 1-(4-benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo [2,3-c]pyridin-3-yl)ethane-1,2-dione, a close analog of BMS-378806, is a small-molecule HIV-1 entry inhibitor that targets gp120. BMS-488043 binds to the HIV-1 gp120 and inhibits its interaction with CD4 at the nanomolar level [56]. It exhibited highly potent inhibitory activity against both laboratory strains and 40 clinical isolates [58]. It showed improved pharmacokinetic properties, particularly its membrane permeability and metabolic stability over BMS-378806 [58]. BMS-488043 exhibited potent in vivo efficacy against HIV-1 in a Phase IIa 8-day monotherapy. However, it also showed limited oral availability and suboptimal pharmacokinetics, resulting in its abandonment from further clinical development [59].
The clinical failure of those earlier compounds resulted in the discovery of the inhibitor BMS-626529 (1-(4-benzoylpiperazin-1-yl)-2-[4-methoxy-7-(3-methyl-1,2,4-triazol-1-yl)-1H-pyrrolo[2,3-c]pyridin-3-yl]ethane-1,2-dione). This new compound showed improved antiviral properties against a panel of laboratory strains representing CCR5, CXCR4 and dual-tropic viruses, as well as a large panel (total of 88) of clinical isolates. This inhibitor showed activity against all subtypes, except subtype A, E and group O viruses [59]. Most interestingly, non-B subtype viruses showed greater susceptibility to BMS-626529 compared to BMS-488043. In another study, the activity of BMS-626529 against CD4-independent viruses was investigated. It was demonstrated that NBD-626529 did, in fact, retain antiviral potency against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors, indicating that viruses are unlikely to become resistant to this inhibitor, which could be a pathway of developing this class of compounds against HIV-1 resistant strains [60]. Despite the improvements discussed above, this compound also suffered from low solubility and poor intrinsic dissolution properties, which were addressed by making the phosphonooxymethyl prodrug BMS-663068, another small-molecule attachment inhibitor targeting HIV-1 gp120 [59]. Its development was based on improved results from an earlier prodrug, BMS-663749, generated from the parent inhibitor BMS-488043 [61]. BMS-663068 was expected to be cleaved by alkaline phosphatase in the small intestine to release BMS-626529, the active component of the prodrug BMS-663068, which showed good membrane permeability [61]. This prodrug was tested on fifty HIV-1-infected subjects for 8 days in an open label, multiple dose parallel study. The drug was shown to be well tolerated, and it substantially declined the plasma RNA level in both antiretroviral-naïve and antiretroviral-experienced subjects. Recent 24-week Phase IIb clinical trial results showed that the prodrug was well tolerated and yielded a similar clinical outcome in treatment-experienced patients taking this prodrug when compared to a protease inhibitor used with boosted doses [62].
In vitro selection studies with BMS-626529 identified mutations L116P, A204D, M426L, M434I-V506M and M475I, which are located in the CD4 binding site in gp120 [63]. A recent study with 85 patients infected with “Non-B” HIV-1, but naïve to BMS-626529 attachment inhibitor, showed the presence of only M426L (in 10 patients) and M434I (in 11 patients) mutations. The M426L mutation was identified in the samples from 10 patients infected with subtype D (46%) and CRF01_AG (7%). The M434I mutation was identified in 15% of CRF02_AG from 11 patients, which was very similar (12.2%) to that found in the Los Alamos National Laboratory (LANL) HIV database [64].
3.2. NBD-556, NBD-09027, JRC-II-191 and their analogs
Using database screening techniques, Debnath and colleagues have identified two N-phenyl-N’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamide analogs, N-(4-Chlorophenyl)-N’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamide (NBD-556, MW=337.8 Da) and N-(4-Bromophenyl)-N’-(2,2,6,6-tetramethylpiperidin-4-yl)-oxalamide (NBD-557, MW=382.3 Da), as novel small-molecule HIV entry inhibitors targeting gp120. These compounds were found to inhibit HIV-1 infection in the low micromolar range [65], and they bound with gp120, but not with the cellular receptor CD4. Like soluble CD4 (sCD4), NBD-556 also binds gp120 with a large entropic change and keeps the conformation of gp120 functionally resembling that of gp120 bound with CD4 [65–67]. Co-crystallographic analysis showed that NBD-556 bound at a highly conserved pocket in gp120 named “Phe43 cavity” at the nexus of inner domain, outer domain, and bridging sheet minidomain of gp120 (Fig. 2b) [44], and its binding to gp120 could promote interaction with the coreceptor CCR5 [68]. Since NBD-556 binding to gp120 could induce thermodynamic changes in gp120 similar to those induced by CD4, NBD-556 has been used as a structure-specific probe to determine the CD4-bound state of gp120 and to assess the conformation of gp120 in the context of the functional viral spike [44].
To investigate the binding position of NBD-556 on gp120, Yoshimura et al [69, 69] selected HIV-1 mutants resistant to NBD-556 and sCD4 in vitro. After more than 20 passages, in the presence of NBD-556, they identified two mutations in C3 (S375N) and C4 (A433T). In the presence of sCD4, they identified seven mutations in gp120 (E211G, P212L, V255E, N280K, S375N, G380R, and G431E). The profiles of the mutations in HIV-1 variants induced in the presence of NBD-556 and sCD4 are highly similar in their three-dimensional positions. Interestingly, combinations of NBD-556 and anti-gp120 MAbs exhibited strong synergistic anti-HIV-1 activity, suggesting that NBD-556 may enhance the neutralizing activities of CD4-induced and anti-V3 antibodies [69]. By adding a fluoro group to the meta position of NBD-556, Sodroski and colleagues have developed an NBD-556 analog, JRC-II-191 [67] and its derivatives [70] (Fig. 2c). These compounds exhibited higher gp120-binding affinity and more potent anti-HIV-1 activity than NBD-556.
Crystal structures of several NBD compounds and analogs of JRC-II-191 have been solved and reported [44, 71–74]. In general, the phenyl oxalamide part in the structures showed a similar trend in binding deep inside the Phe43 cavity. However, there are some differences in the binding of Region III as a result of structural variations of this part. These structures, which provide critical information on the mode of binding of these inhibitors, are expected to help in the design of higher affinity binders and more potent inhibitors.
Most recently, Debnath and colleagues [74, 75] used synthetic chemistry, functional assessment and structure-based analysis to explore variants of each region of these inhibitors for improved antiviral properties. By altering the phenyl group and of the oxalamide linker of NBD-556, they have designed a series of compounds by replacing the tetramethyl piperidine ring with new scaffolds, such as found in compound 6 (NBD-09027) (Fig. 2d). They found that these compounds exhibited much improved anti-HIV-1 activity and stronger binding affinity to the Phe43 cavity in gp120 (Fig. 2e), suggesting that these potential next-generation HIV-1 entry inhibitors targeting gp120 may be further developed as a class of therapeutics and microbicides against HIV-1 [75]. In a recent study, some of these selected inhibitors were tested for their antiviral activity against a large panel of 53 reference HIV-1 Env pseudoviruses representing diverse clades of clinical isolates, and both NBD-09027 and NBD-11018 showed consistently improved activity against most of these clades [74].
The mechanistic study confirmed that NBD-556 and NBD-557 behave as CD4-agonists and can enhance infectivity in CD4-negative cells, an unfavorable therapeutic trait for this class of compounds. However, surprisingly, one of the inhibitors, NBD-09027, showed much reduced agonist properties in both biophysical and functional assays. In order to understand the binding mode of this compound, we first generated the resistant mutants using NBD-09027 and a relatively more soluble analog, NBD-11008. At both 44 μM and 55 μM doses, we identified three amino acid mutations in three sites: two adjacent substitutions (NN301-301KI), a substitution in the CD4 binding site (K432R), and one in gp41 (V782L). On the other hand, with NBD-11008, we identified four amino acid mutations at a dose of 53 μM: V208I located in the gp120 V2 loop, S405L located in the V4 loop and two adjacent substitutions observed with NBD-09027 (NN301-302KI) located in the N-linked glycosylation sites of the V3 loop. At the higher dose of 74 μM, we detected identical amino acid mutations with one additional mutation, S375N, observed with low frequency (2/5) in the CD4-binding site. The resistance study confirmed that the binding site of these inhibitors is the CD4-binding site on gp120.
In order to further confirm the binding mode of these inhibitors and understand the structural basis of improved antiviral potency, we determined the crystal structures of NBD-09027 and NBD-10007 in complex with HIV-1 clade A/E93TH057 H375S gp120 core at 2.5 Å and 2.2 Å, respectively. We had to use the mutated version of gp120 core because NBD compounds do not bind to residue H375, which is located in the Phe43 cavity. As expected, both NBD-09027 and NBD-10007 bound to the Phe43 cavity in a manner similar to that observed for NBD-556. However, the new moiety that replaced the tetramethyl piperidine ring had notably different orientation from that observed for NBD-556. The piperidine N in the newer analogs is now located in close proximity (4.4 Å) to D368, but not quite close enough to have H-bond interaction [74]. Nevertheless, the structures provide the knowledge required to design analogs with much improved activity and with CD4-antagonist properties.
3.3. An acylhydrazone-containing small molecule, 18A, with unique mechanism
Herschhorn et al. recently reported the identification of an acylhydrazone-containing small molecule, 18A (Fig. 2f) by screening 212,285 compounds from known bioactive collections and commercial libraries using a cell-cell fusion assay to identify potential molecules as entry inhibitors [76]. A similar second assay was used to distinguish potential fusion inhibitors from cytotoxic and non-specific inhibitors.
18A, when tested against diverse recombinant HIV-1 pseudotyped with the envelope glycoproteins of different isolates, showed broad-spectrum antiviral activity. The activity was measured against 30 CCR5-dependent HIV-1 isolates representing clades A, B, C, D and AE. The activity range was modest from 1.5–36.8 μM. There was no difference observed in activity against A, B, C and D clade viruses; however, viruses from clade AE showed poor activity. This compound showed inhibition against HIV-2 although the activity was poor. There was no noticeable activity against A-MLV, the control virus used for specificity. 18A was also tested against CXCR4-dependent isolates representing only clade B and its activity was much worse compared to CCR5-dependent isolates (10.5 – 26.2 μM).
Experiments with CD4-negative, CCR5-expressing cells with HIV-1 ADA N197 showed that 18A could protect the infection indicating that the activity of this molecule is not dependent on CD4. Due to low selectivity index 18A could not be used to study resistance which could have helped to map the target of this molecule. However, the study reports the effect of 18A on infectivity of a large number of known gp120 mutants including some from BMS-806 resistance. 18A inhibited all the mutants tested; however, ~5-fold decrease of IC50 values have been shown with mutants at the V1/V2 and β20-β21 regions in gp120, which are located proximal on a HIV Env trimer [77]. Subsequently the authors demonstrated that 18A inhibits the CD4 induced conformational rearrangement of V1/V2 region of gp120 but does not interfere with the binding of CD4 to CCR5.
Despite the fact that inhibitory effect of 18A is in the modest range its unique mechanism demonstrates its value as a probe to study the different conformational states of gp120 and their role in HIV-1 entry. In addition, 18A also represents a reasonable lead for further optimization to convert it to a more potent gp120 targeted entry inhibitor with a unique mechanism never reported before.
4. SMALL-MOLECULE HIV ENTRY INHIBITORS TARGET GP41
Compared to the peptide-based HIV fusion inhibitors, the small-molecule HIV fusion/entry inhibitors targeting gp41 have more advantages in terms of lower cost, longer half-life in vivo and oral availability, making them more convenient to use. Therefore, the development of novel small-molecule HIV fusion inhibitors is urgently needed.
As mentioned above, the deep hydrophobic pocket in the grooves on the surface of the gp41 NHR-trimer accommodates three conserved hydrophobic residues (W628, W631, and I635) in the gp41 CHR region [13, 47, 48], which is the most important target site for identification of small-molecule HIV fusion/entry inhibitors [49, 50].
4.1. ADS-J1 and its resistance profile
Using a virtual screening program, DOCK3.5, against the gp41 pocket, we screened a database from ComGenex consisting of the chemical structures of 20,000 organic compounds. After screening, we selected 200 top-scoring compounds for in-depth inspection of their interactions with the amino acid residues in the hydrophobic cavity and neighboring regions by molecular visualization techniques. Finally, we purchased 16 commercially available compounds to test their inhibitory activity on gp41 6-HB formation using a sandwich enzyme-linked immunosorbent assay (ELISA) with a 6-HB-specific monoclonal antibody (MAb), NC-1 [78, 79], and on HIV-1 Env-mediated cell-cell fusion, as well as HIV-1 replication. We found that two of these compounds, 7-[6-phenylamino-4-[4-[(3,5–disulfo–8-hydroxynaphthyl)azo]–2–methoxy–5-methylphenylamino]-1,3,5–triazine–2-yl]–4–hydroxyl–3-[(2–methoxy–5-sulfophenyl)azo]–2-naphthalene sulfonic acid (ADS-J1) and 5-[(4–chloro–6–phenylamin-1, 3, 5–triazine–2-yl)-amino]–4–hydroxyl–3-[(4–methyl–6-sulfophenyl)azo]-2,7-naphthalene disulfonic acid (ADS-J2) ((Fig (3a)), could effectively block 6-HB formation and inhibit HIV-1 Env-mediated cell fusion and the cytopathic effect of HIV-1 replication [80].
Fig. 3. Chemical structures of HIV entry inhibitors targeting gp41.
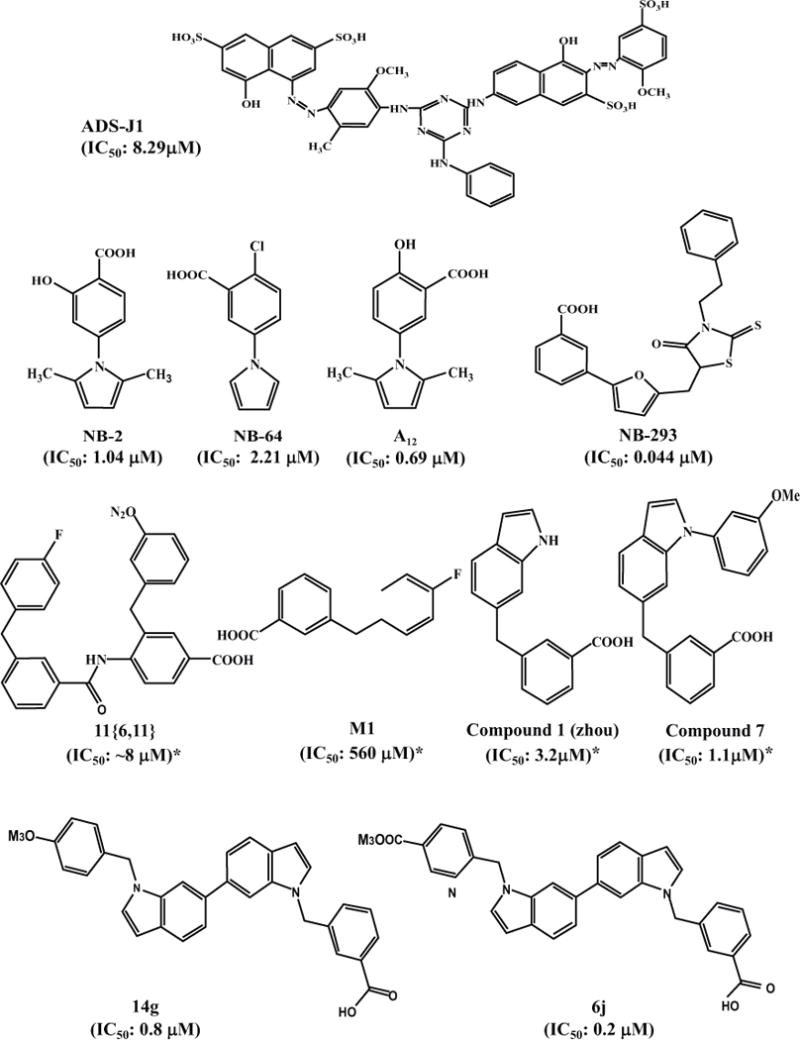
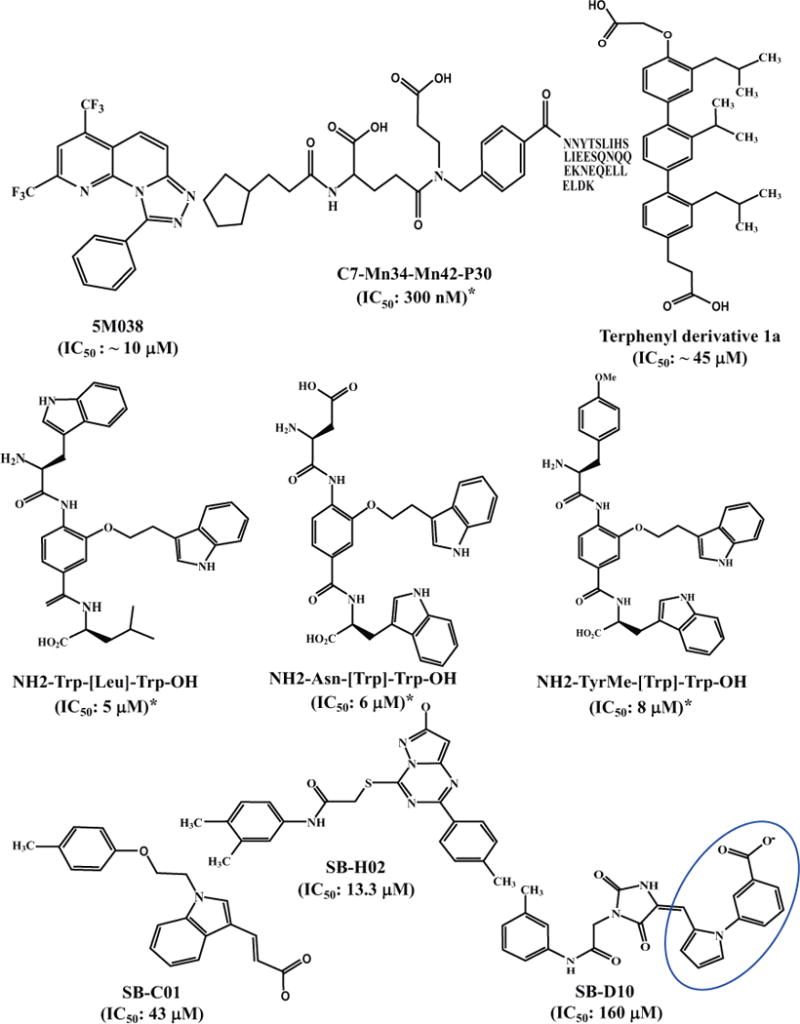
IC50 values with “*” were calculated based on the data from the experiments for inhibiting HIV-1 Env-mediated cell-cell fusion, while those without “*” were calculated based on the data from the experiments for inhibiting HIV-1 infection.
Mechanistic studies suggest that ADS-J1 inhibits HIV-1 Env-mediated membrane fusion by targeting the gp41 NHR domain, particularly the hydrophobic pocket region. First, docking analyses have shown that the hydrophobic groups (phenyl and naphthalene) of ADS-J1 interact with the hydrophobic residues (L568, V570, W571) in the pocket and that one of the negatively charged groups (SO3H) in ADS-J1 is in close proximity to a positively charged residue, K574, located around the pocket. ADS-J2 can interact with K574, but it cannot interact with the hydrophobic residues in the pocket, resulting in lower antiviral activity than ADS-J1. ADS-J13, which has no anti-HIV-1 activity, contains no acidic group for interaction with K574 [80]. Second, ADS-J1 inhibits HIV-1 infection in the early stage of entry by targeting viral Env as shown by time-of-addition and time-of-removal assays. Third, ADS-J1 does not block gp120-CD4 binding, but rather significantly inhibits gp41 6-HB, as demonstrated by ELISA, native polyacrylamide gel electrophoresis and circular dichroism analyses. Fourth, ADS-J1 binds to a trimeric structure formed by the gp41 pocket-forming sequence, IQN17, as shown by surface plasmon resonance assay [81]. All of these results suggest that ADS-J1 inhibits HIV-1 fusion/entry by mainly targeting the gp41 pocket region.
However, Este and colleagues believed that ADS-J1 neither targets the pocket region nor other regions in gp41. Instead, they concluded that ADS-J1 inhibits HIV-1 entry by targeting gp120 based on its failure to induce resistant mutations in the pocket region of gp41, inducing, instead, mutations in multiple regions of gp120, particularly the highly variable V3 loop region [82, 83].
However, we believe that their results may have different interpretation since HIV-1 escape does not necessarily occur at the binding site of a drug. For instance, vicriviroc, the CCR5 antagonist, could induce vicriviroc-resistant mutations in the fusion peptide and NHR domain of gp41 [84, 85]. Unlike T20 that lacks pocket-binding domain (PBD), the CHR-peptides containing PBD, such as T2544 and T2635, have an extremely high barrier to resistance. Although they have difficulty inducing mutations in the highly conserved pocket region of gp41 sequence, they can, instead, elicit mutations in other regions of gp41 or gp120 [86, 87]. For example, Eggink et al. passaged the T20- or T1249-resistant HIV-1 in SupT1 T cell culture in the presence of the PBD-containing CHR-peptide T2635 for 6 months and obtained several T2635-resistant strains with multiple mutations in several regions of gp41, except its pocket region, and some regions in gp120 [87]. We found that T2635-resistant HIV-1 mutants were also resistant to ADS-J1 and that their resistance to T2635 and ADS-J1 was closely correlated [88]. We then constructed pseudoviruses with mutations at positions 64, 66 and 67 in the gp41 pocket region (Q64A, Q64L, A67G, A67S, and Q66R) and compared their sensitivity to T20, a CHR-peptide without PBD; C34 and CP32M, CHR-peptides with PBD; and ADS-J1. We found that these mutants were highly resistant to C34, CP32M, and ADS-J1, but relatively sensitive to T20, suggesting that ADS-J1, C34, CP32M and T2635 share the same target: the pocket region of the HIV-1 gp41 [88–90]. Subsequently, we constructed a gp41 NHR-trimer by conjugating the NHR-peptide N36 with a trimerization motif, foldon (Fd), termed N36Fd, as previously described [91], and several N36Fd mutants, including N36(Q64A)Fd, N36(Q64L)Fd, N36(A67G)Fd, N36(A67S)Fd, and N36(Q66R)Fd. We found that the binding affinity of the mutant N36Fd trimers to ADS-J1 was lower than that of the wild-type N36Fd trimer [88], further confirming that the gp41 pocket region is the main target of ADS-J1.
4.2. NB-2, NB-64, A12, GLS-22, and analogs
To find more potential lead compounds, we have established anELISA [78, 79] for identification of compounds that block gp41 6-HB formation between the NHR-peptide N36 and CHR-peptide C34 using a gp41-specific polyclonal antibody for coating and 6-HB-specific monoclonal antibody (NC-1) for detection [78, 79]. Although this method is sensitive and reliable, it is not very convenient for high-throughput screening (HTS) since this assay took about 6.5 h for a single screening cycle, and most culture steps had to be completed at 37 °C. We therefore developed a fluorescence-linked immunosorbent assay (FLISA)-based HTS using NC-1 to capture 6-HB formed by N36 and fluorescence-labeled C34 [92]. This assay is much more rapid with1.5 h for one screening cycle and more convenient since the experiment can be conducted at room temperature [92].
Using this rapid assay to screen a library consisting of 30,040 “drug-like” compounds, we identified two small-molecule fusion inhibitors, NB-2 (Mr = 231) and NB-64 (Mr = 222) ((Fig (3a)) with inhibitory activity at low μM level against a broad spectrum of primary HIV-1 isolates (clades A to G and group O) and laboratory-adapted HIV-1 strains (IIIB, RF, SF2, and AZT-R)[93]. They were also effective in blocking gp41 6-HB formation and gp41-mediated cell-cell fusion. Both NB-2 and NB-64 are N-substituted pyrrole derivatives and contain a COOH group. Removing the COOH group from these two compounds resulted in complete loss of their inhibitory activity [93], suggesting that this acid group is critical for their anti-HIV-1 activity. Molecular docking analyses suggest that NB-2 or NB-64 bind with the gp41 hydrophobic pocket through their hydrophobic group. At the same time, their COOH group interacts with the positively charged residue K574 on the gp41 NHR domain to form a strong salt bridge.
To optimize the structure of NB-64, a series of small-molecule fusion inhibitors with different functional groups were designed and synthesized. Some of them have shown potent inhibitory activities. Representative compounds include A12 [94], GLS-22, and GLS-23 [95].
4.3. NB-206 and analogs
Since the deep hydrophobic pocket is big enough to accommodate a molecule with a molecular weight of about 600 daltons [50], both NB-2 and NB-64 may be too small to occupy this entire hydrophobic pocket. We thus hypothesized that analogous compounds of NB-2 or NB-64 with increased molecular sizes might occupy much more space in the pocket, resulting in more efficacy than that shown by either NB-2 or NB-64 in blocking 6-HB formation and inhibiting HIV-1 fusion with the target cells. Therefore, we designed and synthesized a series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidiny-lidenemethyl) furans with higher molecular size (437–515 Da). Among the 15 compounds tested, we found that 11a (NB-293), 11b, and 11d (NB-206) showed potent antiviral activity against both primary and laboratory-adapted HIV-1 strains at the nanomolar level ((Fig (3a)) [96].
Based on the chemical structure of NB-206, we then designed and synthesized a series of 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl) phenyl) thiazolidin-4-ones (12a-o) and 2,5-disubstituted furans/pyrroles (5a–h). While all the 5a–h compounds showed much lower anti-HIV-1 activity than NB-206, two of the 12a-o compounds, 12l and 12m, exhibited much better anti-HIV-1 activity at low nanomolar level and high selectivity indexes (SI: > 2,000) ((Fig (3a)) [97].
4.4. Indole-based compounds as small-molecule HIV-1 fusion inhibitors targeting gp41 identified by a fluorescent resonance energy transfer (FRET)-based screening assay
As previously described, the transiently exposed NHR-trimeric conformation at the fusion-intermediate state is very unstable, and N-peptides tend to aggregate under physiological condition [11, 98, 99]. Therefore, establishing an HTS assay using the transiently exposed NHR coiled coil in solution is very challenging. To address this challenge, researchers have employed several strategies, including the design of 5-Helix protein, which contains 5 of the 6 helices that constitute the gp41 trimer-of-hairpins linked into a single polypeptide [100], the addition of a soluble trimeric motif, GCN4 or foldon to a NHR peptide, in order to stabilize the coiled coil [91, 101, 102], and the covalent linking of the peptides in the NHR-trimeric coiled coil [103].
Gochin and colleagues used a fluorescence resonance energy transfer (FRET) technique to screen for HIV-1 fusion inhibitors targeting the gp41 core [104–107]. This HTS assay was designed on the basis of competitive inhibition between an env2.0 as a target and FRET acceptor and a modified C-peptide (C18-Aib-LY) as a ligand and FRET donor. We first designed a 31-mer N-peptide containing the pocket-forming sequence as target and then modified its N-terminus with Tris–2,2′-bipyridine-5-carboxy (BPY) to design BPY-N-peptide. The BPY-N-peptide formed a stable trimer with ferrous ions [FeII-(BPY–N-peptide)3] or with env2.0. Env2.0 is magenta and has an absorbance maximum at 540 nm. It can quench fluorescence emission at this wavelength through FRET. A C18-Aib-LY probe was designed by selecting an 18-mer C-peptide (C18-Aib) that contained the PBD and bound specifically to the N-peptide sequence of env2.0 with an additional C-terminal cysteine for modification. Lucifer yellow dye was modified at the C-terminus of C18-Aib. It has a fluorescence emission maximum at 540 nm when excited at 425 nm. The fluorescence can be quenched by FRET when bound to env2.0, causing a detectable signal change. A competitive assay was developed for small-molecule HIV-1 fusion inhibitors. The assay, which is performed by mixing the acceptor and donor peptides with the compounds to be tested and then measuring fluorescence intensity, exhibits high integrity, specificity and sensitivity.
Using this HTS assay, a focused peptidomimetic small-molecule library of 180 compounds was screened, and several compounds showing inhibitory activity with IC50 at low μM level in HIV-1 Env-mediated cell-cell fusion assays were identified. Compound 11[6,11] was selected for the Structure-Activity Relationships (SAR) study [106]. Based on an active-guided SAR analysis, M1 was identified as the core structure for the anti-HIV-1 activity of compound 11[6,11] (Fig. 3a). Its interaction with the gp41 NHR deep pocket was verified by NMR spectroscopy [108]. Later, this group developed several series of new small-molecule fusion inhibitors with improved anti-HIV potency. The benzyl ring of M1 was substituted by an indole ring based on the rationale that tryptophan residue plays a critical role in the anti-HIV-1 activity of CHR-peptides. The resultant compound 1 showed significantly improved potency, with IC50 at low micromolar level in both FRET binding assay and cell-cell fusion assay. Seven derivatives of compound 1 were then synthesized, and compound 7, the most potent among them, showed an IC50 of 1.1 μM in the cell-cell fusion assay [109].
Based on the nature of gp41protein-protein interaction between NHR and CHR, small molecules with extended structure from compound 7 were designed to increase contact with the gp41 NHR target. Twenty-four compounds were synthesized, and the most potent compound, 14g, showed an IC50 of 0.8 μM in both FRET binding assay and cell-cell fusion assay. However, the anti-HIV activity of 14g was compromised in the presence of serum, possibly because of its interaction with human serum albumin [110]. Further structural optimization was performed to overcome the weakness of these indole compounds and to increase their anti-HIV-1 potency. Thirty new compounds were synthesized by modifying the benzyl rings at both ends. The most potent bis-indole compound, 6j, showed an IC50 of 0.2 μM in cell-cell fusion assay and HIV-1 replication inhibition assay using different HIV-1 strains (Fig. 3a), representing one of the most promising small-molecule HIV-1 fusion inhibitors to target gp41 [111].
4.5. 5M038, 5M041 and analogs identified by fluorescence polarization assay using gp41-5 as the target
Some recombinant proteins, which could simulate the gp41 NHR trimeric structure with the exposed pocket, have been used as templates of the target to screen small-molecule fusion inhibitors. For example, Harrison’s group constructed a five-helix bundle (5-HB), designated gp41-5, which consists of three gp41 NHR-peptides and two CHR-peptides connected by an amino acid linker. The three NHR-peptides form the inner trimeric core, while the two CHR-peptides bind to the two grooves on the NHR-trimer, leaving the pocket in one groove unoccupied to serve as a target for screening of pocket-binding compounds. A fluorescence-labeled CHR-peptide was used as a probe. Upon binding with 5-HB to form a 6HB, a change in fluorescence polarization signal occurs. Using this HTS assay, a library of 38400 compounds was screened, and two compounds, 5M038 and 5M041 ((Fig (3b)), were found to inhibit HIV-1 Env-mediated cell–cell fusion with IC50 values of ~10 μM [112].
4.6. HIV fusion inhibitors targeting gp41 pocket based on alpha-helical mimicry
Harrison and colleagues also used a novel approach to identify gp41 pocket-binding α-helix mimetics [113, 114]. To achieve this goal, they developed a combinatorial library of 61,275 compounds using split-pool synthesis compatible with recursive deconvolution. Each of these compounds consists of three building blocks, including cap (C), monomer 1 (M1) and monomer 2 (M2). Each of the C-M1-M2 compounds was then linked to the N-terminus of CHR-peptide C30, i.e., residues 636–665, corresponding to the gp41 CHR without the PBD. The C-M1-M2 compound and the C30 peptide in each of the hybrid molecules were expected to bind the hydrophobic pocket and the groove on the gp41 NHR-trimer, respectively. One of the hybrid molecules, C7-Mn34-Mn42-P30 (Fig 3b), could inhibit HIV-1 Env-mediated cell-cell fusion with IC50 of 300 nM, while the C30 peptide alone exhibited no significant inhibitory activity [113]. X-ray crystallographic analyses demonstrated that the C7-Mn34-Mn42 motif in the hybrid molecule was able bind to the gp41 pocket via two models [114].
The 3,2′,2″-terphenyl derivative has been reported as an effective mimic of the surface functionality of an α-helix [115]. Based on these findings, Ernst et al. [116] designed a substituted terphenyl scaffold to mimic the surface of an α-helix to screen HIV-1 fusion inhibitors targeting the gp41 pocket. One of the 3,2′,2″-terphenyl derivative compounds, 1a, which mimics the side chains of an i, i+4, i+7 dad hydrophobic surface, was found to inhibit HIV-1-mediated cell-cell fusion and 6-HB formation at low μM concentration (Fig. 3b).
Using a similar approach, Whitby et al. [117] have established a comprehensive α-helix mimetic library. In this template, 20 natural amino acids were substituted on the three positions of the side chains of the α-helix. Together with all combinations, an 8000-member library (20×20×20) was created in order. Using the established FRET-based screening assay, we scanned the library to identify the combinations that could bind with the gp41 NHR hydrophobic pocket and exhibit effective inhibitory activity against HIV-1. Using a step-by-step approach, we found that three of the α-helix mimetics, H2N-Trp-[Trp]-Leu-OH, H2N-Asn-[Trp]-Trp-OH, and H2N-TyrMe-[Trp]-Trp-OH, showed submicromolar Ki in the FRET-based binding assay and IC50 values ranging from 5 to 8 μM in cell-cell fusion assay (Fig. 3b).
4.7. Other approaches
Using the DOCK6 program, Rizzo and colleagues screened about 500,000 compounds from the ZINC database against the gp41 deep pocket [118]. Based on the docking score, they purchased 115 compounds and tested anti-HIV-1 activity. They identified 7 leads with favorable binding affinity and cytotoxicity profiles, as well as good inhibitory activity on HIV-1 Env-mediated cell-cell fusion [119]. For example, compounds SB-C01 and SB-H02 had a binding affinity (Ki) of 13 and 0.46 μM, respectively, and IC50 for inhibiting cell-cell fusion of 43 and 13.3 μM, respectively. Notably, one of the compounds, SB-D10 ((Fig (3b)), contained an N277 substituted pyrrole scaffold, which is remarkably similar in structure to NB-64 [93]. Its acid group could also form a salt bridge with K63, although its anti-HIV-1 activity was found to be much lower than that of NB-64.
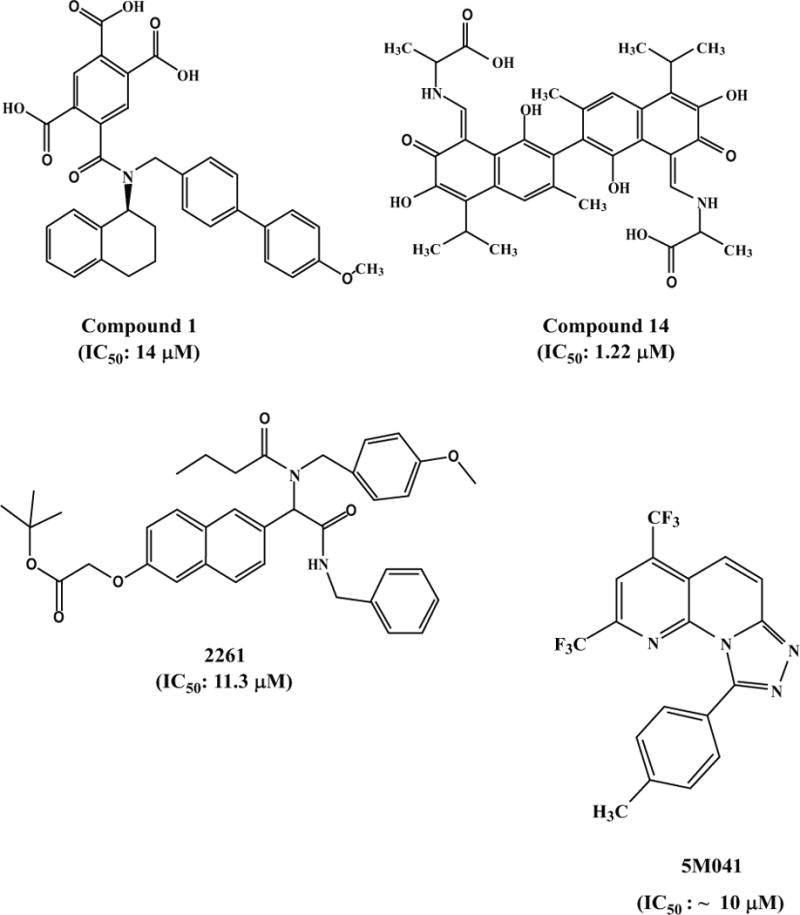
Stewart et al. at Abbott Laboratories [120] used NMR screening to identify anti-HIV-1 benzamide, compound 1, which inhibited HIV-1 Env-mediated cell-cell fusion with an IC50 of 14 μM (Fig. 3c). Yang et al. at Renmin Hospital of Wuhan University, China [121], found that the gossypol derivatives exhibited potent inhibitory activity against HIV-1 by targeting gp41. One of them, compound 14, inhibited HIV-1IIIB infection in TZM-bl cells with IC50 of 1.22 μM (Fig. 3c)[121]. Interestingly, these gossypol derivatives are also effective in inhibiting infection of H5N1 avian influenza virus. Xu et al at the Scripps Research Institute [122] designed and synthesized small-molecule libraries capable of disrupting biologically relevant protein-protein interactions, termed “credit-card” libraries. From these libraries, they identified a compound, 2261, with inhibitory activity against the HIV-1 gp41 6-HB formation, HIV-1 replication and HIV-1 Env-mediated cell-cell fusion at low micromolar concentrations (Fig. 3c).
5. CONCLUSION AND PROPECT
Discovery of the highly potent anti-HIV-1 peptides derived from the HIV-1 gp41 CHR domain in the early 1990s has opened a new avenue for developing viral fusion/entry inhibitors targeting the viral envelope glycoproteins [6, 10]. Since then, a series of peptide viral fusion/entry inhibitors against other enveloped viruses with class I membrane fusion proteins, such as SARS-CoV [28–32], Middle East respiratory syndrome coronavirus (MERS-CoV) [123], Ebola virus [24, 25], and RSV [16], have been identified. However, lack of oral availability is the major weakness of the peptide-based antiviral drugs. Therefore, development of small-molecule HIV fusion/entry inhibitors is urgently needed.
Using HIV-1 gp120 as a target, a series of small-molecule HIV-1 entry inhibitors, e.g., BMS-378806, NBD-556, and compound 6, have been identified. These compounds inhibit HIV-1 entry by blocking the interaction between gp120 and CD4. But none of them has been approved for clinical use.
Using HIV-1 gp41 as a target, we and others have identified several sets of small-molecule compound-based HIV fusion/entry inhibitors, such as ADS-J1, NB-2, NB-64, A12, GLS-22, SB-D10, compound 11d, compound 11(6,11), compound 14g, 5M038, compound 1a, H2N-Asn-[Trp]-Trp-OH, and C7-Mn34-Mn42. Most of these compounds are effective in blocking the interaction between the gp41 NHR and CHR domains to prevent formation of the 6-HB core, thereby inhibiting HIV Env-mediated membrane fusion and viral entry [80, 93–97, 106, 124]. Their main target is the hydrophobic pocket in grooves on the surface of the NHR-trimer at the fusion-intermediate state of gp41 [80, 93, 94, 96, 98, 124–126].
To the best of our knowledge, a number of pharmaceutical companies in European and North American countries, such as Roche, GlaxoSmithKline, Gilead Sciences, Bristol-Myers Squibb, Abbott Laboratories, Tibotec (acquired by Johnson & Johnson), BioChem Pharma (merged with Shire Pharmaceuticals) and Trimeris (merged with Synageva BioPharma) all initiated programs to develop HIV fusion inhibitors targeting the gp41 pocket about a decade ago when enfuvirtide was licensed by the U.S. FDA. Unfortunately, most of these programs were discontinued several years later because of the failure to identify drugable leads. It is generally believed that binding of a small molecule to the deep hydrophobic pocket would not be effective in blocking 6-HB formation between the viral NHR and CHR domains. However, we believe that the deep hydrophobic pocket is still an attractive target for developing small-molecule HIV fusion inhibitors. This hydrophobic pocket is highly conserved; therefore, antiviral drugs targeting the pocket are expected to have potent and broad anti-HIV-1 activity against diverse HIV-1 strains and high genetic barrier for drug resistance. The main challenge is the development of an HTS assay based on a model that can truly mimic the dynamic conformation of the pocket in the grooves on the surface of the NHR-trimer formed transiently at the fusion-intermediate state. Some of the artificially designed constructs with exposed gp41 pocket, to which a CHR-peptide can bind with unusually high binding affinity, such as 5-Helix [100, 127], may not be very practical for the primary screening of lead compounds with relatively low binding affinity to the pocket. The ELISA-, FLISA- and FRET-based HTS are more reliable for screening of small-molecule HIV fusion inhibitors that block 6-HB formation by targeting the hydrophobic pocket of gp41 [78, 92, 106].
All accumulated evidence now suggests that the hydrophobic pocket in the groove on the gp41 N-trimer is the most important target for developing peptide- and small-molecule compound-based HIV fusion inhibitors targeting gp41. However, a question is why most of the active PBD-containing CHR-peptides have a length of more than 30 amino acid residues and these peptides would lose antiviral activity if a few amino acid residues at the C-terminal region are deleted [128], suggesting that other regions in the groove, apart from the pocket, may also be targeted by the CHR-peptides. It is also conceivable that a non-PBD sequence in a CHR-peptide is required for the antiviral activity of the PBD-containing CHR-peptides. For example, the peptide-compound conjugate C7-Mn34-Mn42-P30 contains a small compound C7-Mn34-Mn42 that can effectively bind to the gp41 pocket, as confirmed by the X-ray crystallographic analyses [114], and a 30-mer CHR-peptide P30, which binds to the groove on the gp41 N-trimer, except the pocket. While C7-Mn34-Mn42-P30 exhibits potent HIV-1 inhibitory activity, neither C7-Mn34-Mn42 nor peptide P30 alone display antiviral activity [113], further confirming that both the pocket and other regions in the groove are required for the PBD-containing CHR-peptide-mediated anti-HIV-1 activity. However, some short peptides, such as the 15-mer PIE7 peptide [129, 130], and small-molecule compounds, such as NB-2 (222 Dalton) and NB-206 (470 Dalton) [93, 97], which only bind to the pocket, are effective in blocking gp41 6-HB formation and HIV entry. Furthermore, PIE12-trimer, which is derived from PIE7 and has about 900-fold stronger affinity to the gp41 pocket than PIE7, exhibits about 40-fold higher potency than PIE7 [131]. For short peptides or small-molecule compounds having only PBD, these findings suggest that their binding affinity must be strong enough to compete with the PBD of the viral gp41 CHR to bind to the pocket. Therefore, their potency against HIV fusion/entry is closely associated with their binding affinity to the gp41 pocket, and identification of an organic compound with extremely high binding affinity to the gp41 pocket is the major challenge for developing small-molecule HIV fusion inhibitors targeting gp41.
In addition to the deep hydrophobic packet on the gp41 NHR-trimer, other regions in gp41, such as the fusion peptide (FP) and membrane-proximal external region (MPER), may also serve as targets for development of small molecule HIV fusion inhibitors. Munch et al [132] has reported that a 20-residue peptide, designated VIRIP, derived from the C-proximal region of human alpha1-antitrypsin could interact with FP at the N-terminus of the HIV-1 gp41 and inhibit HIV-1 Env-mediated membrane fusion. Several human monoclonal antibodies targeting the HIV-1 gp41 MPER, such as 2F5 [133], 4E10 [134], and 10E8 [135], exhibited potent and broad HIV-1 neutralizing activity. More interestingly, Huang et al [136] have identified a human monoclonal antibody, 35O22, with broad and potent HIV-1 neutralization activity by binding the gp41-gp120 interface. These findings suggest that like these antibodies, small molecule compounds may also bind to these regions, if they contain pockets, and block HIV-1 fusion with and entry into the target cell.
Compared with the HIV fusion/entry inhibitors targeting gp41, those targeting gp120 are expected to be more effective against HIV infection because the CD4 binding site is exposed at the native state of gp120, while the CHR binding sites in gp41 are exposed only after CD4 binding to gp120, and this exposure is transient, lasting only a few minutes. However, the CD4-binding site in gp120 is less conserved than the CHR-binding sites, especially the hydrophobic pocket region. Therefore, HIV entry inhibitors targeting gp41 pocket region are expected to have broader antiviral spectrum than those targeting gp120. Although BMS-378806 is much more potent than NBD-556/557, it has lower genetic barrier to drug resistance than NBD-556/557 because its binding site in gp120 is less conserved than that of NBD-556 (Phe43 pocket) [53]. Nevertheless, CD4 antagonists, such as NBD-09027 and NBD-11018, are generally more effective against HIV-1 entry than the CD4 agonist, such as NBD-556 and NBD-557, because these CD4 agonists may enhance infectivity in CD4-negative cells. Therefore, Phe43 pocket-binding compounds, particularly the CD4 antagonists, and the gp41 pocket-binding compounds with high binding affinity are expected to be developed as the clinically applicable HIV entry inhibitors targeting gp120 and gp41, respectively.
Acknowledgments
This study was supported by funds from the Natural Science Foundation of China (81173098 and 81361120378 to SJ, 81373456 to LL), and 973 program of China (2012CB519001 to SJ) and Shanghai Pujiang Program (13PJD004) and NIH Grant RO1 AI104416 to AKD.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Fischl MA, Richman DD, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, Leedom JM, Groopman JE, Mildvan D, Schooley RT. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N Engl J Med. 1987;317(4):185–191. doi: 10.1056/NEJM198707233170401. [DOI] [PubMed] [Google Scholar]
- 2.Brook I. Approval of zidovudine (AZT) for acquired immunodeficiency syndrome. A challenge to the medical and pharmaceutical communities. JAMA. 1987;258(11):1517. [PubMed] [Google Scholar]
- 3.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA, Deutsch P, Holder D, Schleif WA, Condra JH. Treatment with Indinavir, Zidovudine, and Lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337(11):734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 4.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387(6629):188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 5.New York Blood Center. Synthetic peptides as inhibitors of HIV-1. 1992 US5,444,044. [Google Scholar]
- 6.Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365(6442):113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 7.Jiang S, Lin K, Strick N, Neurath AR. Inhibition of HIV-1 infection by a fusion domain binding peptide from HIV-1 envelope glycoprotein gp41. Biochem Biophys Res Commun. 1993;195(2):533–538. doi: 10.1006/bbrc.1993.2078. [DOI] [PubMed] [Google Scholar]
- 8.Duke University. Synthetic peptide inhibitors of HIV transmission. 1993 US5,464,933. [Google Scholar]
- 9.Wild C, Greenwell T, Matthews T. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res Hum Retroviruses. 1993;9(11):1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 10.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91(21):9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2(12):1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 12.Lu M, Kim PS. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J Biomol Struct Dyn. 1997;15(3):465–471. doi: 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- 13.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 14.Rapaport D, Ovadia M, Shai Y. A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 1995;14(22):5524–5531. doi: 10.1002/j.1460-2075.1995.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR., Jr Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc Natl Acad Sci USA. 1996;93(5):2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang E, Sun X, Qian Y, Zhao L, Tien P, Gao GF. Both heptad repeats of human respiratory syncytial virus fusion protein are potent inhibitors of viral fusion. Biochem Biophys Res Commun. 2003;302(3):469–475. doi: 10.1016/s0006-291x(03)00197-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J, Li P, Wu T, Gao F, Ding Y, Zhang CW, Rao Z, Gao GF, Tien P. Design and analysis of post-fusion 6-helix bundle of heptad repeat regions from Newcastle disease virus F protein. Protein Eng. 2003;16(5):373–379. doi: 10.1093/protein/gzg041. [DOI] [PubMed] [Google Scholar]
- 18.Zhu JQ, Zhang CW, Rao Z, Tien P, Gao GF. Biochemical and biophysical analysis of heptad repeat regions from the fusion protein of Menangle virus, a newly emergent paramyxovirus. Arch Virol. 2003;148(7):1301–1316. doi: 10.1007/s00705-003-0105-x. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Zhu J, Feng MG, Tien P, Gao GF. Six-helix bundle assembly and analysis of the central core of mumps virus fusion protein. Arch Biochem Biophys. 2004;421(1):143–148. doi: 10.1016/j.abb.2003.09.037. [DOI] [PubMed] [Google Scholar]
- 20.Blacklow SC, Lu M, Kim PS. A trimeric subdomain of the simian immunodeificiency virous envelope glycoprotein. Biochemistry. 1995;34(46):114955–114962. doi: 10.1021/bi00046a001. [DOI] [PubMed] [Google Scholar]
- 21.Lombardi S, Massi C, Indino E, La Rosa C, Mazzetti P, Falcone ML, Rovero P, Fissi A, Pieroni O, Bandecchi P, Esposito F, Tozzini F, Bendinelli M, Garzelli C. Inhibition of feline immunodeficiency virus infection in vitro by envelope glycoprotein synthetic peptides. Virology. 1996;220(2):274–284. doi: 10.1006/viro.1996.0315. [DOI] [PubMed] [Google Scholar]
- 22.Lee KK, Pessi A, Gui L, Santoprete A, Talekar A, Moscona A, Porotto M. Capturing a Fusion Intermediate of Influenza Hemagglutinin with a Cholesterol-conjugated Peptide, a New Antiviral Strategy for Influenza Virus. J Biol Chem. 2011;286(49):42141–42149. doi: 10.1074/jbc.M111.254243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein DE, Choi JL, Harrison SC. Structure of a dengue virus envelope protein late-stage fusion intermediate. J Virol. 2013;87(4):2287–2293. doi: 10.1128/JVI.02957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weissenhorn W, Carfi A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2(5):605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 25.Miller EH, Harrison JS, Radoshitzky SR, Higgins CD, Chi X, Dong L, Kuhn JH, Bavari S, Lai JR, Chandran K. Inhibition of Ebola Virus Entry by a C-peptide Targeted to Endosomes. J Biol Chem. 2011;286(18):15854–15861. doi: 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y, Gao S, Cole DK, Zhu J, Su N, Wang H, Gao GF, Rao Z. Basis for fusion inhibition by peptides: analysis of the heptad repeat regions of the fusion proteins from Nipah and Hendra viruses, newly emergent zoonotic paramyxoviruses. Biochem Biophys Res Commun. 2004;315(3):664–670. doi: 10.1016/j.bbrc.2004.01.115. [DOI] [PubMed] [Google Scholar]
- 27.Porotto M, Doctor L, Carta P, Fornabaio M, Greengard O, Kellogg GE, Moscona A. Inhibition of hendra virus fusion. J Virol. 2006;80(19):9837–9849. doi: 10.1128/JVI.00736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, Xiong H, Farmar J, Debnath AK, Tien P, Jiang S. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363(9413):938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripet B, Howard MW, Jobling M, Holmes RK, Holmes KV, Hodges RS. Structural characterization of the SARS-coronavirus spike S fusion protein core. J Biol Chem. 2004;279(20):20836–20849. doi: 10.1074/jbc.M400759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Y, Lou Z, Liu Y, Pang H, Tien P, Gao GF, Rao Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J Biol Chem. 2004;279(47):49414–49419. doi: 10.1074/jbc.M408782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu J, Xiao G, Xu Y, Yuan F, Zheng C, Liu Y, Yan H, Cole DK, Bell JI, Rao Z, Tien P, Gao GF. Following the rule: formation of the 6-helix bundle of the fusion core from severe acute respiratory syndrome coronavirus spike protein and identification of potent peptide inhibitors. Biochem Biophys Res Commun. 2004;319(1):283–288. doi: 10.1016/j.bbrc.2004.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ingallinella P, Bianchi E, Finotto M, Cantoni G, Eckert DM, Supekar VM, Bruckmann C, Carfi A, Pessi A. Structural characterization of the fusion-active complex of severe acute respiratory syndrome (SARS) coronavirus. Proc Natl Acad Sci U S A. 2004;101(23):8709–8714. doi: 10.1073/pnas.0402753101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazzarin A, Clotet B, Cooper D, Reynes J, Arasteh K, Nelson M, Katlama C, Stellbrink HJ, Delfraissy JF, Lange J, Huson L, DeMasi R, Wat C, Delehanty J, Drobnes C, Salgo M. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348(22):2186–2195. doi: 10.1056/NEJMoa035211. [DOI] [PubMed] [Google Scholar]
- 34.Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ, Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in north and south America. N Engl J Med. 2003;348(22):2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 35.FDA. FDA approves Fuzeon. 2003 http://www.fda.gov/ForConsumers/ByAudience/ForPatientAdvocates/HIVandAIDSActivities/ucm125088.htm.
- 36.Flores A, Quesada E. Entry inhibitors directed towards glycoprotein gp120: an overview on a promising target for HIV-1 therapy. Curr Med Chem. 2012;20(6):751–771. [PubMed] [Google Scholar]
- 37.Baba M. Recent advances of CCR5 antagonists. Curr Opin HIV AIDS. 2006;1(5):367–372. doi: 10.1097/01.COH.0000239848.13205.2a. [DOI] [PubMed] [Google Scholar]
- 38.Westby M, van der RE. CCR5 antagonists: host-targeted antiviral agents for the treatment of HIV infection, 4 years on. Antivir Chem Chemother. 2010;20(5):179–192. doi: 10.3851/IMP1507. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Zhan P, De CE, Liu X. Recent progress in small molecule CCR5 antagonists as potential HIV-1 entry inhibitors. Curr Pharm Des. 2012;18(1):100–112. doi: 10.2174/138161212798919084. [DOI] [PubMed] [Google Scholar]
- 40.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vezinet-Brun F, Rouzioux C, Rozenbaum W, Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 41.Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, Palker TJ, Redfield R, Oleske J, Safai B. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984;224(4648):500–503. doi: 10.1126/science.6200936. [DOI] [PubMed] [Google Scholar]
- 42.Luciw PA, Potter SJ, Steimer K, Dina D, Levy JA. Molecular cloning of AIDS-associated retrovirus. Nature. 1984;312(5996):760–763. doi: 10.1038/312760a0. [DOI] [PubMed] [Google Scholar]
- 43.Starcich BR, Hahn BH, Shaw GM, McNeely PD, Modrow S, Wolf H, Parks ES, Parks WP, Josephs SF, Gallo RC. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell. 1986;45(5):637–648. doi: 10.1016/0092-8674(86)90778-6. [DOI] [PubMed] [Google Scholar]
- 44.Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci U S A. 2012;109(15):5663–5668. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Cc, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310(5750):1025–1028. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pancera M, Majeed S, Ban YEA, Chen L, Huang Cc, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proceedings of the National Academy of Sciences. 2010;107(3):1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic Structure of the Ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–428. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 48.Tan K, Liu J, Wang J, Shen S, Liu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94(23):12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ji H, Shu W, Burling T, Jiang S, Lu M. Inhibition of HIV-1 infectivity by the gp41 core: role of a conserved hydrophobic cavity in membrane fusion. J Virol. 1999;73(10):8578–8586. doi: 10.1128/jvi.73.10.8578-8586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA. 1998;95(26):15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dwyer JJ, Hasan A, Wilson KL, White JM, Matthews TJ, Delmedico MK. The hydrophobic pocket contributes to the structural stability of the N-terminal coiled coil of HIV gp41 but is not required for six-helix bundle formation. Biochemistry. 2003;42(17):4945–4953. doi: 10.1021/bi027283n. [DOI] [PubMed] [Google Scholar]
- 52.Guo Q, Ho HT, Dicker I, Fan L, Zhou N, Friborg J, Wang T, McAuliffe BV, Wang HG, Rose RE, Fang H, Scarnati HT, Langley DR, Meanwell NA, Abraham R, Colonno RJ, Lin PF. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J Virol. 2003;77(19):10528–10536. doi: 10.1128/JVI.77.19.10528-10536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100(19):11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433(7028):834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- 55.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci U S A. 2004;101(14):5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho HT, Fan L, Nowicka-Sans B, McAuliffe B, Li CB, Yamanaka G, Zhou N, Fang H, Dicker I, Dalterio R, Gong YF, Wang T, Yin Z, Ueda Y, Matiskella J, Kadow J, Clapham P, Robinson J, Colonno R, Lin PF. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J Virol. 2006;80(8):4017–4025. doi: 10.1128/JVI.80.8.4017-4025.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou N, Fan L, Ho HT, Nowicka-Sans B, Sun Y, Zhu Y, Hu Y, McAuliffe B, Rose B, Fang H, Wang T, Kadow J, Krystal M, Alexander L, Colonno R, Lin PF. Increased sensitivity of HIV variants selected by attachment inhibitors to broadly neutralizing antibodies. Virology. 2010;402(2):256–261. doi: 10.1016/j.virol.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 58.Wang T, Yin Z, Zhang Z, Bender JA, Yang Z, Johnson G, Yang Z, Zadjura LM, D’Arienzo CJ, DiGiugno PD, Gesenberg C, Yamanaka GA, Gong YF, Ho HT, Fang H, Zhou N, McAuliffe BV, Eggers BJ, Fan L, Nowicka-Sans B, Dicker IB, Gao Q, Colonno RJ, Lin PF, Meanwell NA, Kadow JF. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment. 5. An evolution from indole to azaindoles leading to the discovery of 1-(4-benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane -1,2-dione (BMS-488043), a drug candidate that demonstrates antiviral activity in HIV-1-infected subjects. J Med Chem. 2009;52(23):7778–7787. doi: 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- 59.Nowicka-Sans B, Gong YF, McAuliffe B, Dicker I, Ho HT, Zhou N, Eggers B, Lin PF, Ray N, Wind-Rotolo M, Zhu L, Majumdar A, Stock D, Lataillade M, Hanna GJ, Matiskella JD, Ueda Y, Wang T, Kadow JF, Meanwell NA, Krystal M. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother. 2012;56(7):3498–3507. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Z, Zhou N, Sun Y, Ray N, Lataillade M, Hanna GJ, Krystal M. Activity of the HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068, against CD4-independent viruses and HIV-1 envelopes resistant to other entry inhibitors. Antimicrob Agents Chemother. 2013;57(9):4172–4180. doi: 10.1128/AAC.00513-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kadow JF, Ueda Y, Meanwell NA, Connolly TP, Wang T, Chen CP, Yeung KS, Zhu J, Bender JA, Yang Z, Parker D, Lin PF, Colonno RJ, Mathew M, Morgan D, Zheng M, Chien C, Grasela D. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment 6. Preclinical and human pharmacokinetic profiling of BMS-663749, a phosphonooxymethyl prodrug of the HIV-1 attachment inhibitor 2-(4-benzoyl-1-piperazinyl)-1-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)-2-oxo ethanone (BMS-488043) J Med Chem. 2012;55(5):2048–2056. doi: 10.1021/jm201218m. [DOI] [PubMed] [Google Scholar]
- 62.Highleyman L. HIV attachment inhibitor BMS-663068 shows good safety and efficacy in phase 2b study. The 21st Conference on Retroviruses and Opportunistic Infections (CROI 2014) took place in Boston, USA, 3rd-6th March 2014. 2015 [Google Scholar]
- 63.Nowicka-Sans B, Gong YF, Ho HT. Antiviral activity of a new small molecule HIV-1 attachment inhibitor, BMS-626529, the parent of BMS663068 (Abstract 518). Poster session presented at: 18th Conference on Retroviruses and Opportunistic Infections; 2012 February 27–March 3; Boston, MA. 2015. [Google Scholar]
- 64.Charpentier C, Larrouy L, Visseaux B, Landman R, Levittas M, Storto A, Damond F, Yazdanpanah Y, Yeni P, Brun-Vezinet F, Descamps D. Prevalence of subtype-related polymorphisms associated with in vitro resistance to attachment inhibitor BMS-626529 in HIV-1 ’non-B’-infected patients. J Antimicrob Chemother. 2012;67(6):1459–1461. doi: 10.1093/jac/dks067. [DOI] [PubMed] [Google Scholar]
- 65.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N’-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339(2):213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 66.Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, III, Sodroski J, Freire E. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry. 2006;45(36):10973–10980. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Madani N, Sch÷n A, Princiotto AM, LaLonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Do Kwon Y, Huang Cc, Wyatt R, Kwong PD, Freire E;, Smith AB, III, Sodroski J. Small-Molecule CD4 Mimics Interact with a Highly Conserved Pocket on HIV-1 gp120. Structure. 2008;16(11):1689–1701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shrivastava I, LaLonde JM. Enhanced dynamics of HIV gp120 glycoprotein by small molecule binding. Biochemistry. 2011;50(19):4173–4183. doi: 10.1021/bi2002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshimura K, Harada S, Shibata J, Hatada M, Yamada Y, Ochiai C, Tamamura H, Matsushita S. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J Virol. 2010;84(15):7558–7568. doi: 10.1128/JVI.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.LaLonde JM, Elban MA, Courter JR, Sugawara A, Soeta T, Madani N, Princiotto AM, Kwon YD, Kwong PD, Schon A, Freire E, Sodroski J;, Smith AB., III Design, synthesis and biological evaluation of small molecule inhibitors of CD4-gp120 binding based on virtual screening. Bioorg Med Chem. 2011;19(1):91–101. doi: 10.1016/j.bmc.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LaLonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu X, Schon A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N;, Smith AB., III Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem. 2012;55(9):4382–4396. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.LaLonde JM, Le-Khac M, Jones DM, Courter JR, Park J, Schon A, Princiotto AM, Wu X, Mascola JR, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB., III Structure-Based Design and Synthesis of an HIV-1 Entry Inhibitor Exploiting X-Ray and Thermodynamic Characterization. ACS Med Chem Lett. 2013;4(3):338–343. doi: 10.1021/ml300407y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwon YD, LaLonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB, III, Debnath AK, Kwong PD. Crystal structures of HIV-1 gp120 envelope glycoprotein in complex with NBD analogues that target the CD4-binding site. PLoS ONE. 2014;9(1):e85940. doi: 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Curreli F, Kwon YD, Zhang H, Yang Y, Scacalossi D, Kwong PD, Debnath AK. Binding mode characterization of NBD series CD4-mimetic HIV-1 entry inhibitors by X-ray structure and resistance study. Antimicrob Agents Chemother. 2014;58(9):5478–5491. doi: 10.1128/AAC.03339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Curreli F, Choudhury S, Pyatkin I, Zagorodnikov VP, Bulay AK, Altieri A, Kwon YD, Kwong PD, Debnath AK. Design, synthesis and antiviral activity of entry inhibitors that target the CD4-binding site of HIV-1. J Med Chem. 2012;55(10):4764–4775. doi: 10.1021/jm3002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Herschhorn A, Gu C, Espy N, Richard J, Finzi As, Sodroski JG. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat Chem Biol. 2014;10(10):845–852. doi: 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013 Oct 31; doi: 10.1126/science.1245625. [Epub ahead of print]( [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang S, Lin K, Lu M. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the HIV-1 envelope glycoprotein. J Virol. 1998;72(12):10213–10217. doi: 10.1128/jvi.72.12.10213-10217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang S, Lin K, Zhang L, Debnath AK. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J Virol Methods. 1999;80(1):85–96. doi: 10.1016/s0166-0934(99)00041-5. [DOI] [PubMed] [Google Scholar]
- 80.Debnath AK, Radigan L, Jiang S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodecifiency virus type 1. J Med Chem. 1999;42(17):3203–3209. doi: 10.1021/jm990154t. [DOI] [PubMed] [Google Scholar]
- 81.Wang H, Qi Z, Guo A, Mao Q, Lu H, An X, Xia C, Li X, Debnath AK, Wu S, Liu S, Jiang S. ADS-J1 inhibits HIV-1 entry by interacting with the gp41 pocket region and blocking the fusion-active gp41 core formation. Antimicrob Agents Chemother. 2009;53(12):4987–4998. doi: 10.1128/AAC.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Armand-Ugon M, Clotet-Codina I, Tintori C, Manetti F, Clotet B, Botta M, Este JA. The anti-HIV activity of ADS-J1 targets the HIV-1 gp120. Virology. 2005;343(1):141–149. doi: 10.1016/j.virol.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 83.Gonzalez-Ortega E, Mena MP, Permanyer M, Ballana E, Clotet B, Este JA. ADS-J1 inhibits HIV-1 entry by interacting with gp120 and does not block fusion-active gp41 core formation. Antimicrob Agents Chemother. 2010;54(10):4487–4492. doi: 10.1128/AAC.00359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anastassopoulou CG, Ketas TJ, Klasse PJ, Moore JP. Resistance to CCR5 inhibitors caused by sequence changes in the fusion peptide of HIV-1 gp41. Proc Natl Acad Sci USA. 2009;106(13):5318–5323. doi: 10.1073/pnas.0811713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anastassopoulou CG, Ketas TJ, Depetris RS, Thomas AM, Klasse PJ, Moore JP. Resistance of a human immunodeficiency virus type 1 isolate to a small molecule CCR5 inhibitor can involve sequence changes in both gp120 and gp41. Virology. 2011;413(1):47–59. doi: 10.1016/j.virol.2010.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc Natl Acad Sci USA. 2007;104(31):12772–12777. doi: 10.1073/pnas.0701478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eggink D, Bontjer I, Langedijk JP, Berkhout B, Sanders RW. Resistance of human immunodeficiency virus type 1 to a third-generation fusion inhibitor requires multiple mutations in gp41 and is accompanied by a dramatic loss of gp41 function. J Virol. 2011;85(20):10785–10797. doi: 10.1128/JVI.05331-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu F, Lu L, Liu Q, Yu X, Wang L, He E, Zou P, Du L, Sanders RW, Liu S, Jiang S. ADS-J1 inhibits HIV-1 infection and membrane fusion by targeting the highly conserved pocket in the gp41 NHR-trimer. Biochim Biophys Acta. 2014;1838(5):1296–1305. doi: 10.1016/j.bbamem.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 89.Yu X, Lu L, Cai L, Tong P, Tan S, Zou P, Meng F, Chen YH, Jiang S. Mutations of Gln64 in the HIV-1 gp41 N-terminal heptad repeat render viruses resistant to peptide HIV fusion inhibitors targeting gp41 pocket. J Virol. 2012;86(1):589–593. doi: 10.1128/JVI.05066-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu L, Tong P, Yu X, Pan C, Zou P, Chen YH, Jiang S. HIV-1 variants with a single-point mutation in the gp41 pocket region exhibiting different susceptibility to HIV fusion inhibitors with pocket- or membrane-binding domain. Biochim Biophys Acta. 2012;1818(12):2950–2957. doi: 10.1016/j.bbamem.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 91.Chen X, Lu L, Qi Z, Lu H, Wang J, Yu X, Chen Y, Jiang S. Novel recombinant engineered gp41 N-terminal heptad repeat trimers and their potential as anti-HIV-1 therapeutics or microbicides. J Biol Chem. 2010;285(33):25506–25515. doi: 10.1074/jbc.M110.101170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu S, Boyer-Chatenet L, Lu H, Jiang S. Rapid and automated fluorescence-linked immunosorbent assay for high throughput screening of HIV-1 fusion inhibitors targeting gp41. J Biomol Screening. 2003;8(6):685–693. doi: 10.1177/1087057103259155. [DOI] [PubMed] [Google Scholar]
- 93.Jiang S, Lu H, Liu S, Zhao Q, He Y, Debnath AK. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob Agents Chemother. 2004;48(11):4349–4359. doi: 10.1128/AAC.48.11.4349-4359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu K, Lu H, Hou L, Qi Z, Teixeira C, Barbault F, Fan BT, Liu S, Jiang S, Xie L. Design, synthesis, and biological evaluation of N-carboxyphenylpyrrole derivatives as potent HIV fusion inhibitors targeting gp41. J Med Chem. 2008;51(24):7843–7854. doi: 10.1021/jm800869t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y, Lu H, Zhu Q, Jiang S, Liao Y. Structure-based design, synthesis and biological evaluation of new N-carboxyphenylpyrrole derivatives as HIV fusion inhibitors targeting gp41. Bioorg Med Chem Lett. 2010;20(1):189–192. doi: 10.1016/j.bmcl.2009.10.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Katritzky AR, Tala SR, Lu H, Vakulenko AV, Chen QY, Sivapackiam J, Pandya K, Jiang S, Debnath AK. Design, synthesis, and structure-activity relationship of a novel series of 2-Aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidin-ylidenemethyl)furans as HIV-1 entry inhibitors. J Med Chem. 2009;52(23):7631–7639. doi: 10.1021/jm900450n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang S, Tala SR, Lu H, bo-Dya NE, Avan I, Gyanda K, Lu L, Katritzky AR, Debnath AK. Design, synthesis, and biological activity of novel 5-((arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phenyl)thi azolidin-4-ones as HIV-1 fusion inhibitors targeting gp41. J Med Chem. 2011;54(2):572–579. doi: 10.1021/jm101014v. [DOI] [PubMed] [Google Scholar]
- 98.Cai L, Jiang S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem. 2010;5(11):1813–1824. doi: 10.1002/cmdc.201000289. [DOI] [PubMed] [Google Scholar]
- 99.Cai L, Gochin M, Liu K. Biochemistry and biophysics of HIV-1 gp41 – membrane interactions and implications for HIV-1 envelope protein mediated viral-cell fusion and fusion inhibitor design. Curr Top Med Chem. 2011;11(24):2959–2984. doi: 10.2174/156802611798808497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Root MJ, Kay MS, Kim PS. Protein design of an HIV-1 entry inhibitor. Science. 2001;291(5505):884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 101.Weissenhorn W, Calder LJ, Dessen A, Laue T, Skehel JJ, Wiley DC. Assembly of a rod-shaped chimera of a trimeric GCN4 zipper and the HIV-1 gp41 ectodomain expressed in Escherichia coli. Proc Natl Acad Sci U S A. 1997;94(12):6065–6069. doi: 10.1073/pnas.94.12.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eckert DM, Malashkevich VN, Kim PS. Crystal structure of GCN4-pIQI, a trimeric coiled coil with buried polar residues. J Mol Biol. 1998;284(4):859–865. doi: 10.1006/jmbi.1998.2214. [DOI] [PubMed] [Google Scholar]
- 103.Louis JM, Nesheiwat I, Chang L, Clore GM, Bewley CA. Covalent trimers of the internal N-terminal trimeric coiled-coil of gp41 and antibodies directed against them are potent inhibitors of HIV envelope-mediated cell fusion. J Biol Chem. 2003;278(22):20278–20285. doi: 10.1074/jbc.M301627200. [DOI] [PubMed] [Google Scholar]
- 104.Gochin M, Kiplin GR, Case MA. A metallopeptide assembly of the HIV-1 gp41 coiled coil is an ideal receptor in fluorescence detection of ligand binding. Angew Chem Int Ed Engl. 2003;42(43):5325–5328. doi: 10.1002/anie.200352006. [DOI] [PubMed] [Google Scholar]
- 105.Gochin M, Savage R, Hinckley S, Cai L. A fluorescence assay for rapid detection of ligand binding affinity to HIV-1 gp41. Biol Chem. 2006;387(4):477–483. doi: 10.1515/BC.2006.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cai L, Gochin M. A novel fluorescence intensity screening assay identifies new low molecular weight inhibitors of the gp41 coiled coil domain of HIV-1. Antimicrob Agents Chemother. 2007;51(7):2388–2395. doi: 10.1128/AAC.00150-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cai L, Balogh E, Gochin M. Stable extended human immunodeficiency virus type 1 gp41 coiled coil as an effective target in an assay for high-affinity fusion inhibitors. Antimicrob Agents Chemother. 2009;53(6):2444–2449. doi: 10.1128/AAC.00150-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Balogh E, Wu D, Zhou G, Gochin M. NMR second site screening for structure determination of ligands bound in the hydrophobic pocket of HIV-1 gp41. J Am Chem Soc. 2009;131(8):2821–2823. doi: 10.1021/ja8094558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou G, Wu D, Hermel E, Balogh E, Gochin M. Design, synthesis and evaluation of indole compounds as novel inhibitors targeting gp41. Bioorganic & Medicinal Chemistry Letters. 2010;20(5):1500–1503. doi: 10.1016/j.bmcl.2010.01.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou G, Wu D, Snyder B, Ptak RG, Kaur H, Gochin M. Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J Med Chem. 2011;54(20):7220–7231. doi: 10.1021/jm200791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou G, Sofiyev V, Kaur H, Snyder BA, Mankowski MK, Hogan PA, Ptak RG, Gochin M. Structure-Activity Relationship Studies of Indole-Based Compounds as Small Molecule HIV-1 Fusion Inhibitors Targeting Glycoprotein 41. J Med Chem. 2014;57(12):5270–5281. doi: 10.1021/jm500344y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Frey G, Rits-Volloch S, Zhang XQ, Schooley RT, Chen B, Harrison SC. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc Natl Acad Sci U S A. 2006;103(38):13938–13943. doi: 10.1073/pnas.0601036103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ferrer M, Kapoor TM, Strassmaier T, Weissenhorn W, Skehel JJ, Oprian D, Schreiber SL, Wiley DC, Harrison SC. Selection of gp41-mediated HIV-1 cell entry inhibitors from biased combinatorial libraries of non-natural binding elements. Nat Struct Biol. 1999;6(10):953–960. doi: 10.1038/13324. [DOI] [PubMed] [Google Scholar]
- 114.Zhou G, Ferrer M, Chopra R, Kapoor TM, Strassmaier T, Weissenhorn W, Skehel JJ, Oprian D, Schreiber SL, Harrison SC, Wiley DC. The structure of an HIV-1 specific cell entry inhibitor in complex with the HIV-1 gp41 trimeric core. Bioorg Med Chem. 2000;8(9):2219–2227. doi: 10.1016/s0968-0896(00)00155-3. [DOI] [PubMed] [Google Scholar]
- 115.Orner BP, Ernst JT, Hamilton AD. Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of an alpha-helix. J Am Chem Soc. 2001;123(22):5382–5383. doi: 10.1021/ja0025548. [DOI] [PubMed] [Google Scholar]
- 116.Ernst JT, Kutzki O, Debnath AK, Jiang S, Lu H, Hamilton AD. Design of a protein surface antagonist based on alpha-helix mimicry: inhibition of gp41 assembly and viral fusion. Angew Chem Int Ed Engl. 2002;41(2):278–281. doi: 10.1002/1521-3773(20020118)41:2<278::aid-anie278>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 117.Whitby LR, Ando Y, Setola V, Vogt PK, Roth BL, Boger DL. Design, synthesis, and validation of a beta-turn mimetic library targeting protein-protein and peptide-receptor interactions. J Am Chem Soc. 2011;133(26):10184–10194. doi: 10.1021/ja201878v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15(6):1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Holden PM, Kaur H, Goyal R, Gochin M, Rizzo RC. Footprint-based identification of viral entry inhibitors targeting HIVgp41. Bioorganic & Medicinal Chemistry Letters. 2012;22(8):3011–3016. doi: 10.1016/j.bmcl.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stewart KD, Huth JR, Ng TI, McDaniel K, Hutchinson RN, Stoll VS, Mendoza RR, Matayoshi ED, Carrick R, Mo H, Severin J, Walter K, Richardson PL, Barrett LW, Meadows R, Anderson S, Kohlbrenner W, Maring C, Kempf DJ, Molla A, Olejniczak ET. Non-peptide entry inhibitors of HIV-1 that target the gp41 coiled-coil pocket. Bioorg Med Chem Lett. 2010;20(2):612–617. doi: 10.1016/j.bmcl.2009.11.076. 612-7(2) [DOI] [PubMed] [Google Scholar]
- 121.Yang J, Zhang F, Li J, Chen G, Wu S, Ouyang W, Pan W, Yu R, Yang J, Tien P. Synthesis and antiviral activities of novel gossypol derivatives. Bioorg Med Chem Lett. 2012;22(3):1415–1420. doi: 10.1016/j.bmcl.2011.12.076. [DOI] [PubMed] [Google Scholar]
- 122.Xu Y, Lu H, Kennedy JP, Yan X, McAllister LA, Yamamoto N, Moss JA, Boldt GE, Jiang S, Janda KD. Evaluation of “credit card” libraries for inhibition of HIV-1 gp41 fusogenic core formation. J Comb Chem. 2006;8(4):531–539. doi: 10.1021/cc0600167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lu L, Liu Q, Zhu Y, Chan KH, Qin L, Li Y, Wang Q, Chan JF, Du L, Yu F, Ma C, Ye S, Yuen KY, Zhang R, Jiang S. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun. 2014;5:3067. doi: 10.1038/ncomms4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jiang S, Tala SR, Lu H, Zou P, Avan I, Ibrahim TS, bo-Dya NE, Abdelmajeid A, Debnath AK, Katritzky AR. Design, synthesis, and biological activity of a novel series of 2,5-disubstituted furans/pyrroles as HIV-1 fusion inhibitors targeting gp41. Bioorg Med Chem Lett. 2011;21(22):6895–6898. doi: 10.1016/j.bmcl.2011.08.081. [DOI] [PubMed] [Google Scholar]
- 125.Jiang S, Siddiqui P, Liu S. Blocking viral entry: a complementary strategy for HIV therapy. Drug Discovery Today: Therapeutic Strategies. 2004;1(4):497–503. [Google Scholar]
- 126.Liu S, Jiang S. High throughput screening and characterization of HIV-1 entry inhibitors targeting gp41: theories and techniques. Curr Pharm Des. 2004;10(15):1827–1843. doi: 10.2174/1381612043384466. [DOI] [PubMed] [Google Scholar]
- 127.Root MJ, Hamer DH. Targeting therapeutics to an exposed and conserved binding element of the HIV-1 fusion protein. Proc Natl Acad Sci U S A. 2003;100(9):5016–5021. doi: 10.1073/pnas.0936926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu S, Jing W, Cheung B, Lu H, Sun J, Yan X, Niu J, Farmar J, Wu S, Jiang S. HIV gp41 C-terminal heptad repeat contains multifunctional domains: relation to mechanisms of action of anti-HIV peptides. J Biol Chem. 2007;282(13):9612–9620. doi: 10.1074/jbc.M609148200. [DOI] [PubMed] [Google Scholar]
- 129.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99(1):103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 130.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci USA. 2007;104(43):16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Welch BD, Francis JN, Redman JS, Paul S, Weinstock MT, Reeves JD, Lie YS, Whitby FG, Eckert DM, Hill CP, Root MJ, Kay MS. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J Virol. 2010;84(21):11235–11244. doi: 10.1128/JVI.01339-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Munch J, Standker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, Pohlmann S, Chaipan C, Biet T, Peters T, Meyer B, Wilhelm D, Lu H, Jing W, Jiang S, Forssmann WG, Kirchhoff F. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell. 2007;129(2):263–275. doi: 10.1016/j.cell.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 133.Purtscher M, Trkola A, Grassauer A, Schulz PM, Klima A, Dopper S, Gruber G, Buchacher A, Muster T, Katinger H. Restricted antigenic variability of the epitope recognized by the neutralizing gp41 antibody 2F5. AIDS. 1996;10(6):587–593. doi: 10.1097/00002030-199606000-00003. [DOI] [PubMed] [Google Scholar]
- 134.Zwick MB, Jensen R, Church S, Wang M, Stiegler G, Kunert R, Katinger H, Burton DR. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. The Journal of Virology. 2005;79(2):1252–1261. doi: 10.1128/JVI.79.2.1252-1261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491(7424):406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang GY, Druz A, Doria-Rose NA, Laub L, Sliepen K, van Gils MJ, de la Pena AT, Derking R, Klasse PJ, Migueles SA, Bailer RT, Alam M, Pugach P, Haynes BF, Wyatt RT, Sanders RW, Binley JM, Ward AB, Mascola JR, Kwong PD, Connors M. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature. 2014;515(7525):138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]