

Abstract
Despite the introduction of at least 20 new antiepileptic drugs (AEDs) into clinical practice over the past decades, about one third of all epilepsies remain refractory to conventional forms of treatment. In addition, currently used AEDs have been developed to suppress neuronal hyperexcitability, but not necessarily to address pathogenic mechanisms involved in epilepsy development or progression (epileptogenesis). For those reasons endogenous seizure control mechanisms of the brain may provide alternative therapeutic opportunities. Adenosine is a well characterized endogenous anticonvulsant and seizure terminator of the brain. Several lines of evidence suggest that endogenous adenosine-mediated seizure control mechanisms fail in chronic epilepsy, whereas therapeutic adenosine augmentation effectively prevents epileptic seizures, even those that are refractory to conventional AEDs. New findings demonstrate that dysregulation of adenosinergic mechanisms are intricately involved in the development of epilepsy and its comorbidities, whereas adenosine-associated epigenetic mechanisms may play a role in epileptogenesis. The first goal of this review is to discuss how maladaptive changes of adenosinergic mechanisms contribute to the expression of seizures (ictogenesis) and the development of epilepsy (epileptogenesis) by focusing on pharmacological (adenosine receptor dependent) and biochemical (adenosine receptor independent) mechanisms as well as on enzymatic and transport based mechanisms that control the availability (homeostasis) of adenosine. The second goal of this review is to highlight innovative adenosine-based opportunities for therapeutic intervention aimed at reconstructing normal adenosine function and signaling for improved seizure control in chronic epilepsy. New findings suggest that transient adenosine augmentation can have lasting epigenetic effects with disease modifying and antiepileptogenic outcome.
Keywords: Epilepsy, Adenosine, Adenosine kinase, Astrocytes, Ictogenesis, Epileptogenesis, Epigenetics, Adenosine augmentation therapy
1. Introduction
Thirty-five years ago it was first demonstrated that endogenous adenosine may act as an endogenous anticonvulsant of the brain (Dunwiddie, 1980). Since then a large number of studies have validated the concept that adenosine acts as endogenous anticonvulsant and seizure terminator of the brain (Ault and Wang, 1986; Dragunow, 1991; Dragunow et al., 1985; Dunwiddie and Fredholm, 1984; Lee et al., 1984), not at least supported by studies showing a rise of seizure-induced endogenous adenosine coinciding with seizure termination (During and Spencer, 1992; Van Gompel et al., 2014). Neuronal excitability in the brain is modulated by activation of G protein coupled adenosine receptors (A1, A2A, A2B, A3) (Fredholm et al., 2001; Fredholm et al., 2005; Fredholm et al., 2011). Therefore, excitability depends on the equilibrium of different receptor-mediated effects, receptor expression levels, and availability of endogenous adenosine to activate the receptors. In addition, adenosine has receptor independent effects that regulate biochemical enzyme reactions and that affect epigenetic functions (Williams-Karnesky et al., 2013). Whereas different sources of adenosine from neurons and astrocytes affect synaptic versus homeostatic adenosine signaling (Cunha, 2001, 2008), overall levels of adenosine are largely under the control of metabolic clearance through the astrocyte-based enzyme adenosine kinase (ADK) (Boison, 2013; Fedele et al., 2004; Lloyd and Fredholm, 1995; Pak et al., 1994). Maladaptive processes that determine adenosine availability and signaling have been associated with the development of epilepsy and – consequently – therapeutic approaches aimed at restoring normal adenosinergic function hold promise for the therapy of epilepsy (Boison, 2008, 2012a). The following sections will review maladaptive changes of the adenosine system in epilepsy and discuss the therapeutic potential of adenosine augmentation therapies.
2. Imbalance of adenosine receptor activation
Several lines of evidence suggest that maladaptive changes in adenosine receptor signaling contribute to the pathophysiology of epilepsy. It is conceivable that any shift in the ratio of inhibitory A1R vs. stimulatory A2ARs directly affects neuronal excitability. Material covered in subsequent sections summarizes findings that show that the epileptic state is indeed characterized by decreased A1R signaling and increased A2AR signaling. However, it is currently unknown whether changes in adenosine receptor expression are cause for or consequence of epilepsy.
2.1. Adenosine A1R
Experimentally, a decrease in A1R density and the failure of endogenous adenosine-based seizure control mechanisms have been described in the rat kindling model of epilepsy suggesting the failure of endogenous seizure control mechanisms in epilepsy (Rebola et al., 2003). Receptor knockout studies have shown that mice lacking the A1R have spontaneous electrographic seizures (Li et al., 2007a) and develop lethal status epilepticus following the intrahippocampal injection of kainic acid, or a traumatic brain injury (Fedele et al., 2006; Kochanek et al., 2006). These studies directly show that A1R activation is needed to prevent seizure spread. Histopathological and biochemical analyses from specimen surgically resected from patients with intractable epilepsy show decreased expression levels of A1 receptors, suggesting that decreased A1R expression may contribute to seizure generation in human chronic epilepsy (Glass et al., 1996). Experimentally, dynamic changes in A1R signaling or expression have been described as a direct consequence of acute seizures. Desensitization of A1R responses but normal receptor levels have been described in the hippocampus of rats after status epilepticus elicited by performant path stimulation (Hamil et al., 2012), whereas upregulation of the A1R in the entorhinal cortex has been described as a response to spontaneous seizures induced by electrical stimulation (Hargus et al., 2012). In a human genomic study variants in the A1R gene have been associated with the development of posttraumatic seizures after a severe traumatic brain injury, suggesting that deficiency in A1R signaling might be associated with posttraumatic epileptogenesis (Wagner et al., 2010). Together, these data suggest that dysregulation of A1R signaling is intricately linked to the pathophysiology of epilepsy.
2.2. Adenosine A2AR
The synaptic fraction of A2ARs can mediate synaptotoxic effects of the synaptic pool of adenosine (Matos et al., 2012; Popoli et al., 2003; Silva et al., 2007), which is largely dependent on the neuronal release of adenosine or its precursor ATP (Lovatt et al., 2012). Thereby, neuronal hyperexcitability in epilepsy likely leads to enhanced synaptic A2AR activation, which could aggravate synaptotoxicity and thereby further the degeneration of normal circuitry contributing to the progressive course of epilepsy. Interestingly, genetic variants of the A2AR gene have been associated with acute encephalopathy with biphasic seizures and late reduced diffusion in children, suggesting that A2AR dysregulation promotes seizures and excitotoxic brain damage in those patients (Shinohara et al., 2013). In Wistar Albino Glaxo/Rijswijk (WAG/Rij) rats, a model of human absence epilepsy, increased expression of A2ARs in epileptic rats, but not in pre-symptomatic animals has been described suggesting that increased A2AR expression in this model supports the epileptic phenotype (D’Alimonte et al., 2009). In line with a potentially pro-convulsive role of the A2AR, A2AR knockout mice were partially resistant to limbic seizures. In conclusion, increased A2AR activation may promote the epileptic state.
2.3. Adenosine A2B and A3Rs
Little is known about the contribution of A2BRs and A3Rs in epilepsy. Using an in vitro system of tissue from human resected epileptogenic foci, which was microtransplanted into Xenopus oocytes, it was shown that the A2BR selective antagonist MRS1706 as well as the A3R selective antagonist MRS1334 reduced the run-down of GABA currents (Roseti et al., 2008). These findings suggest that cortical adenosine A2B and A3 receptors alter the stability of GABAA receptors and thereby fine-tune neuronal excitability.
3. Adenosine receptor-independent effects
In addition to adenosine receptor dependent effects, adenosine influences biochemical enzyme reactions through mass action. Adenosine is an obligatory end product of S-adenosylmethionine (SAM) dependent transmethylation reactions, which also include methyl group transfers onto DNA, catalyzed by DNA methyltransferases (Boison et al., 2002; Mato et al., 2008; Williams-Karnesky et al., 2013). ADK, by removing adenosine drives the flux of methyl groups through the transmethylation pathway; therefore, increased ADK expression directly leads to hypermethylated DNA (Williams-Karnesky et al., 2013) (Figure 1). Conversely, increased adenosine (or reduced ADK), as well as increased homocysteine, which is under tight metabolic control in the brain, shift the equilibrium of the S-adenosylhomocysteine (SAH) hydrolase reaction towards the formation of SAH (Mandaviya et al., 2014; Williams-Karnesky et al., 2013). SAH in turn is known to block DNA methyltransferase activity by product inhibition (James et al., 2002). Consequently, the intracebroventricular injection of adenosine as well as of homocysteine decreased global DNA methylation levels in the hippocampus of rats, whereas the injection of SAM increased global hippocampal DNA methylation (Williams-Karnesky et al., 2013). Through those biochemical adenosine-receptor independent effects, adenosine contributes to the homeostasis of the DNA methylome, and thereby assumes a novel function as epigenetic regulator. In support of a role of SAM/SAH-dependent regulation of seizure activity and the underlying role of methylation reactions, the convulsant L-methionine-dl-sulfoximine (MSO) leads to an increased SAM/SAH ratio, to an increase in the methylation flux, and to seizures, which can be blocked by adenosine and homocysteine (Gill and Schatz, 1985; Schatz et al., 1983; Sellinger et al., 1984). Although not directly shown, those studies suggest that SAM might have a pro-convulsive profile, a consideration that needs further investigation because SAM is widely used as a neutraceutical with antidepressive and precognitive properties (Cantoni et al., 1989; Chavez, 2000; Lu, 2000).
Figure 1. The epigenetics of epileptogenesis.
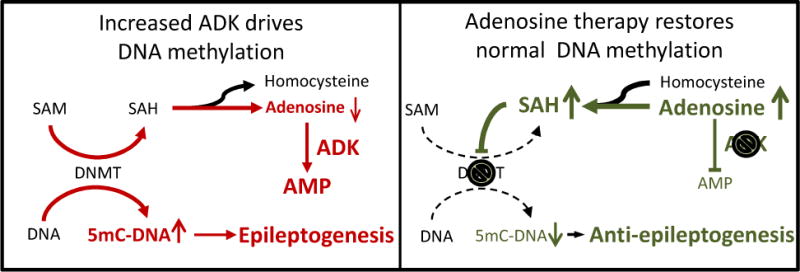
Increased ADK expression drives increased DNA methylation as a prerequisite for progressive epileptogenesis. Conversely, adenosine therapy restores normal DNA methylation nad thereby prevents epileptogenesis. For further details, please refer to the main text. SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; DNMT: DNA-methyltransferase; 5mC: 5-methylcytidine.
Importantly, epileptogenic areas of the brain are characterized by hypermethylation of the DNA (Kobow et al., 2013; Miller-Delaney et al., 2015; Williams-Karnesky et al., 2013). The ‘methylation hypothesis of epileptogenesis’ (Kobow and Blumcke, 2011) suggests that methylation changes in DNA contribute to epileptogenesis and are implicated in disease progression and maintenance of the epileptic phenotype. We recently demonstrated that a transient 10-day dose of adenosine delivered to the brain ventricles of rats after the onset of epilepsy, reversed the hypermethylated state of DNA back to methylation levels in the normal range, and prevented mossy fiber sprouting, disease progression, and progressive epileptogenesis long-term for at least three months (Williams-Karnesky et al., 2013). These findings demonstrate that changes in DNA methylation patterns are a key determinant of the progression of epilepsy and that adenosine augmentation therapies may reverse DNA hypermethylation and break the cycle of increasing seizure severity.
4. Disruption of adenosine homeostasis
It is now well-established that adenosine homeostasis is disrupted both in animal models of epilepsy (Gouder et al., 2004; Rebola et al., 2003) as well as in human epilepsies (Aronica et al., 2011; de Groot et al., 2012; Masino et al., 2011). Consequently, adenosine deficiency can be considered a pathological hallmark of epilepsy. Several mechanisms contribute to the disruption of adenosine homeostasis in epilepsy as will be outlined in the subsequent sections.
4.1. Astroglial dysfunction in epilepsy
Hippocampal sclerosis is a characteristic pathological hallmark of temporal lobe epilepsy (TLE) (Malmgren and Thom, 2012) and surgical resections of epileptogenic brain tissue have demonstrated that onset zones for chronic temporal lobe-derived and post-traumatic seizures correlate with gliotic scarring. Astrocytes influence the pathogenesis and pathophysiology of epilepsy by the homeostatic control of synaptic transmission via the release of gliotransmitters such as glutamate, ATP, and D-serine (Haydon and Carmignoto, 2006), in addition to the reuptake of neurotransmitters such as glutamate (Coulter and Eid, 2012) or neuromodulators such as adenosine (Boison, 2012b). An “astrocytic basis of epilepsy” was proposed based on findings suggesting that prolonged episodes of neuronal depolarization evoked by the astrocytic release of glutamate contributes to epileptiform discharges (Tian et al., 2005). Astrocytes play important ‘upstream’ homeostatic role in controlling uptake, degradation and recycling of neurotransmitters. In addition, glial dysfunction in the blood brain barrier, as well as neuroimmunological functions governed by glia have been implicated not only in seizure generation (i.e. ictogenesis) but most importantly in the pathophysiological processes that lead to the development of epilepsy (i.e. epileptogenesis) (Cacheaux et al., 2009; Devinsky et al., 2013; Heinemann et al., 2012; Ivens et al., 2007; Ravizza et al., 2011; Vezzani et al., 2013). Because glia communicate with each other and assume a role that is upstream of neuronal function, perturbations of glial homeostasis can affect entire neuronal networks. Those network effects of glia might indeed be a reason why neuronal networks in epilepsy synchronize; similarly, fluctuations in homeostatic functions of glia might explain why seizures are sporadic. In the adult brain the metabolic clearance of adenosine is largely mediated by astrocytes and the astroglial enzyme ADK. Astrogliosis in the epileptic brain is associated with increased expression of ADK and resulting adenosine deficiency (Aronica et al., 2013; Aronica et al., 2011; Boison, 2012b; de Groot et al., 2012; Gouder et al., 2004; Li et al., 2008; Masino et al., 2011). Consequently, astrogliosis in epilepsy is tightly linked to the disruption of adenosine homeostasis.
4.2. The adenosine kinase hypothesis of epileptogenesis
Interestingly, ADK undergoes biphasic expression changes during epileptogenesis (Boison, 2013; Gouder et al., 2004; Li et al., 2008), which form the basis of the ADK hypothesis of epileptogenesis originally proposed in 2008 (Boison, 2008) and amended in the following by incorporating biphasic epigenetic changes associated with epileptogenesis (Williams-Karnesky et al., 2013): Acquired epilepsies including temporal lobe epilepsy are thought to be triggered by an insult to the brain, which can be a brain injury, an acute seizure, a stroke or hypoxic period, high fever or an infection, as well as unknown or unconfirmed triggers (Dube et al., 2007; Frey, 2003; Kwan, 2010; Pardo et al., 2014). Scientific evidence as discussed further below suggests the existence of two distinct phases of epileptogenesis, an acute, initiating phase followed by chronic processes needed for the progression and maintenance of epileptogenesis. Phase I, acute, initiating: Acute insults to the brain such as traumatic brain injury (Clark et al., 1997), seizures (During and Spencer, 1992; Gouder et al., 2004), or a stroke (Pignataro et al., 2008) lead to an acute surge in adenosine associated with transient downregulation of ADK within the first two to three hours after the injury (Gouder et al., 2004; Pignataro et al., 2008). High levels of adenosine in turn can lead to reduced DNA methylation (Williams-Karnesky et al., 2013). We propose that the injury-induced adenosine surge may precipitate epileptogenesis through an epigenetic mechanism whereby the resulting acute hypomethylation of DNA may permit the transcription and expression of epileptogenesis initiating genes. Future studies are needed to identify genes that might be regulated by the injury-induced adenosine surge. Phase II, epilepsy progression: During the ‘latent period’ of epileptogenesis which occurs during the first few days or weeks after an insult in rodent models, or weeks and months in humans, inflammatory processes are activated that lead to microglial and astroglial activation (Devinsky et al., 2013; Nabbout et al., 2011; Vezzani et al., 2011). As mentioned earlier, astrogliosis is associated with increases in ADK expression and consequential development of adenosine deficiency. We have shown that seizures originate in areas of astrogliosis with overexpression of ADK (Li et al., 2012; Li et al., 2008), that seizure onset during epileptogenesis temporally coincides with the emergence of astrogliosis and overexpressed ADK (Li et al., 2007a), that overexpression of ADK as such is sufficient to generate electrographic seizures (Li et al., 2007a; Li et al., 2008), and that overexpression of ADK associates with hypermethylation of DNA (Williams-Karnesky et al., 2013). Since therapeutic adenosine augmentation restores normal DNA methylation levels and prevents epilepsy progression long-term (Williams-Karnesky et al., 2013) we propose that increased ADK and increased DNA methylation status form a vicious cycle implicated in the progression and maintenance of the epileptic state. Therefore, dysregulation of ADK plays a significant role in the processes that turn a normal brain into an epileptic brain (Figure 1).
4.3. The triad of ADK, A2AR, and GFAP dysregulation
Astrogliosis is not only associated with upregulation of ADK, but also with increased expression of the astroglial A2AR in GFAP-positive astrocytes (Matos et al., 2012; Orr et al., 2015). As early as three and seven days after kainate-induced neurotoxicity, prominent increases in the levels of the astrocytic A2AR were found (Orr et al., 2015). This is an important finding because astrocytic A2ARs play a role in the control of astrocyte physiology. A2AR activation stimulates astrogliosis via an Akt/NF-kappaB dependent pathway in vitro (Ke et al., 2009), whereas the pharmacological blockade of A2ARs prevented BDNF-induced reactive astrogliosis in rat striatal primary astrocytes (Brambilla et al., 2003). Since A2ARs, as well as ADK, are both upregulated in GFAP-positive reactive astrocytes, the intriguing possibility exists that both systems undergo interdependent maladaptive changes during epileptogenesis. Increased A2AR expression may trigger compensatory upregulation of ADK expression to limit activation of the gliosis-promoting A2AR; whereas increased A2AR expression might be a compensatory response to adapt to the reduced adenosine tone, which is a consequence of increased ADK expression. Therefore, a self-reinforcing maladaptive circuit might be generated that promotes reactive astrocytosis and progressive adenosine deficiency. It remains to be determined which factors trigger this maladaptive process and whether ADK, the A2AR, and GFAP share common regulatory elements that control gene expression.
4.4. Ecto-5′-nucleotidase (CD73)
Ecto-5′-nucleotidase (CD73) is the major adenosine producing enzyme in the extracellular space (Augusto et al., 2013; Chu et al., 2014; Cunha et al., 1992; Koszalka et al., 2004) and implicated in reactive synaptogenesis (Lie et al., 1999; Schoen et al., 1999). Interestingly, increased CD73 expression was found in the dentate gyrus molecular layer in human resective surgery specimen from TLE patients, implicating TLE-associated reactive synaptogenesis in this brain region (Lie et al., 1999). Consistent with these human data, increases in CD73 activity were also found in rodent models of epilepsy (Bonan et al., 2000a; Bonan et al., 2000b; Schoen et al., 1999). More recently, genetic variants of both CD73 and ADK were associated with the development of posttraumatic epilepsy in a human study involving samples from 162 subjects (Diamond et al., 2015).
Epilepsy-associated expression changes in the largely synapse-associated adenosine producing enzyme CD73 and the largely astrocytic adenosine metabolizing enzyme ADK need to be discussed within the context of spatio-temporal mechanisms of adenosine signaling in epilepsy: In the brain adenosine fulfills two very different, seemingly opposing roles. As a homeostatic regulator and retaliatory metabolite adenosine sets the inhibitory and general neuroprotective ‘tone’ via activation of widespread inhibitory A1Rs (Meghji and Newby, 1990). On the synaptic level however, adenosine facilitates synaptic function via activation of stimulatory A2ARs (Cunha, 2001, 2008). Whereas the tonic inhibitory pool of adenosine is thought to be largely under the control of astrocytes, the stimulatory pool of adenosine acting at the synapse level is likely derived from neurons to allow a highly localized modulation of individual synapses (Cunha, 2001, 2008; Lovatt et al., 2012; Meghji and Newby, 1990). These two parallel, but different functions of adenosine have likely evolved to increase salience of synaptic transmission in a tonically inhibited network, a mechanistic strategy to enhance the signal to noise ratio (Cunha, 2001). Thus, increased expression of CD73 in the epileptic brain might be a compensatory response to inhibit synaptic transmission in mossy fibers, but is unlikely to control the homeostatic pool of adenosine, controlled by astrocytic ADK, which determines the activation status of the anticonvulsive and neuroprotective A1R.
4.5. Adenosine transporters
Adenosine homeostasis in the brain also depends on adenosine transport, which is mediated by nucleoside transporters (NTs), either equilibrative transporters (ENTs) or cotransporters (CNTs) (Parkinson et al., 2011). Blockade of NTs leads to a rise in extracellular adenosine leading to increased A1R activation, and consequently an anti-epileptic effect of NT inhibitors has been reported (Zhang et al., 1993). ENT1 expression appears to be under the influence of seizure activity since an increase in ENT1 binding site density was observed as a consequence of seizures (Pagonopoulou and Angelatou, 1998). More recently it was shown that cannabidiol is a potent inhibitor of ENT1 (Carrier et al., 2006). Therefore, an increase in extracellular adenosine through ENT1 blockade may contribute to the well-known anti-seizure activity of plant based cannabinoids. A potential role for NT inhibitors in the treatment of seizure disorders has been discussed (Boison, 2005).
5. Comorbidities of epilepsy
Patients with epilepsy have a high prevalence of cognitive and psychiatric comorbidities, which may even precede the diagnosis of epilepsy Psychiatric disorders, including cognitive changes, attention deficits, psychosis, and personality changes, as well as depression, and anxiety occur more frequently in people with epilepsy than in the general population, particularly in patients with refractory epilepsy (Gaitatzis et al., 2004a; Gaitatzis et al., 2012; Gaitatzis et al., 2004b; LaFrance et al., 2008). Clinically, comorbid cognitive impairments are among the most debilitating and persistent concerns of chronic epilepsy (Elger et al., 2004; Hermann et al., 2008; LaFrance et al., 2008). A recent survey study indicates that 54% of epilepsy patients exhibit at least one abnormal score on a battery of cognitive tests, twice the rate in healthy volunteers (Taylor et al., 2010). From a patient’s perspective cognitive co-morbidities that include impairment of memory, concentration, and ability to think clearly are perceived as the most debilitating complication associated with epilepsy (Fisher et al., 2000). In particular, episodic memory impairment is now recognized as a key feature of temporal lobe epilepsy (TLE) and cognitive impairment has been described as “the most problematic of the comorbidities of epilepsy” (Bell et al., 2011). Apart from mediating seizure control, adenosine is a crucial regulator of behavior. Importantly, disruption of adenosine homeostasis has been linked with cognitive and psychiatric phenotypes as shown in our previous work (Boison et al., 2012; Shen et al., 2012a; Wei et al., 2011; Yee et al., 2007). The brain-wide transgenic overexpression of ADK in mice was sufficient to trigger adenosine-deficiency (Shen et al., 2011) and impairment of cognitive function (Boison et al., 2012; Yee et al., 2007), in particular severe learning deficits in the Morris water maze task and in Pavlovian conditioning (Yee et al., 2007). Thus, reconstruction of adenosine homeostasis emerges as a rational and innovative approach to restore cognitive function under conditions of increased metabolic adenosine-clearance (as found in TLE). We recently demonstrated that therapeutic adenosine augmentation with the ADK inhibitor ABT-702 exerted antipsychotic-like activity in the pre-pulse inhibition paradigm for psychiatric gating deficits typical for schizophrenia, whereas adenosine releasing cell grafts to the hippocampal formation restored cognitive performance in Adk-transgenic mice (Shen et al., 2012b). Together these findings suggest that maladaptive changes in adenosine homeostasis can not only give rise to epileptic seizures, but also give rise to comorbidities commonly associated with epilepsy.
6. Adenosine augmentation therapies for epilepsy
Experimental and clinical findings outlined in the sections above provide a neurochemical rationale to use therapeutic adenosine augmentation for the treatment of seizures and associated comorbidities in epilepsy. Several approaches have been tested, which all demonstrate a potent antiictogenic and antiepileptogenic role of adenosine therapy.
6.1. Pharmacological approaches
Adenosine augmentation therapies (AATs) make rational therapeutic use of an endogenous anticonvulsant and neuroprotectant of the brain with the potential to not only suppress seizures, but also to prevent epileptogenesis (Boison, 2009, 2012a). ADK inhibitors are the most efficient therapeutic agents to raise the tissue tone of endogenous adenosine (Boison, 2013). By reducing the metabolic clearance of adenosine, ADK inhibitors can potentiate an endogenous adenosine response, for example a seizure-induced adenosine release, in a site- and event-specific manner (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000; McGaraughty et al., 2001; McGaraughty et al., 2005). Because ADK is pathologically overexpressed in epileptogenic brain regions (Aronica et al., 2011; Boison, 2012b; Gouder et al., 2004; Li et al., 2008) and because overexpression of ADK is sufficient to promote seizures (Etherington et al., 2009; Li et al., 2012; Li et al., 2008; Theofilas et al., 2011) the use of ADK inhibitors for the treatment of epilepsy is based on a strong neurochemical rationale. Importantly, the ADK inhibitor 5-ITU effectively suppresses seizures in a mouse model of pharmacoresistant temporal lobe epilepsy (Gouder et al., 2004) suggesting that ADK inhibitors might be superior to conventional antiepileptic drugs. In addition, several lines of evidence indicate that ADK, rather than adenosine deaminase (ADA) is the major adenosine metabolizing enzyme in the brain. In particular, ADK inhibition but not ADA inhibition increased endogenous adenosine and depressed neuronal activity in hippocampal slices (Pak et al., 1994) and a genetic knockout of ADK was more efficient in inducing adenosine secretion from cells than a genetic knockout of ADA (Huber et al., 2001). In line with those findings the ADK inhibitors 5′-amino-5′-deoxyadenosine or 5-ITU, but not by the adenosine deaminase (ADA) inhibitor 2′-deoxycoformycin suppressed bicuculline-induced seizures in rats suggesting that the antiictogenic activity of ADK inhibition is superior to ADA inhibition (Zhang et al., 1993). Several ADK inhibitors have subsequently been developed for seizure control (Ugarkar et al., 2000a; Ugarkar et al., 2000b). One of those agents, GP-3269, showed enhanced pharmacokinetic properties, attenuation of the seizure response in the rat maximum electroshock (MES) and kindling models, and lack of profound cardiovascular side effects (Erion et al., 1997; McGaraughty et al., 2005). However, despite an improved side effect profile, the chronic, systemic use of ADK inhibitors for epilepsy therapy might not be an option due to liver toxicity (Boison et al., 2002) and the occurrence of cognitive and sedative side effects (Boison, 2013).
6.2. Cell-based adenosine delivery
An alternative approach to avoid systemic side effects of a drug is its focal delivery to the brain (Nilsen and Cock, 2004). One strategy for the focal augmentation of adenosine signaling in the brain is an ex vivo gene therapy approach to first delete the Adk gene in cultured cells to induce therapeutic adenosine release, and then to transplant the resulting adenosine-releasing cells into the host brain to therapeutically exploit locally enhanced adenosine release. The first successful cell therapy approach was achieved with baby hamster kidney (BHK) cells that were engineered to lack the Adk gene (Huber et al., 2001). As a result of this manipulation, these ADK-deficient BHK cells released about 40 ng adenosine per 105 cells per day. When encapsulated into semipermeable polymer membranes and transplanted into the brain ventricles of kindled epileptic rats the implants almost completely suppressed any seizures in an A1R dependent manner (Huber et al., 2001). Unfortunately, seizure suppression was limited to two weeks due to the reduced longevity of the encapsulated cells. To develop a more versatile cell-based system for seizure control, both alleles of the Adk gene were disrupted in mouse embryonic stem (ES) cells by homologous recombination; the resulting Adk−/− ES cells yielded glial populations with an adenosine release profile comparable to Adk-deficient BHK cells (Fedele et al., 2004). When differentiated into neural precursor cells and grafted into the infrahippocampal fissure of rats, the adenosine cells profoundly suppressed kindling epileptogenesis (Li et al., 2007b). Likewise, when grafted into the infrahippocampal fissure of mice 24 hours after a status epilepticus, the same cells prevented the development of epilepsy in a post status model of epileptogenesis (Li et al., 2008). Adk−/− cells attenuated astrogliosis, prevented overexpression of ADK, and led to a complete lack of any seizures, whereas animals with control grafts underwent normal epileptogenic pathology and developed spontaneous electrographic seizures at a rate of about 4 seizures per hour (Li et al., 2008). In an attempt to engineer human stem cells for therapeutic adenosine release, human mesenchymal stem cells were infected with a lentivirus engineered to express a micro RNA directed against Adk. This approach reduced ADK expression to 20% of its normal levels and triggered the release of about 1 ng adenosine per 105 cells per hour (Ren et al., 2007). When transplanted into the infrahippocampal fissure of mice, these implants reduced acute seizure-induced cell death (Ren et al., 2007) and led to a partial suppression of epileptogenesis (Li et al., 2009). This partial therapeutic effect is most likely due to the 40-times lower amounts of adenosine released by those cells as compared to the engineered ES cells that completely lacked any ADK expression. Together, these reports demonstrate that disruption of ADK expression in cells is a promising therapeutic strategy to augment adenosine signaling at a local site within the brain with potent therapeutic effects resulting in seizure suppression as well as prevention of epileptogenesis.
6.3. Gene therapy
A different strategy for targeted and cell-type selective adenosine augmentation is gene therapy. Whereas conventional gene therapies overexpress a transgene to produce a therapeutic agent, the therapeutic goal here is to use a gene therapy approach that reduces the expression of the endogenous Adk-gene. This can best be achieved with antisense approaches (Boison, 2010) designed to knock down gene expression. An adeno associated virus vector was constructed to expresses an Adk cDNA in antisense orientation under the control of an astrocyte specific gfaABC1D promoter (Lee et al., 2008). Injection of this virus into the hippocampus of transgenic mice with spontaneous electrographic seizures resulted in a substantial unilateral decrease in seizure activity ipsilateral to the virus injection site with 0.6 ± 0.6 seizures/h, compared to 5.8 ± 0.5 seizures/h on the contralateral (non-injected) side (Theofilas et al., 2011). This study constitutes a proof of feasibility that a gene therapy targeting ADK, restricted to a specific brain area (hippocampus) and to a specific cell type (astrocyte), can have potent therapeutic effects based on boosting the anticonvulsive properties of adenosine. Additional work is needed to evaluate whether anti-ADK gene therapies are effective in clinically relevant models of temporal lobe epilepsy.
6.4. Dietary interventions
The high fat low carbohydrate ketogenic diet is a metabolic intervention, which provides effective seizure control in many forms of pharmacoresistant epilepsy (Freeman, 2009; Kossoff and Rho, 2009; Kossoff et al., 2009; Neal et al., 2008; Yellen, 2008). Although it has been used clinically for over 80 years, the mechanisms underlying the therapeutic actions of a ketogenic diet have not been fully explored. A ketogenic diet induces the brain to use ketones as primary energy source instead of glucose and it is those metabolic changes that are thought to underlie the therapeutic effects of this type of metabolic intervention (Bough, 2008; Bough et al., 2006; Kalapos, 2007; Ma et al., 2007; Yellen, 2008). Although several mechanisms of ketogenic diet therapy may synergistically contribute to antiepileptic outcome, a large body of evidence supports the notion that a ketogenic diet increases adenosine signaling in the brain (Masino and Geiger, 2008, 2009; Masino et al., 2012; Masino et al., 2009). Indeed, it was recently shown that a ketogenic diet reduced the expression of ADK in mice (Masino et al., 2011). In line with this finding, the ketogenic diet suppressed seizures in adenosine deficient Adk-tg mice, but not in A1R deficient mice, demonstrating that functional A1R activation is necessary for the antiepileptic effects of the diet (Masino et al., 2011). Although anecdotal clinical data suggest an antiepileptogenic activity of ketogenic diet therapy, further studies are needed to explore whether a ketogenic diet is antiepileptogenic and to identify the underlying mechanisms.
6.5. Antiepileptogenesis
Several lines of evidence suggest that adenosine might prevent epileptogenesis. Transgenic mice with an engineered reduction of ADK expression in forebrain did not develop epilepsy, even when an epileptogenesis-triggering status epilepticus was coupled with transient blockade of the A1R (Li et al., 2008). As mentioned above, adenosine-releasing stem cells – implanted into the hippocampal formation after triggering epileptogenesis – dose-dependently attenuated astrogliosis, suppressed ADK-increases, and attenuated the development of spontaneous seizures (Li et al., 2008). In an independent therapeutic approach, the transient delivery of adenosine by intraventricular silk for only 10 days provided long-lasting (beyond adenosine-release) antiepileptogenic effects in the rat kindling model (Szybala et al., 2009). More recent findings that the antiepileptogenic effects of adenosine are based on an epigenetic mechanism will be discussed in more detail below.
As mentioned earlier, increased DNA methylation has been considered to play a key role in epileptogenesis (Kobow and Blumcke, 2011). Theoretically, DNA methylation inhibitors might be of therapeutic value to treat epilepsy by restoring non-pathological epigenetic homeostasis. Current DNMT inhibitors such as 5-azacytidine or zebularine are antimetabolites, which are incorporated into DNA, and which therefore are clinically used for the treatment of cancers (Christman, 2002). The use of those drugs for the treatment of persons with epilepsy must be approached with caution due to risks of cell death and mutagenesis. As an alternative to conventional pharmacological DNMT inhibitors, focal adenosine therapy may act as an effective epigenetic medicine. Recently, we described a novel antiepileptogenic role for adenosine whereby a transient dose of adenosine administered to epileptic rats after the onset of epilepsy not only suppressed seizures during active adenosine release, but also prevented further disease progression long-term even after the therapy was suspended. Adenosine treatment restored normal DNA methylation levels in the otherwise hypermethylated hippocampus of the epileptic rat (Figure 1). More specifically, genome wide analysis using a methylated DNA immunoprecipitation array revealed that out of the 125 genes which showed increased DNA methylation in epilepsy, 66 also showed reduced DNA methylation after adenosine therapy in treated epileptic rats. Interestingly, multiple targets that function to either interact with DNA or play a role in gene transcription and translation (PolD1, Polr1e, Rps6kl1, Snrpn, Znf524, Znf541, Znf710) responded to adenosine therapy. Consequently, those targets are of interest as likely candidates to mediate adenosine-dependent changes in major homeostatic functions of the epigenome (Williams-Karnesky et al., 2013).
7. Conclusions and outlook
The excitability of the brain is determined by neurons, which in turn are connected into networks, which in turn are broadly controlled by the homeostatic environment of their surroundings, which to a large degree is under the control of glial cells. It becomes clear that complex neurological syndromes, such as epilepsy, which are not only defined by a dominant symptom (i.e. a seizure), but also by a growing number of associated comorbidities, can best be explained by the disruption of network homeostasis. Disruption of network homeostasis implies the simultaneous and concordant dysregulation of several molecular pathways, which in turn can affect each other and lead to a progression of the disease in the sense that ‘seizures beget seizures’. This old concept has a lot of truth and can best be explained by the self-reinforcing interplay of several homeostatic systems that become progressively dysregulated during disease progression. Conventional antiepileptic drugs with a target-centric mode of action are unlikely to affect network homeostasis and to prevent the progressive maladaptive changes occurring during epileptogenesis, which can be considered a lifelong process of disease progression. Novel therapeutic interventions based on adenosine, epigenetic mechanisms, or dietary and lifestyle interventions might hold promise to affect network homeostasis as a novel conceptual strategy to treat and prevent epilepsy on the network level. Adenosine emerges as a prototype homeostatic network regulator with the proven capability to control network activity both through receptor-dependent as well as through receptor-independent epigenetic and bioenergetic mechanisms (Boison, 2013). Consequently, therapeutic adenosine augmentation has been demonstrated to suppress epileptic seizures (Li et al., 2008; Li et al., 2007b), to prevent disease progression and epileptogenesis (Li et al., 2008; Williams-Karnesky et al., 2013), but also to prevent psychosis and to improve cognition (Shen et al., 2012b) without any known adverse effects. The robust antipsychotic and pro-cognitive effect of adenosine in mice (Shen et al., 2012b) suggests that therapeutic adenosine augmentation might combine anticonvulsant with cognition-enhancing effects. In preclinical toxicity studies of intrathecal adenosine in dogs, no side effects were observed with intrathecal adenosine infused chronically for 26 days (Chiari et al., 1999). Likewise, intrathecal adenosine was tested in humans in escalating doses of up to 2 mg without any adverse effects (Eisenach et al., 2002a, b). Several therapeutic adenosine augmentation strategies ranging from gene therapy to dietary intervention are currently in preclinical development. It is within the scope of possibilities that adenosine augmenting therapies are introduced into the clinic as a novel class of ‘homeostatic network therapy’ within the next ten years. Challenges will be to develop strategies to confine adenosine’s action to identifiable target areas and cells; the advent of cell-type specific gene therapy vectors might offer a promising strategy to manipulate adenosine homeostasis in a localized and cell-type selective manner.
Highlights.
Adenosine deficiency is a pathological hallmark of epilepsy
Hypermethylation of DNA is associated with the epileptic state
Adenosine deficiency causes increased DNA methylation
Adenosine therapy effectively suppresses seizures
Adenosine therapy restores normal DNA methylation levels & prevents epileptogenesis
Acknowledgments
D.B. is funded through grants from the National Institutes of Health R01 NS084920, R21 NS088024, R01MH83973, from Citizens United for Research in Epilepsy (CURE), and the Legacy Hospital Foundations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
D.B. wrote the manuscript.
Competing financial interests
The author declares no competing financial interests.
References
- Aronica E, Sandau US, Iyer A, Boison D. Glial adenosine kinase - A neuropathological marker of the epileptic brain. Neurochem Int. 2013;63:688–695. doi: 10.1016/j.neuint.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Zurolo E, Iyer A, de Groot M, Anink J, Carbonell C, van Vliet EA, Baayen JC, Boison D, Gorter JA. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia. 2011;52:1645–1655. doi: 10.1111/j.1528-1167.2011.03115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto E, Matos M, Sevigny J, El-Tayeb A, Bynoe MS, Muller CE, Cunha RA, Chen JF. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33:11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ault B, Wang CM. Adenosine inhibits epileptiform activity arising in hippocampal area CA3. Br J Pharmacol. 1986;87:695–703. doi: 10.1111/j.1476-5381.1986.tb14587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell B, Lin JJ, Seidenberg M, Hermann B. The neurobiology of cognitive disorders in temporal lobe epilepsy. Nat Rev Neurol. 2011;7:154–164. doi: 10.1038/nrneurol.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine and epilepsy: from therapeutic rationale to new therapeutic strategies. Neuroscientist. 2005;11:25–36. doi: 10.1177/1073858404269112. [DOI] [PubMed] [Google Scholar]
- Boison D. The adenosine kinase hypothesis of epileptogenesis. Progress in Neurobiology. 2008;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res. 2009;85:131–141. doi: 10.1016/j.eplepsyres.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Inhibitory RNA in epilepsy: Research tool and therapeutic perspectives. Epilepsia. 2010;51:1659–1668. doi: 10.1111/j.1528-1167.2010.02672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine augmentation therapy for epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Oxford University Press; Oxford: 2012a. pp. 1150–1160. [PubMed] [Google Scholar]
- Boison D. Adenosine dysfunction in epilepsy. Glia. 2012b;60:1234–1243. doi: 10.1002/glia.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacological Reviews. 2013;65:906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA. 2002;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Singer P, Shen HY, Feldon J, Yee BK. Adenosine hypothesis of schizophrenia-opportunities for pharmacotherapy. Neuropharmacology. 2012;62:1527–1543. doi: 10.1016/j.neuropharm.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonan CD, Amaral OB, Rockenbach IC, Walz R, Battastini AM, Izquierdo I, Sarkis JJ. Altered ATP hydrolysis induced by pentylenetetrazol kindling in rat brain synaptosomes. Neurochem Res. 2000a;25:775–779. doi: 10.1023/a:1007557205523. [DOI] [PubMed] [Google Scholar]
- Bonan CD, Walz R, Pereira GS, Worm PV, Battastini AM, Cavalheiro EA, Izquierdo I, Sarkis JJ. Changes in synaptosomal ectonucleotidase activities in two rat models of temporal lobe epilepsy. Epilepsy Res. 2000b;39:229–238. doi: 10.1016/s0920-1211(00)00095-4. [DOI] [PubMed] [Google Scholar]
- Bough K. Energy metabolism as part of the anticonvulsant mechanism of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):91–93. doi: 10.1111/j.1528-1167.2008.01846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol. 2006;60:223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Cottini L, Fumagalli M, Ceruti S, Abbracchio MP. Blockade of A2A adenosine receptors prevents basic fibroblast growth factor-induced reactive astrogliosis in rat striatal primary astrocytes. Glia. 2003;43:190–194. doi: 10.1002/glia.10243. [DOI] [PubMed] [Google Scholar]
- Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, Heinemann U, Friedman A, Kaufer D. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009;29:8927–8935. doi: 10.1523/JNEUROSCI.0430-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni GL, Mudd SH, Andreoli V. Affective disorders and S-adenosylmethionine: a new hypothesis. Trends Neurosci. 1989;12:319–324. doi: 10.1016/0166-2236(89)90038-6. [DOI] [PubMed] [Google Scholar]
- Carrier EJ, Auchampach JA, Hillard CJ. Inhibition of an equilibrative nucleoside transporter by cannabidiol: A mechanism of cannabinoid immunosuppression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7895–7900. doi: 10.1073/pnas.0511232103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez M. SAMe: S-Adenosylmethionine. Am J Health Syst Pharm. 2000;57:119–123. doi: 10.1093/ajhp/57.2.119. [DOI] [PubMed] [Google Scholar]
- Chiari A, Yaksh TL, Myers RR, Provencher J, Moore L, Lee CS, Eisenach JC. Preclinical toxicity screening of intrathecal adenosine in rats and dogs. Anesthesiology. 1999;91:824–832. doi: 10.1097/00000542-199909000-00035. [DOI] [PubMed] [Google Scholar]
- Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- Chu S, Xiong W, Parkinson FE. Effect of ecto-5′-nucleotidase (eN) in astrocytes on adenosine and inosine formation. Purinergic Signal. 2014;10:603–609. doi: 10.1007/s11302-014-9421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Carcillo JA, Kochanek PM, Obrist WD, Jackson EK, Mi Z, Wisneiwski SR, Bell MJ, Marion DW. Cerebrospinal fluid adenosine concentration and uncoupling of cerebral blood flow and oxidative metabolism after severe head injury in humans. Neurosurgery. 1997;41:1284–1292. doi: 10.1097/00006123-199712000-00010. discussion 1292–1293. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A(1) receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A(2A) receptor-mediated facilitation of plasticity. Neurochemistry International. 2008;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastiao AM, Ribeiro JA. Ecto-5′-nucleotidase is associated with cholinergic nerve terminals in the hippocampus but not in the cerebral cortex of the rat. J Neurochem. 1992;59:657–666. doi: 10.1111/j.1471-4159.1992.tb09420.x. [DOI] [PubMed] [Google Scholar]
- D’Alimonte I, D’Auro M, Citraro R, Biagioni F, Jiang S, Nargi E, Buccella S, Di Iorio P, Giuliani P, Ballerini P, Caciagli F, Russo E, De Sarro G, Ciccarelli R. Altered distribution and function of A2A adenosine receptors in the brain of WAG/Rij rats with genetic absence epilepsy, before and after appearance of the disease. Eur J Neurosci. 2009;30:1023–1035. doi: 10.1111/j.1460-9568.2009.06897.x. [DOI] [PubMed] [Google Scholar]
- de Groot M, Iyer A, Zurolo E, Anink J, Heimans JJ, Boison D, Reijneveld CJ, Aronica E. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia. 2012;53:58–66. doi: 10.1111/j.1528-1167.2011.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–184. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Diamond ML, Ritter AC, Jackson EK, Conley YP, Kochanek PM, Boison D, Wagner AK. Genetic Variation in the Adenosine Regulatory Cycle is Associated with Post-traumatic Epilepsy Development. Epilepsia. 2015 doi: 10.1111/epi.13044. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragunow M. Adenosine and seizure termination. Ann Neurol. 1991;29:575. doi: 10.1002/ana.410290524. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Goddard GV, Laverty R. Is adenosine an endogenous anticonvulsant? Epilepsia. 1985;26:480–487. doi: 10.1111/j.1528-1157.1985.tb05684.x. [DOI] [PubMed] [Google Scholar]
- Dube CM, Brewster AL, Richichi C, Zha Q, Baram TZ. Fever, febrile seizures and epilepsy. Trends Neurosci. 2007;30:490–496. doi: 10.1016/j.tins.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV. Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia. 1980;21:541–548. doi: 10.1111/j.1528-1157.1980.tb04305.x. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Fredholm BB. Adenosine receptors mediating inhibitory electrophysiological responses in rat hippocampus are different from receptors mediating cyclic AMP accumulation. Naunyn Schmiedebergs Arch Pharmacol. 1984;326:294–301. doi: 10.1007/BF00501433. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol. 1992;32:618–624. doi: 10.1002/ana.410320504. [DOI] [PubMed] [Google Scholar]
- Eisenach JC, Hood DD, Curry R. Phase I safety assessment of intrathecal injection of an American formulation of adenosine in humans. Anesthesiology. 2002a;96:24–28. doi: 10.1097/00000542-200201000-00010. [DOI] [PubMed] [Google Scholar]
- Eisenach JC, Hood DD, Curry R. Preliminary efficacy assessment of intrathecal injection of an American formulation of adenosine in humans. Anesthesiology. 2002b;96:29–34. doi: 10.1097/00000542-200201000-00011. [DOI] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3:663–672. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- Erion MD, Ugarkar BG, Dare J, Castellino AJ, Fujitaki JM, Dixon R, Appleman JR, Wiesner JB. Design, Synthesis and Anticonvulsant Activity Of the Potent Adenosine Kinase Inhibitor Gp3269. Nucleosides & Nucleotides. 1997;16:1013–1021. [Google Scholar]
- Etherington LA, Patterson GE, Meechan L, Boison D, Irving AJ, Dale N, Frenguelli B. Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology. 2009;56:429–437. doi: 10.1016/j.neuropharm.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Brüstle O, Scheurer L, Simpson EM, Mohler H, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Vickrey BG, Gibson P, Hermann B, Penovich P, Scherer A, Walker S. The impact of epilepsy from the patient’s perspective I. Descriptions and subjective perceptions. Epilepsy Res. 2000;41:39–51. doi: 10.1016/s0920-1211(00)00126-1. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Ijzerman AP, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JM. Seizures, EEG events, and the ketogenic diet. Epilepsia. 2009;50:329–330. doi: 10.1111/j.1528-1167.2008.01757.x. [DOI] [PubMed] [Google Scholar]
- Frey LC. Epidemiology of posttraumatic epilepsy: A critical review. Epilepsia. 2003;44:11–17. doi: 10.1046/j.1528-1157.44.s10.4.x. [DOI] [PubMed] [Google Scholar]
- Gaitatzis A, Carroll K, Majeed A, J WS. The epidemiology of the comorbidity of epilepsy in the general population. Epilepsia. 2004a;45:1613–1622. doi: 10.1111/j.0013-9580.2004.17504.x. [DOI] [PubMed] [Google Scholar]
- Gaitatzis A, Sisodiya SM, Sander JW. The somatic comorbidity of epilepsy: a weighty but often unrecognized burden. Epilepsia. 2012;53:1282–1293. doi: 10.1111/j.1528-1167.2012.03528.x. [DOI] [PubMed] [Google Scholar]
- Gaitatzis A, Trimble MR, Sander JW. The psychiatric comorbidity of epilepsy. Acta Neurol Scand. 2004b;110:207–220. doi: 10.1111/j.1600-0404.2004.00324.x. [DOI] [PubMed] [Google Scholar]
- Gill MW, Schatz RA. The effect of diazepam on brain levels of S-adenosyl-L-methionine and S-adenosyl-L-homocysteine: possible correlation with protection from methionine sulfoximine seizures. Res Commun Chem Pathol Pharmacol. 1985;50:349–363. [PubMed] [Google Scholar]
- Glass M, Faull RL, Bullock JY, Jansen K, Mee EW, Walker EB, Synek BJ, Dragunow M. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res. 1996;710:56–68. doi: 10.1016/0006-8993(95)01313-x. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamil NE, Cock HR, Walker MC. Acute down-regulation of adenosine A(1) receptor activity in status epilepticus. Epilepsia. 2012;53:177–188. doi: 10.1111/j.1528-1167.2011.03340.x. [DOI] [PubMed] [Google Scholar]
- Hargus NJ, Jennings C, Perez-Reyes E, Bertram EH, Patel MK. Enhanced actions of adenosine in medial entorhinal cortex layer II stellate neurons in temporal lobe epilepsy are mediated via A(1)-receptor activation. Epilepsia. 2012;53:168–176. doi: 10.1111/j.1528-1167.2011.03337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiological Reviews. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGF-beta signaling and astrocyte dysfunction in epilepsy. Glia. 2012 doi: 10.1002/glia.22311. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann B, Seidenberg M, Jones J. The neurobehavioural comorbidities of epilepsy: can a natural history be developed? Lancet Neurol. 2008;7:151–160. doi: 10.1016/S1474-4422(08)70018-8. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Deglon N, Aebischer P, Mohler H, Boison D. Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA. 2001;98:7611–7616. doi: 10.1073/pnas.131102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, Seiffert E, Heinemann U, Friedman A. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130:535–547. doi: 10.1093/brain/awl317. [DOI] [PubMed] [Google Scholar]
- James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA. Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr. 2002;132:2361S–2366S. doi: 10.1093/jn/132.8.2361S. [DOI] [PubMed] [Google Scholar]
- Kalapos MP. Possible mechanism for the effect of ketogenic diet in cases of uncontrolled seizures. The reconsideration of acetone theory. Med Hypotheses. 2007;68:1382–1388. doi: 10.1016/j.mehy.2006.10.041. [DOI] [PubMed] [Google Scholar]
- Ke RH, Xiong J, Liu Y, Ye ZR. Adenosine A2a receptor induced gliosis via Akt/NF-kappaB pathway in vitro. Neurosci Res. 2009;65:280–285. doi: 10.1016/j.neures.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Kobow K, Blumcke I. The methylation hypothesis: do epigenetic chromatin modifications play a role in epileptogenesis? Epilepsia. 2011;52(Suppl 4):15–19. doi: 10.1111/j.1528-1167.2011.03145.x. [DOI] [PubMed] [Google Scholar]
- Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, Fritzsche I, Hauke J, Hahnen E, Coras R, Muhlebner A, El-Osta A, Blumcke I. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013;126:741–756. doi: 10.1007/s00401-013-1168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–575. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Rho JM. Ketogenic diets: evidence for short- and long-term efficacy. Neurotherapeutics. 2009;6:406–414. doi: 10.1016/j.nurt.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossoff EH, Zupec-Kania BA, Rho JM. Ketogenic Diets: An Update for Child Neurologists. J Child Neurol. 2009;24:979–988. doi: 10.1177/0883073809337162. [DOI] [PubMed] [Google Scholar]
- Koszalka P, Ozuyaman B, Huo Y, Zernecke A, Flogel U, Braun N, Buchheiser A, Decking UK, Smith ML, Sevigny J, Gear A, Weber AA, Molojavyi A, Ding Z, Weber C, Ley K, Zimmermann H, Godecke A, Schrader J. Targeted disruption of cd73/ecto-5′-nucleotidase alters thromboregulation and augments vascular inflammatory response. Circ Res. 2004;95:814–821. doi: 10.1161/01.RES.0000144796.82787.6f. [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Bhagwat SS, Jarvis MF. Adenosine kinase inhibitors. Current Pharmaceutical Design. 1998;4:403–416. [PubMed] [Google Scholar]
- Kowaluk EA, Jarvis MF. Therapeutic potential of adenosine kinase inhibitors. Expert Opin Investig Drugs. 2000;9:551–564. doi: 10.1517/13543784.9.3.551. [DOI] [PubMed] [Google Scholar]
- Kwan J. Stroke: predicting the risk of poststroke epilepsy-why and how? Nat Rev Neurol. 2010;6:532–533. doi: 10.1038/nrneurol.2010.140. [DOI] [PubMed] [Google Scholar]
- LaFrance WC, Jr, Kanner AM, Hermann B. Psychiatric comorbidities in epilepsy. Int Rev Neurobiol. 2008;83:347–383. doi: 10.1016/S0074-7742(08)00020-2. [DOI] [PubMed] [Google Scholar]
- Lee KS, Schubert P, Heinemann U. The anticonvulsive action of adenosine: a postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Res. 1984;321:160–164. doi: 10.1016/0006-8993(84)90694-2. [DOI] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Li T, Lan JQ, Fredholm BB, Simon RP, Boison D. Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biology. 2007a;3:353–366. doi: 10.1017/S1740925X0800015X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Lytle N, Lan JQ, Sandau US, Boison D. Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia. 2012;60:83–95. doi: 10.1002/glia.21250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Kaplan DL, Boison D. Human mesenchymal stem cell grafts engineered to release adenosine reduce chronic seizures in a mouse model of CA3-selective epileptogenesis. Epilepsy Res. 2009;84:238–241. doi: 10.1016/j.eplepsyres.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Inv. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007b;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Lie AA, Blumcke I, Beck H, Wiestler OD, Elger CE, Schoen SW. 5′-Nucleotidase activity indicates sites of synaptic plasticity and reactive synaptogenesis in the human brain. J Neuropathol Exp Neurol. 1999;58:451–458. doi: 10.1097/00005072-199905000-00004. [DOI] [PubMed] [Google Scholar]
- Lloyd HG, Fredholm BB. Involvement of adenosine deaminase and adenosine kinase in regulating extracellular adenosine concentration in rat hippocampal slices. Neurochem Int. 1995;26:387–395. doi: 10.1016/0197-0186(94)00144-j. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, Tieu K, Nedergaard M. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc Natl Acad Sci U S A. 2012;109:6265–6270. doi: 10.1073/pnas.1120997109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32:391–395. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- Ma W, Berg J, Yellen G. Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci. 2007;27:3618–3625. doi: 10.1523/JNEUROSCI.0132-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmgren K, Thom M. Hippocampal sclerosis–origins and imaging. Epilepsia. 2012;53(Suppl 4):19–33. doi: 10.1111/j.1528-1167.2012.03610.x. [DOI] [PubMed] [Google Scholar]
- Mandaviya PR, Stolk L, Heil SG. Homocysteine and DNA methylation: a review of animal and human literature. Mol Genet Metab. 2014;113:243–252. doi: 10.1016/j.ymgme.2014.10.006. [DOI] [PubMed] [Google Scholar]
- Masino SA, Geiger JD. Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends in Neurosciences. 2008;31:273–278. doi: 10.1016/j.tins.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Geiger JD. The ketogenic diet and epilepsy: is adenosine the missing link? Epilepsia. 2009;50:332–333. doi: 10.1111/j.1528-1167.2008.01771.x. [DOI] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Ruskin DN, Geiger JD, Boison D. Purines and Neuronal Excitability: Links to the Ketogenic Diet. Epilepsy Res. 2012;100:229–238. doi: 10.1016/j.eplepsyres.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Kawamura M, Wasser CA, Pomeroy LT, Ruskin DN. Adenosine, ketogenic diet and epilepsy: the emerging therapeutic relationship between metabolism and brain activity. Curr Neuropharmacol. 2009;7:257–268. doi: 10.2174/157015909789152164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J Clin Inv. 2011;121:2679–2683. doi: 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato JM, Martinez-Chantar ML, Lu SC. Methionine metabolism and liver disease. Annual Review of Nutrition. 2008;28:273–293. doi: 10.1146/annurev.nutr.28.061807.155438. [DOI] [PubMed] [Google Scholar]
- Matos M, Augusto E, Machado NJ, dos Santos-Rodrigues A, Cunha RA, Agostinho P. Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis. 2012;31:555–567. doi: 10.3233/JAD-2012-120469. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF. Recent developments in the discovery of novel adenosine kinase inhibitors: mechanism of action and therapeutic potential. CNS Drug Rev. 2001;7:415–432. doi: 10.1111/j.1527-3458.2001.tb00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF, Berman RF. Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem. 2005;5:43–58. doi: 10.2174/1568026053386845. [DOI] [PubMed] [Google Scholar]
- Meghji P, Newby AC. Sites of adenosine formation, action and inactivation in the brain. Neurochem Int. 1990;16:227–232. doi: 10.1016/0197-0186(90)90095-b. [DOI] [PubMed] [Google Scholar]
- Miller-Delaney SF, Bryan K, Das S, McKiernan RC, Bray IM, Reynolds JP, Gwinn R, Stallings RL, Henshall DC. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain. 2015;138:616–631. doi: 10.1093/brain/awu373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol. 2011;10:99–108. doi: 10.1016/S1474-4422(10)70214-3. [DOI] [PubMed] [Google Scholar]
- Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 2008;7:500–506. doi: 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- Nilsen KE, Cock HR. Focal treatment for refractory epilepsy: hope for the future? Brain Res Brain Res Rev. 2004;44:141–153. doi: 10.1016/j.brainresrev.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W, Kang J, Yu GQ, Adame A, Devidze N, Dubal DB, Masliah E, Conklin BR, Mucke L. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci. 2015;18:423–434. doi: 10.1038/nn.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagonopoulou O, Angelatou F. Time development and regional distribution of [3H]nitrobenzylthioinosine adenosine uptake site binding in the mouse brain after acute Pentylenetetrazol-induced seizures. J Neurosci Res. 1998;53:433–442. doi: 10.1002/(SICI)1097-4547(19980815)53:4<433::AID-JNR5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Pak MA, Haas HL, Decking UKM, Schrader J. Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacol. 1994;33:1049–1053. doi: 10.1016/0028-3908(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Nabbout R, Galanopoulou AS. Mechanisms of epileptogenesis in pediatric epileptic syndromes: Rasmussen encephalitis, infantile spasms, and febrile infection-related epilepsy syndrome (FIRES) Neurotherapeutics. 2014;11:297–310. doi: 10.1007/s13311-014-0265-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson FE, Damaraju VL, Graham K, Yao SY, Baldwin SA, Cass CE, Young JD. Molecular biology of nucleoside transporters and their distributions and functions in the brain. Curr Top Med Chem. 2011;11:948–972. doi: 10.2174/156802611795347582. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Maysami S, Studer FE, Wilz A, Simon RP, Boison D. Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J Cereb Blood Flow Metab. 2008;28:17–23. doi: 10.1038/sj.jcbfm.9600499. [DOI] [PubMed] [Google Scholar]
- Popoli P, Frank C, Tebano MT, Potenza RL, Pintor A, Domenici MR, Nazzicone V, Pezzola A, Reggio R. Modulation of glutamate release and excitotoxicity by adenosine A2A receptors. Neurology. 2003;61:S69–71. doi: 10.1212/01.wnl.0000095216.89483.a2. [DOI] [PubMed] [Google Scholar]
- Ravizza T, Balosso S, Vezzani A. Inflammation and prevention of epileptogenesis. Neurosci Lett. 2011;497:223–230. doi: 10.1016/j.neulet.2011.02.040. [DOI] [PubMed] [Google Scholar]
- Rebola N, Coelho JE, Costenla AR, Lopes LV, Parada A, Oliveira CR, Soares-da-Silva P, de Mendonca A, Cunha RA. Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci. 2003;18:820–828. doi: 10.1046/j.1460-9568.2003.02815.x. [DOI] [PubMed] [Google Scholar]
- Ren G, Li T, Lan JQ, Wilz A, Simon RP, Boison D. Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control. Exp Neurol. 2007;208:26–37. doi: 10.1016/j.expneurol.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseti C, Martinello K, Fucile S, Piccari V, Mascia A, Di Gennaro G, Quarato PP, Manfredi M, Esposito V, Cantore G, Arcella A, Simonato M, Fredholm BB, Limatola C, Miledi R, Eusebi F. Adenosine receptor antagonists alter the stability of human epileptic GABAA receptors. Proc Natl Acad Sci U S A. 2008;105:15118–15123. doi: 10.1073/pnas.0807277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz RA, Wilens TE, Tatter SB, Gregor P, Sellinger OZ. Possible role of increased brain methylation in methionine sulfoximine epileptogenesis: effects of administration of adenosine and homocysteine thiolactone. J Neurosci Res. 1983;10:437–447. doi: 10.1002/jnr.490100410. [DOI] [PubMed] [Google Scholar]
- Schoen SW, Ebert U, Loscher W. 5′-Nucleotidase activity of mossy fibers in the dentate gyrus of normal and epileptic rats. Neuroscience. 1999;93:519–526. doi: 10.1016/s0306-4522(99)00135-9. [DOI] [PubMed] [Google Scholar]
- Sellinger OZ, Schatz RA, Porta R, Wilens TE. Brain methylation and epileptogenesis: the case of methionine sulfoximine. Ann Neurol. 1984;16(Suppl):S115–120. doi: 10.1002/ana.410160717. [DOI] [PubMed] [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J Cereb Blood Flow Metab. 2011;31:1648–1659. doi: 10.1038/jcbfm.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Singer P, Lytle N, Wei CJ, Lan J, Williams-Karnesky RL, Chen JF, Yee BK, Boison D. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia in mice. J Clin Inv. 2012a;122:2567–2577. doi: 10.1172/JCI62378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Singer P, Lytle N, Wei CJ, Lan JQ, Williams-Karnesky RL, Chen JF, Yee BK, Boison D. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest. 2012b;122:2567–2577. doi: 10.1172/JCI62378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Saitoh M, Nishizawa D, Ikeda K, Hirose S, Takanashi J, Takita J, Kikuchi K, Kubota M, Yamanaka G, Shiihara T, Kumakura A, Kikuchi M, Toyoshima M, Goto T, Yamanouchi H, Mizuguchi M. ADORA2A polymorphism predisposes children to encephalopathy with febrile status epilepticus. Neurology. 2013;80:1571–1576. doi: 10.1212/WNL.0b013e31828f18d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva CG, Porciuncula LO, Canas PM, Oliveira CR, Cunha RA. Blockade of adenosine A(2A) receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Szybala C, Pritchard EM, Wilz A, Kaplan DL, Boison D. Antiepileptic effects of silk-polymer based adenosine release in kindled rats. Exp Neurol. 2009;219:126–135. doi: 10.1016/j.expneurol.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J, Kolamunnage-Dona R, Marson AG, Smith PE, Aldenkamp AP, Baker GA. Patients with epilepsy: cognitively compromised before the start of antiepileptic drug treatment? Epilepsia. 2010;51:48–56. doi: 10.1111/j.1528-1167.2009.02195.x. [DOI] [PubMed] [Google Scholar]
- Theofilas P, Brar S, Stewart KA, Shen HY, Sandau US, Poulsen DJ, Boison D. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia. 2011;52:589–601. doi: 10.1111/j.1528-1167.2010.02947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu QW, Peng WG, Lin J, Oberheim N, Lou NH, Wang XH, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nature Medicine. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarkar BG, Castellino AJ, DaRe JM, Kopcho JJ, Wiesner JB, Schanzer JM, Erion MD. Adenosine kinase inhibitors. 2. Synthesis, enzyme inhibition, and antiseizure activity of diaryltubercidin analogues. J Med Chem. 2000a;43:2894–2905. doi: 10.1021/jm0000259. [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE, 3rd, Schanzer JM, Wiesner JB, Erion Md. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem. 2000b;43:2883–2893. doi: 10.1021/jm000024g. [DOI] [PubMed] [Google Scholar]
- Van Gompel JJ, Bower MR, Worrell GA, Stead M, Chang SY, Goerss SJ, Kim I, Bennet KE, Meyer FB, Marsh WR, Blaha CD, Lee KH. Increased cortical extracellular adenosine correlates with seizure termination. Epilepsia. 2014;55:233–244. doi: 10.1111/epi.12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. doi: 10.1016/j.expneurol.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AK, Miller MA, Scanlon J, Ren D, Kochanek PM, Conley YP. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res. 2010;90:259–272. doi: 10.1016/j.eplepsyres.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CJ, Singer P, Boison D, Yee BK, Chen JF. Selective inactivation of adenosine A2A receptors in striatal neurons enhances working memory and reversal learning. Learn Mem. 2011;18:459–474. doi: 10.1101/lm.2136011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Inv. 2013;123:3552–3563. doi: 10.1172/JCI65636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee BK, Singer P, Chen JF, Feldon J, Boison D. Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs. Eur J Neurosci. 2007;26:3237–3252. doi: 10.1111/j.1460-9568.2007.05897.x. [DOI] [PubMed] [Google Scholar]
- Yellen G. Ketone bodies, glycolysis, and KATP channels in the mechanism of the ketogenic diet. Epilepsia. 2008;49(Suppl 8):80–82. doi: 10.1111/j.1528-1167.2008.01843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Franklin PH, Murray TF. Manipulation of endogenous adenosine in the rat prepiriform cortex modulates seizure susceptibility. J Pharmacol Exp Ther. 1993;264:1415–1424. [PubMed] [Google Scholar]