

Abstract
Glycogen synthase kinase-3 (GSK-3) is associated with various key biological processes, including glucose regulation, apoptosis, protein synthesis, cell signaling, cellular transport, gene transcription, proliferation, and intracellular communication. Accordingly, GSK-3 has been implicated in a wide variety of diseases and specifically targeted for both therapeutic and imaging applications by a large number of academic laboratories and pharmaceutical companies. Here, we review the structure, function, expression levels, and ligand-binding properties of GSK-3 and its connection to various diseases. A selected list of highly potent GSK-3 inhibitors, with IC50 <20 nM for adenosine triphosphate (ATP)-competitive inhibitors and IC50 <5 μM for non-ATP-competitive inhibitors, were analyzed for structure activity relationships. Furthermore, ubiquitous expression of GSK-3 and its possible impact on therapy and imaging are also highlighted. Finally, a rational perspective and possible route to selective and effective GSK-3 inhibitors is discussed.
Keywords: Glycogen synthase kinase-3, GSK-3 inhibitors
Introduction
Glycogen synthase kinase-3 (GSK-3) is expressed in all tissues and is a member of the protein kinase family, a group of enzymes that catalyze the transfer of a phosphate group from adenosine triphosphate (ATP) to target substrates. GSK-3 is a serine/threonine kinase, and thus transfers a phosphate group to either the serine or threonine residues of its substrates.1 The mechanism of phosphorylation regulates various complex biological processes, including metabolism (glucose regulation),2-6 cell signaling,7-9 cellular transport,10-12 apoptosis,13-15 proliferation, and intracellular communication.16 Additional roles for GSK-3 in biological processes are likely to be identified in the future.16 Phosphorylation is a key regulatory step that initiates, enhances, or inhibits the function of a target substrate. Given the breadth of processes associated with GSK-3, it is not surprising that GSK-3 has emerged as an important target for drug development and medical imaging in various diseases.
Since the isolation of GSK-3 from rabbit skeletal muscle in 1980,17 its role and participation in glucose metabolism has been extensively studied. However, GSK-3 was considered a therapeutic target for diabetes only after the discovery of its participation in insulin signal transduction in the mid-1990s.18 A primary function of GSK-3 is the regulation of the activity of glycogen synthase (GS), an enzyme that mediates the conversion of glucose to glycogen.19, 20 Under basal conditions, GS is maintained in its inactive form due to phosphorylation by GSK-3. Under conditions of high glucose and insulin availability, GSK-3 activity is inhibited via the action of insulin through PI3 kinase (phosphatidylinositol-4, 5-bisphosphate 3-kinase)/PKB (protein kinase B) pathways. This action results in the activation of GS and the formation of glycogen.21-23 Elevated expression and over-activity of GSK-3 are associated with insulin resistance in type 2 diabetes. Therefore, GSK-3 inhibitors are under development for the treatment of type 2 diabetes.6,20
Elevated GSK-3 levels in bipolar disorder (BD) have been documented in both preclinical and clinical studies.24-27 Specifically, higher levels of GSK-3α and GSK-3β were found in peripheral blood mononuclear cells (PBMC) of BD subjects than in those of healthy controls.26 After treatment of BD patients with lithium, valproate, or atypical antipsychotics, significant increases in the inhibitory serine phosphorylation of GSK-3 were observed in PBMCs. However, the total level of GSK-3 remained unchanged.26 This finding suggests that the ratio of active/inactive GSK-3 has a greater regulatory effect in BD than the total levels of GSK-3.
GSK-3β is also associated with several neurodegenerative diseases,28 including Parkinson's disease (PD),29 Alzheimer's disease (AD),30-33 and Huntington's disease (HD).34 A recent review by Li et al.29 summarized the involvement of GSK-3 in PD and the hypothesis that inhibition of GSK-3β activity may protect dopaminergic neurons. Similarly, recent findings by Llorens-Maritin et al.30-33 highlighted the role of GSK-3β in AD pathobiology, including a predominant role in the phosphorylation of tau protein.18 Furthermore, Lim et al.34 observed higher levels of phosphorylated GSK-3 in post-mortem human HD brain samples correlating with disrupted energy metabolism in two transgenic mouse models.
GSK-3 regulatory activity is associated with acquired immunodeficiency syndrome (AIDS),35,36 malaria,37,38 inflammation,39-41 apoptosis,13-15 neuronal growth,32, 42-44 and canonical Wnt/β-catenin45-47 pathways. GSK-3 also appears to play an important role in cancer progression.48 In some tumor types, GSK-3 exerts tumor suppressor functions, whereas in other tumor types, GSK-3 is associated with tumor progression by stabilizing the beta-catenin complex.49-53 Clearly, GSK-3 is an important regulator of metabolic and signaling functions that are associated with a broad spectrum of disease areas, motivating extensive efforts on inhibitor development and, more recently, imaging probe development.
This review summarizes the recent progress in understanding the structure and function of GSK-3 and the development of high-affinity inhibitors for clinical applications in therapy and imaging. For ATP-competitive inhibitors, our focus is on those with IC50 values <20 nM are discussed. For non-ATP-competitive inhibitors, in general only those with IC50 values < 5 uM are included. Recent progress in the development of GSK-3 imaging probes for positron emission tomography (PET) is also reviewed. Finally, we discuss our perspective on the continued development of GSK-3 targeting molecules for therapy and imaging.
Structure and Functions of GSK-3
Almost all eukaryotic organisms, ranging from flies to humans, contain homologous forms of GSK-3 with a high degree of similarity. In mammals, there are two isoforms of GSK-3, including GSK-3α and GSK-3β, which are encoded by separate genes.54 GSK-3α (51 KDa) has a slightly higher molecular weight than that of GSK-3β (47 KDa). The extra length of GSK-3α is due to the presence of an additional 63 amino acid residues at the N-terminus. Although both isoforms are structurally quite similar, they are functionally different. For example, mice carrying a selective deletion of exon 2 of GSK-3β undergo embryonic death due to extensive hepatocyte apoptosis despite the presence of GSK-3α.55
A detailed structural and functional analysis of GSK-3β reported by ter Haar et al.56 is depicted in Figure 1.56 There are two major GSK-3β domains: a β-strand domain present at the N-terminus between amino acid residues 25-138 (Figure 1) and an α-helical domain present at the C-terminus between amino acid residues 139-343. An ATP-binding site is present at the interface of the two domains that is adjoined by a glycine-rich loop and hinge region. The catalytic activity of GSK-3β is regulated by phosphorylation at two different sites, including Ser 9 and Tyr 216. Phosphorylation of the Ser 9 site inactivates GSK-3β, whereas phosphorylation at Tyr 216 within the activation loop increases its catalytic activity.1, 56 For GSK-3α, phosphorylation at Ser 21 renders it inactive.57
Figure 1.

The different regions of GSK-3β. (Reproduced from ter Haar et al.56 with permission from the Nature Publishing office.)
Before phosphorylation can be catalyzed by GSK-3, both the β-strand and α-helical domains must align in a catalytically active conformation for effective binding of the substrate. GSK-3β phosphorylates various substrates with different efficiencies and mechanisms. In certain cases, GSK-3β directly phosphorylates the substrate, whereas under other circumstances, priming of the substrate by another kinase is required before phosphorylating the target. As a result, the function and mechanism of action of GSK-3β is substrate-specific. Typically, phosphorylation through the priming mechanism is 100 to 1000 times more efficient than phosphorylation without priming.58 Additionally, the action of GSK-3β is regulated by two different mechanisms, including phosphorylation of Ser 9 and formation of a GSK-3β protein complex. Bijur and Jope59 suggested an alternative mechanism of GSK-3β regulation via subcellular compartmentalization.59 Previous studies reported that GSK-3β is a cytosolic protein and predominantly exists in an active form, but may also be present in nuclei60, 61 and mitochondria.62 Bijur and Jope59 noted that induced apoptotic signaling increases the active form of GSK-3β in the nucleus and mitochondria by several-fold, but does not affect the cytosolic levels.59 Interestingly, inhibition of GSK-3β by lithium causes a reduction in the active form of GSK-3β in all three pools.59
Importance of Selectivity in Kinase Inhibition
Due to the pivotal role of intracellular kinases in cell signaling, protein regulation, metabolism, and cellular transport, kinase inhibition has become an attractive target for therapeutic intervention. However, the design and discovery of drugs that effectively target specific kinases (e.g., GSK-3β) is challenging due to the conserved nature of the ATP-binding sites across the kinase family. Typically, small-molecule kinase inhibitors inhibit multiple protein kinases.63, 64 Therefore, it is important to examine the selectivity of drug candidates against a wide array of well-characterized protein kinases to mitigate any possibility of off-target interactions. Two excellent studies by Davis et al.64 and Anastassiadis et al.63 address target kinase selectivity by profiling kinase inhibitors against a wide array of kinases using functional assays. Their findings suggest evaluation of every new inhibitor over a broad range of kinases to map their off-target interactions. There is a long list of known bona fide GSK-3 substrates but not possible to include that in the present review. However, readers are directed to read a recent review on GSK-3 bona fide substrates compiled by Calum Sutherland.65
Different Classes of GSK-3 Inhibitors
A large number of heterocyclic inhibitors of GSK-3 have been identified. ATP-competitive and non-ATP-competitive inhibitors will be discussed separately.
I. ATP-Competitive GSK-3 Inhibitors
Pyrazolopyrimidines
The identification of a pyrazolopyrimidine derivative (1, Table 1a) as a promising GSK-3 inhibitor via high-throughput screening led Peat et al.20,66 to perform detailed molecular modeling to define the structure-activity relationship (SAR) and potentially enhance the affinity of other GSK-3 inhibitors. During this SAR analysis (compounds 2-7), it was noted that the presence of hydrogen at positions R1 and R2 and a phenyl group at R3 were critical for high potency.20 Additionally, the hydrazone hydrogen and proximal nitrogen of pyrimidine interact with GSK-3 by hydrogen bonding to the carbonyl and NH groups of the valine 135 residue, respectively, within the hinge region of the ATP-binding pocket (Figure 2).
Table 1a.
Pyrazolopyrimidines Inhibitors of GSK-3β

*Log of the concentration, in nM, that inhibits 50% of GSK-3 activity.
Figure 2.

The interactions of pyrazolopyrimidine (8) with different regions of GSK-3 during silico docking analysis. (Reproduced from Peat et al.20 with permission from Elsevier Science.)
Further analysis of the docking results (5-7, Table 1a) showed that the substituted phenyl ring at R3 was tightly bound deep in the cavity. Hydrogen bonding between the C-2 hydrogen of pyrimidine and the backbone carbonyl of Asp 133 was also evident. The R4 substituent extended into solvent, suggesting a higher tolerance for modification at this site. Various substitutions were intoduced at R4 to increase the solubility and cell permeability (8-13, Table 1b). It was found that a 4-pyridyl group at R4 increased the cell permeability (8, 9, and 12), and that a benzimidazole moiety at R3, in addition to 4-pyridyl, further enhanced cell permeability (12, Table 1b). In another study by the same group, the S-cis conformation of hydrazine and ring planarity of 3-methoxypheny and pyrazolopyrimidine were shown to enhance the inhibitory potency of this group of inhibitors.67 The authors reported that the pyrazolopyrimidines are ATP-competitive inhibitors of GSK-3β.
Table 1b.
Selected Pyrazolopyrimidine Inhibitors with High Cell Permeability

*The number written inside the parenthesis corresponds to the number/code of the respective compound provided in the original publication.
**Log of the concentration, in nM, that inhibits 50% of GSK-3 activity.
Benzimidazoles
Shin et al.68 reported new analogs of 7-hydroxy-1H-benzimidazole as novel ATP-competitive GSK-3β inhibitors. Some of these inhibitors (Table 2) exhibited IC50 values in the nanomolar (nM) range. Shin et al.68 introduced additional hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) groups in this series to improve GSK-3 binding. These modifications resulted in an increase in the IC50 to 15 to 25 nM (Table 2). Because the ATP-binding site contains Val 135 and Asp 133 residues, Shin et al.68 introduced a hydroxyl group at C7 of the benzimidazole to create hydrogen bonds to the NH group of Val 135,and the carbonyl group of the Asp 133 residue (Figure 3). Additional hydrogen bonding between the carbonyl oxygen at Val 135 and the benzimidazole NH enhanced the activity of the new analogs (Figure 3). The potency of novel 7-hydroxy benzimidazole analogs was compared to that of the known inhibitor 14. Introduction of a p-methanesulfoneamidephenethyl amine group at the fourth position (R2) of the benzimidazoles (15 and 16) produced IC50 values between 15 to 25 nM, whereas when R2 was benzyl or propyl benzene, the IC50 values were observed to be 870 nM and 580 nM, respectively.68 The role of the p-methanesulfoneamidephenethyl amine group at R2, which is important for high activity, was not explained by the authors.
Table 2.
Selected Benzimidazole Analogs and Their IC50 Values

*The number inside the parenthesis is the number/code given to the compound in the original publication.
Figure 3.

Docking result of a benzimidazole inhibitor to GSK-3β, and the hydrogen-bond network within the hinge region. (Reproduced from Shin et al.68 with permission from Elsevier Science.)
Pyridinones
Coffman et al.69 developed a new series of GSK-3β inhibitors using high-throughput screening of a chemical library. These authors discovered that 4-(4-hydroxy-3-methylphenyl)-6-phenyl pyrimidin-2-ol (17, Table 3, Figure 4) was a promising inhibitor with an IC50 value of 17.2 nM observed in a cell-free enzyme assay. The co-crystal structure of 17 and GSK-3β protein was reported. Compound 17 binds within the ATP site of GSK-3, where the phenolic hydroxyl forms hydrogen bonds with the Val 135 and Asp 133 residues of the hinge region. Additionally, the pyrimidine hydroxyl group forms a hydrogen bond with the Lys 85 residue of the catalytic triad of GSK-3β (Figure 4). Coffman et al.69 carried out a detailed SAR study to improve the therapeutic potency of analogs of 17 by mitigating the metabolic liability associated with the phenolic group, and reducing the overall number of hydrogen bond donors. The resulting series of inhibitors and their IC50 values are listed in Table 3 (18-20). The modifications introduced in the series include replacement of a phenolic moiety by a pyrimidine ring, a hydroxypyrimidine replaced by a pyridine, and an aryl group replaced by a variety of simple amines. A docking study of the new pyridone inhibitors showed a good overlap with 17, and predicted their high potency as GSK-3β inhibitors. These structural modifications improved the IC50 value of some of the new pyridone analogs (19-20, Table 3) in a cell-free enzyme assay. However, the new analogs (18-20) exhibited IC50 values higher than 600 nM in a cell-based assay, but showed better central nervous system (CNS) multi-parameter optimization (MPO) scores (Table 3)70.
Table 3.
Selected Pyridinone Analogs Acting as GSK-3β Inhibitors

*The number written inside the parenthesis corresponds to the number/code given to the compounds in their original publication.
aGSK-3β cell-free enzyme inhibition assay.
bGSK-3β whole-cell enzyme inhibition assay.
cCNS multi-parameter optimization (MPO) score. A score >4.5 indicates an increased probability of CNS penetration.
Figure 4.

The structure of 17 and its co-crystal structure with GSK-3β showing the hydrogen bonding sites. (Reproduced from Coffman et al.69 with permission from Elsevier Science.)
In addition to addressing the metabolic liability present in the parent compound (17), the newer inhibitors (19-20) have improved IC50 values in both cell-based and cell-free assays. Previous studies investigated the potency, selectivity, and suitability of pyridinone analogs as therapeutics in the CNS. Inhibitors 18 and 20 are selective for GSK-3β versus casein kinase II (CK2) and dual-specificity protein kinase (CLK1).
Pyrimidines
Nuss et al.71, 72 reported a highly potent pyrimidine series of GSK-3β inhibitors that target the ATP-binding pocket. An interesting finding of their work was the use of an ethylenediamine linker at the pyrimidine (21-24, Table 4). Neither shorter nor longer linkers were well tolerated at the same position, possibly due to changing the planarity of the compound and blocking its transport to the target site. Additionally, the presence of ortho and para substituents on the phenyl ring were well tolerated, and the 2,4-dichlorophenyl substitution produced the highest-affinity analogs (Table 4). The imidazole moiety at the R1 position provided an optimal result. Furthermore, alteration of the point of attachment of the imidazole moiety (attached to -N/ C atom) to the pyrimidine core did not change the potency of the compounds (Table 4). A variety of different substituents were investigated at the R2 and R1 positions, demonstrating that the presence of a combination of 1,3-imidazole and 2-amino, 3-nitropyridine moieties is critical for the enhanced GSK-3 inhibition (21-24, Table 4).
Table 4.
Selected Pyrimidine GSK-3β Inhibitors

*The number inside the parenthesis corresponds to the number/code given to the compounds in their original publication.
The pyrimidine inhibitors 21 and 23 were reported to be highly selective for GSK-3 in comparison to 20 different kinases, including structurally similar kinases such as CDK1 (cyclin-dependent kinase 1) and ERK2 (extracellular signal-regulated kinase 2).73
Indolylmaleimide
Various attempts have been made to develop novel series of indolylmaleimide analogs that can act as ATP-competitive GSK-3 inhibitors.74-77 Zhang et al.74 investigated macrocyclic bisindolylmaleimides with linkers composed of various heteroatoms connecting two indolyl moieties (25-29, Table 5a). Based on the IC50 values listed in Table 5a, the presence of a nitrogen atom at position “J” appears to be critical for enhanced activity as a GSK-3β inhibitor. The importance of this atom is further demonstrated by the loss of activity after replacement of the nitrogen with oxygen (25 versus 26-29, Table 5a). Zhang et al. investigated the selectivity of potent macrocyclic bisindolylmaleimide analogs compared to various isoforms of protein kinase C -α, -γ, -δ, -ε and -ζ. The authors reported that 25 is selective versus other kinases in the panel.74 Additionally, some of the macrocyclic bisindolylmaleimide analogs (25 and 26) were effective at blocking PKC-βII-induced IL-8 release via GSK-3β inhibition.74
Table 5a.
Selected Macrocyclic Bisindolylmaleimide Analogs Acting as GSK-3β Inhibitors

*The number inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Engler et al.76 reported on a new series of ATP-competitive GSK-3β inhibitors based on imidazo[1,2-a]pyridine-3-yl and 4-(1,2,3,4-tetrahydro-[1,4]diazepino-[6,7,1-hi)indol-7-yl substituted pyrrole-2,5-dione (maleimide) scaffolds (30-35, Table 5b). A variety of R1 substituents, including piperidinyl (31-32, 35), morpholinyl (33), and dimethylaminyl (34) gave high-affinity analogs.
Table 5b.
Selected Bisarylmaleimide Analogs Acting as GSK-3β Inhibitors

*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Inhibitors in this series were shown to have high selectivity for GSK-3β in comparison to a panel of other kinases in a cell-based assay. Compound 30 was over 15,000 fold selective for GSK-3β versus AKT-1, KDR, TGFβ-R2, hPKAβ, MLK7, CAMKII, and p38, and 600 - 2,500 fold selective versus CDK2, CDK4, and PKCβ-II (Table 5b). Most of the other inhibitors of the series showed a similar degree of selectivity.76 Among these, inhibitors 33 and 35 (IC50 = 1.3±0.1 nM and 1.1±0.3 nM, respectively) were subsequently evaluated in in vivo studies. Compound 35 was selected for evaluation in an oral glucose tolerance test in ZDF rats. In this dose-response study, rats were treated with 35 (0.1 - 3 mg/Kg) by gavage for 8 days. On the eighth day, they received a 1.25 g/Kg oral glucose challenge, and blood glucose was measured over 2 hours. Inhibitor 35 had a threshold minimum effective dose (TMED) of 0.49 mg/Kg for reduction of blood glucose, confirming robust in vivo activity.
Engler et al.75 developed another series of non-macrocyclic analogs of bisarylmaleimide that can act as GSK-3β inhibitors. The authors changed one of the indolyl moieties to diazepino-indolyl and another indolyl moiety with a variety of different aryl and heterocyclic aryl analogs. The IC50 values of the new series of compounds 36-45 (Table 5c) were lower than those of the macrocyclic bisindolylmaleimdes.74 The presence of a 2,3-dihydro-1-benzofuran substituent in 36 produced an IC50 value of 1.1 nM (Table 5c). However, imidazo[1,2-b]pyridazine and imidazo[1,2-a]pyridine substitutions at the R2 position in 43 and 45 gave the lowest IC50 values for the series at 0.7 and 0.8 nM, respectively (Table 5c). The selectivity of new analogs (36-45) for GSK-3 inhibition over other kinases like CDK2, CDK4, and PKCβ were evaluated (Table 5c). Compound 41 was found to be 8,575 times more potent as an inhibitor of GSK-3 than CDK4, and 11,800 times more selective over CDK2 and PKCβ.75 The remarkable specificity of 41 was attributed to the presence of imidazo[1,2-a]pyrimidine moiety. Engler et al.75 reported that most of the analogs (36-45) showed a >160- to 10,000-fold higher selectivity for GSK-3 inhibition relative to other kinases. Additionally, some of these compounds produced lower plasma glucose in ZDF rat models of type 2 diabetes and a strong inhibition of tau phosphorylation.
Table 5c.
Selected Bisarylmaleimide Analogs Acting as GSK-3β Inhibitors

*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Imidazopyridines
Yngve et al.78 reported a new series of imidazopyridine analogs that act as GSK-3β inhibitors (Table 6). These authors utilized the hydrogen acceptor and donor motif to achieve effective interactions with kinase residues and binding. Among these inhibitors, 46 and 47 showed the highest inhibition with IC50 values of 3.0 and 1.4 nM, respectively. An analysis of the structure of these analogs suggests that their additional hydrogen bond-accepting capacity is likely to be responsible for their higher potency. Imidazopyridine analogs inhibited GSK-3β activity through competitive binding to the ATP domain of the enzyme. Despite the promising inhibitory activities of imidazopyridine analogs, their selectivity compared with other kinases has not been reported.78
Table 6.
Selected Imidazopyridine Analogs Acting as GSK-3β Inhibitors

*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Oxadiazoles
After a 1,3,4-oxadiazole-containing compound was identified to be a GSK-3β inhibitor through high-throughput screening, Saitoh et al.79 synthesized a series of novel analogs of 1,3,4-oxadiazole (48-52, Table 7). Among these, 52 showed the highest selectivity and potency for GSK-3β inhibition with an IC50 value of 2.3 nM. The selectivity of 52 for inhibition of GSK-3β was compared with 23 different kinases, including CDK2, CDK5, PKA, ERK1, and JNK1, and the results showed a >1,000-fold higher selectivity in all cases except for CDK1, MEKK-1, and PKCΘ, where selectivity was 460, 810, and 350 times, respectively. The co-crystal structure of 52 and GSK-3β showed hydrogen-bonding interactions between the benzimidazole moiety and the hinge region of GSK-3β. An additional interaction was suggested for the oxadiazole nucleus and the Arg 141 residue on GSK-3β. The co-crystal structure analysis confirmed the competitive binding of 1,3,4-oxadiazole analogs with the ATP-binding site of GSK-3β as the inhibitory mode of action.
Table 7.
Selected 1,3,4-Oxadiazole Analogs Acting as GSK-3β Inhibitors

*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Pyrazines
Based on a high-throughput screen and co-crystal structure analysis of pyrazine-containing derivatives with GSK-3, Berg et al.80 reported a new series of pyrazine analogs (53-62, Table 8). The X-ray crystal structure of some of these analogs confirmed their ability to act as ATP-competitive inhibitors of GSK-3β. In a SAR analysis, it was found that the presence of small substituents like F, CH3, or CF3 ortho to the pyrazine ring (53-56) caused a considerable increase in the GSK-3β inhibition potency and selectivity.80 Compounds 54 and 58-62 showed IC50 values for GSK-3β <1.0 nM. The selectivity of individual compounds varied. However, among the potent pyrazine analogs, 54 and 56 showed 3,700- and >20,000-fold selectivity versus CDK2, respectively. Compounds 57 and 59 were tested for blood-brain barrier permeability in a bovine endothelial cell assay, and all three of these compounds showed good permeability in this assay. The permeability of other analogs was examined by Caco2 assay for oral suitability. The results are listed in Table 8. Among these, 55 and 56 showed the highest permeability. Furthermore, some of these analogs (53-59) showed a substantial reduction in tau phosphorylation in a dose-dependent manner, having IC50 5-87 nM in 3T3 fibroblast cells. Therefore, these pyrazine analogs show promise as candidate molecules for therapy and imaging in AD.
Table 8.
Selected Analogs of Pyrazines Acting as GSK-3β Inhibitors

Abbreviations: ΨKi, is an inhibitory constant; nd, not determined.
*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Perez et al.81 reported on the ability of a halomethylketone addition to convert a reversible inhibitor into a more potent irreversible GSK-3 inhibitor. A few examples (63-70) are listed in Table 9 showing improved IC50 after halomethylketone addition. Remarkably, small addition of halomethylketone on compounds 63, 65, 67 and 69 caused 4-178 fold decrease in their IC50 values (Table 9). Perez et al. 81 demonstrated that the halomethylketone moiety reacts with the sulfur atom of cysteine-199 present in the ATP binding site of GSK-3. Irreversible binding was also confirmed through model reactions, docking studies and mass spectrometric analysis of the resultant product 81. This approach may be useful in designing more potent GSK-3 inhibitors.
Table 9.
Comparison of IC50 and Binding Mode Before and After Addition of Halomethylketone

* Number written inside the parenthesis corresponds to the original number /code given to the respective compound in their original publication.
II. Non-ATP-Competitive GSK-3 Inhibitors
In early 2002, Martinez et al.82 were the first to report thiadiazolidinones (TDZDs, 71-81, Table 10) to be non-ATP-competitive GSK-3β inhibitors. It was hypothesized that these compounds recognize and interact with the oxyanion binding site of GSK-3β. These authors also suggested that the negative charge of TDZD was important for oxyanion recognition.82 Different analogs of TDZD were synthesized and evaluated for GSK-3β-inhibitory properties (71-81, Table 10). On a careful SAR analysis, it is evident that a substitution of the ethyl group (71-73) with benzyl (74-76) or substituted phenyl moieties (77-81) at the R1 position have considerably increased the GSK-3β-inhibition potency of thiadiazolidinone analogs. Additionally, the presence of a methyl group (71, 74, and 77-81) was preferred over ethyl (75), propyl (72), cyclohexyl (73), and benzyl (76) groups at the R2 position to retain the potency of the thiadiazolidinone analogs. One of these TDZD analogs, 74 (TDZD-8), was screened against a variety of other kinases, including protein kinase A and C (PKA, PKC), casein kinase II (CK2), and cyclin-dependent kinase 1 (CDK1/cyclin B). Compound 74 showed high selectivity for GSK-3β with a >100 μM IC50.
Table 10.
Select Analogs of Thiadiazolidinones (TDZDs) Acting as GSK-3β Inhibitors

Abbreviations: CDK1/cycline B, cyclin-dependent kinase 1; CK2, casein kinase II; PKA, protein kinase A; PKC, protein kinase C.
*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Even though TDZD analogs have shown GSK-3β-inhibition constants in the μM range, there remains an interest to test them in human subjects as a neurodegenerative disease treatment. One of the TDZD analogs, later named Tideglusib (NP031112, NP-12, 94), was under clinical investigation and further discussed in the section describing the compounds used in clinical trials.
Emodin and 4-Aminoethylamino Emodin
Gebhardt et al.83 showed application of emodin (82) and its ethylenediamine analog 83 as non-ATP competitive inhibitors of GSK-3 (Table 11). Addition of the ethylenediamine group on the emodin nucleus increased potency of inhibition (IC50 0.56±0.02 µM, 83), reduced cytotoxicity and generated an insulin sensitizing effect mediated by increasing hepatocellular glycogen and fatty acid biosynthesis. Selectivity's of compounds 82 and 83 were evaluated against twelve protein kinases including eleven of human protein kinases. Compound 83 showed high selectivity towards GSK-3β but 82 failed to do so.
Table 11.
Emodin Analogs Acting as GSK-3 inhibitors

* Number written inside the parenthesis corresponds to the original number /code given to the respective compound in their original publication.
5-Imino-1,2,4-Thiadiazoles (ITDZs)
Polomo et al.84 reported 5-imino-1,2,4-thiadiazoles (ITDZs) as a first class of compounds able to inhibit GSK-3 in a substrate competitive manner. The class of ITDZs was identified by performing a detailed structure activity relationship study on a thiadiazolidinedione family. Thirty one ITDZ variants were synthesized, thirty of which were shown to have more than 60 % of enzymatic (GSK-3) inhibition at 10 µM concentrations. The IC50 values of these compounds were found in the low micromolar-submicromolar range (0.3-7.0 µM). The most potent compounds are listed in Table 12. No major difference in the IC50 values of the compounds 84-88 were found even by changing various functional groups at the positions R1 or R2 or R3 (Table 12). However, a slightly negative change was observed when Br- in 86 was replaced with Cl- in 89 as a counter ion. Due to small differences in IC50 values of the various ITDZs analogs, it is hard to draw any conclusive structure activity relationship among these ITDZs.
Table 12.
Selected Analogs of 5-Imino-1,2,4-Thiadiazoles Acting as GSK-3 inhibitors

* Number written inside the parenthesis corresponds to the original number /code given to the respective compound in their original publication.
Through a kinetic study, Polomo et al. 84 confirmed that ITDZs bind reversibly to GSK-3. ITDZs are non-ATP competitive inhibitors and compete with the primed substrate in the GSK-3 inhibition. Utilizing a docking study, Polomo et al. 84 concluded that ITDZs interact with GSK-3 protein through: (i) hydrogen bonding between the proton of the imino charged group and backbone oxygen of Phe67 and (ii) Aromatic S- π interaction between the thiadiazole nucleus and the aromatic ring of the Phe67. The blood brain barrier (BBB) permeability of these analogs were investigated using the parallel artificial membrane permeability assay (PAMPA) and the results are presented in Table 12, showing that several of the ITDZs analogs are able to cross the BBB by passive permeation.
Conde et al.85 identified two new classes of molecules as non-ATP competitive inhibitors of GSK-3β: chloromethyl thienyl ketones and halomethyl phenyl ketones. In addition to the forementioned compounds there are a few natural products (90-92) reported as non-ATP competitive inhibitors as listed in Table 13 86-88. Among these, the β-carboline alkaloid Manzamine A (92) showed a high degree of selectivity towards GSK-3β as demonstrated by Hamann et al.89 Manzamine was isolated from the sponges found in the Indo-pacific regions86, 90. Palinurin (90) and Tricantin (91) are furano sesquiterpenoids isolated from Mediterranean sponges28. Interestingly, Bidon-Chanal et al.91 in their recent study demonstrated allosteric modulation of GSK-3β with 90.
Table 13.
Selected miscellaneous Non-ATP-competitive GSK-3β inhibitors

* Number written inside the parenthesis corresponds to the original number /code given to the respective compound in their original publication.
In a recent study, Palomo et al.92 identified available binding sites using the geometry-based algorithm fpocket (http://fpocket.sourceforge.net) and hpocket programs to map the surface of GSK-3. Palomo et al. identified seven conserved binding sites (Figure 5). These binding sites could be targeted for selective and effective modulation of protein-catalytic functions. Out of the seven identified binding sites on the surface of GSK-3, three consisted of the previously reported (Figure 5, sites 1-3) ATP, substrate, and peptides axin/fratide-binding sites. The four additional binding sites were newly identified to be non-ATP sites.92
Figure 5.

The binding sites available on the surface of GSK-3. (Reproduced from Palomo et al.92 with permission from the American Chemical Society.)
One of the four additional sites is situated on the C-terminal lobe (carboxyl terminal) of the protein and is exposed to the solvent. Another site is located on the hinge region. The last two sites are located on the N-terminal lobe (amino terminal) of the protein.92
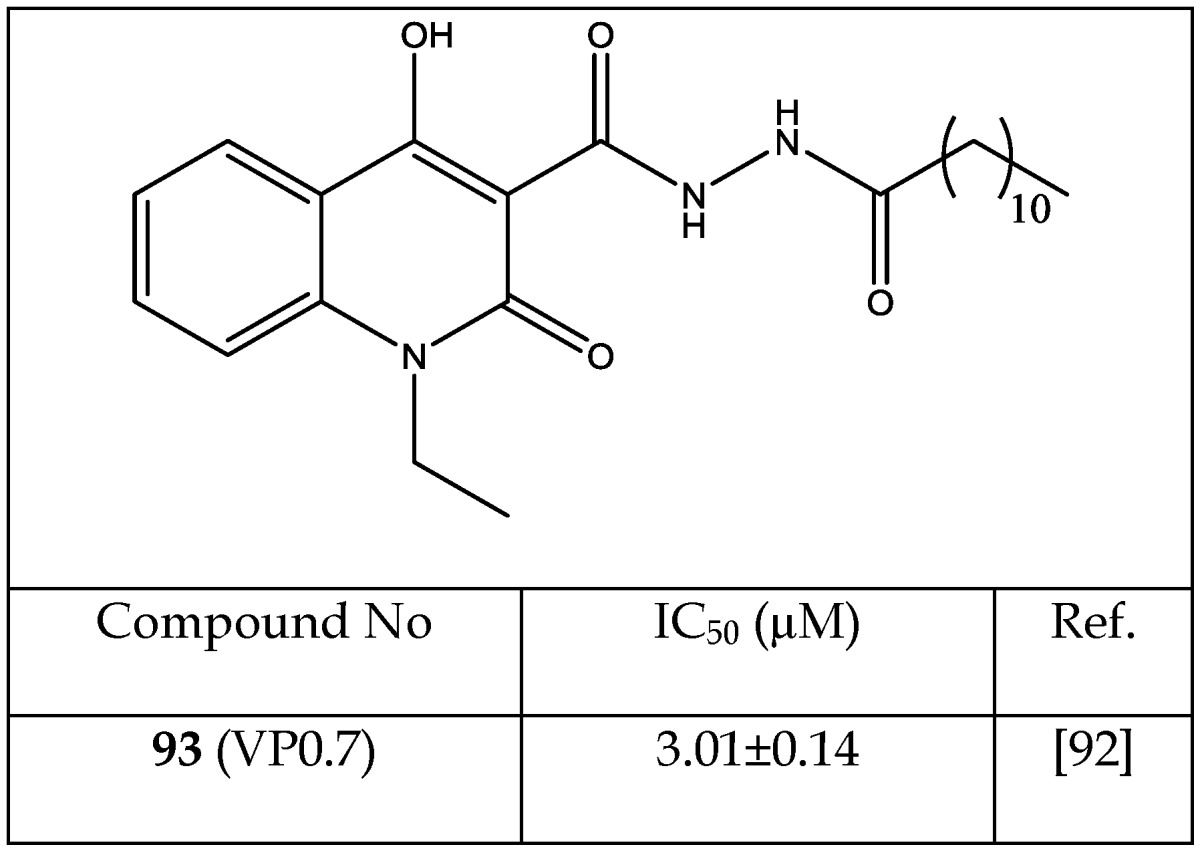
A quinolone moiety (N'-dodecanoyl-1-ethyl-4-hydroxy-2-oxo-1,2-dihydroquinoline-3-carbohydrazide (93), Table 14), IC50= 3.01±0.15 μM for GSK-3, was identified as a non-ATP-competitive inhibitor of GSK-3. The non-ATP-competitive nature of quinolone moiety was evidenced by a lack of sensitivity to increased ATP concentration on enzymatic inhibition. A docking study of the quinolone moiety and GSK-3 supported the notion of an ATP-independent interaction. It was proposed that a change in the activation loop of the enzyme resulted from allosteric binding of the quinolone to GSK-3.92 In our opinion, the discovery of ATP-independent binding sites on GSK-3 by Palomo et al.92 is valuable and may lead to the design and development of new GSK-3 modulators that bind to allosteric sites. Forthcoming GSK-3 modulators may be able to fine-tune the selectivity of anti-GSK-3 activity.
Table 14.
First Quinoline Analog Acting as a Non-ATP-Competitive Inhibitor
Number written inside the parenthesis corresponds to the original number /code given to the respective compound in their original publication.
Clinical Prospects of GSK-3 in Imaging and Therapy
The involvement and association of GSK-3 in the pathogenesis of a wide variety of diseases makes it an important therapeutic and imaging target. The potential of GSK-3β inhibition in diabetes, bipolar disorder, and AD has already been identified, and considerable progress has been made in the development of various GSK-3β-inhibitory molecules. The use of GSK-3 in the noninvasive imaging of various pathological conditions has yet to be explored; however, some researchers have begun developing imaging probes targeted to GSK-3.93-98
Clinical Studies on GSK-3 Inhibitors
Of the many developed GSK-3 inhibitors, only a few have reached clinical trials in human subjects. Here, we review clinical studies that have resulted in peer-reviewed articles along with studies listed on clinicaltrials.gov for GSK-3 inhibitors. We included Tideglusib (94), LY2090314 (95), Enzastaurin (96), and lithium chloride (97) in our discussion (Table 15).
Table 15.
GSK-3β Inhibitors Under Clinical Investigation

*The number written inside the parenthesis corresponds to the number/code given to the in the original publication.
Tideglusib (94), also known as NP031112/NP-12, was designed for the treatment of AD and progressive supranuclear palsy (PSP).99-101 The latter is characterized by the deterioration of cells in areas of brain that control body movement and thinking. Tideglusib binds irreversibly to the non-ATP-competitive GSK-3β site, as demonstrated in a binding study using radiolabeled Tideglusib (35S-tideglusib) and GSK-3β C199A.112 An IC50 value of 60 nM was reported.112 The safety and efficacy characteristics of Tideglusib were investigated in human patients with AD and progressive supranuclear palsy.99, 101 This small study (n= 30 mild-moderate AD subjects) showed Tideglusib to be safe.101 Importantly, AD patients treated with Tideglusib showed a considerable improvement in the mini-mental status examination (MMSE), a test that measures cognitive function, and thus encourages further clinical trials.101 Two other clinical studies were conducted using Tideglusib in patients with PSP.99,100 The drug was reported to be safe with some minor side effects, but it did not show any clinical improvement in the PSP patients.
The GSK-3 inhibitor LY2090314 (95) was developed by Eli Lilly for the treatment of cancer.103 It selectively targets the ATP-binding site as a competitive inhibitor. The clinical study was focused on the pharmacokinetics, metabolism, and excretion of the GSK-3 inhibitor 95 in humans. The clearance of 95 in cancer patients was very rapid in the first hour, less rapid in next 8 hours, and ever slower at the 24-hour time point. The half-life of 95 in humans was reported to be 1.8 to 3.35 hours, and the primary site of metabolism was identified as the piperidine ring, opening to aminopropionic acid and piperidine N-dealkylation.102, 103 However, the authors did not report any therapeutic effects of 95.102, 103
Enzastaurin, also known as LY317615 (96), was developed as a selective inhibitor for PKC-β with an IC50 of 0.6 nM. However, it indirectly reduces the phosphorylation of GSK-3β.113 A clinical trial was carried out using enzastaurin in patients with recurrent gliomas and ovarian cancer.104, 105 The drug showed unacceptable toxicity at the higher doses required for clinical efficacy and was therefore discontinued.104, 105
Lithium (97) has been utilized for decades as a clinical treatment of bipolar disorder and is known to act at least in part through GSK-3 inhibition.111, 114, 115 A major role of GSK-3β in neurologic disorders has been implicated due to its phosphorylation of tau protein. Hyperphosphorylation of tau is a pathological characteristic of Alzheimer's disease. Accordingly, various clinical studies have evaluated the therapeutic effect of lithium on AD.106-108 The results of these trials appear mixed. Some studies reported no effects, whereas other reported mild affects. However, there remains an interest to continue further clinical studies with lithium as a treatment option for AD to obtain more meaningful information.
Therapeutic Prospects
To date, involvement of GSK-3 in the pathophysiology of a wide variety of diseases, including type 2 diabetes,2-6 bipolar disorder,116 schizophrenia,117-121 AD,24, 30, 31, 122, 123 PD,124 developmental disorders24, 27, 125 and cancer,126-130 has been implicated. Among these, the most studied example of a GSK-3β-targeted therapy is obesity/diabetes. Various pharmaceutical companies have developed a variety of high-affinity GSK-3β inhibitory molecules as anti-diabetics.2-6 Nikoulina et al.6 reported higher expression levels of GSK-3 in the skeletal muscle of type 2 diabetic patients relative to healthy subjects. Inhibition of GSK-3 in obese mouse models resulted in reductions in body weight and serum glucose levels and increases in serum insulin, C-peptide, and adiponectin.131
The second most studied GSK-3β-targeted therapy is for the treatment of BD. Lithium (a known inhibitor of GSK-3), valproic acid, and atypical antipsychotics have produced mild improvement of BD patients' cognitive condition.132-137 Li et al.26 reported considerably higher levels of GSK-3 (non-phosphorylated) in bipolar manic subjects than in healthy controls. Kozlovsky et al.120 found substantially lower levels of GSK-3β in the post-mortem brains of schizophrenic patients in comparison to the levels in normal human brains.
The frequent dysregulation of the canonical Wnt/β-catenin and PI3K/Akt signaling pathways in cancer and the intrinsic role of GSK-3 in these pathways have made a strong connection between GSK-3 and cancer.126-130 Dysregulation or silencing of GSK-3 has been documented in advanced prostate cancer.138 Therefore, therapeutic modulation of GSK-3 could be advantageous in cancer therapy.
Evidence is growing for a connection of GSK-3 dysregulation in neurodegenerative diseases, such as AD and PD.139 AD has been suggested to be both directly and indirectly associated with GSK-3 dysregulation.122 Hooper et al.122 proposed a “GSK-3 hypothesis of AD” and emphasized hyperphosphorylation of tau protein mediated by GSK-3 as a mechanism leading to neuronal apoptosis. With regard to PD, Morales-Garcia et al.124 reported a neuroprotective effect in PD using GSK-3 inhibitors in an in vitro model of dopaminergic cell death.
Imaging Prospects: To develop an imaging probe, various critical and essential factors must be considered. Among these, biochemical, physiological, and chemistry aspects are essential for a successful imaging probe development. In 2010, Cohen summarized the obligatory criteria for the study of protein kinase inhibitors.140 He emphasized that, due to the conservation of ATP-binding sites across various protein kinases, there is a high probability of inhibition of additional protein kinases other than the targeted protein kinase. Therefore, he suggested a mandatory evaluation of protein kinase inhibitors against a wide array of different kinases (50-100) to assess the specificity and selectivity of a particular inhibitor.140 Several studies have been published on displacement assays141-143 and tissue based assays,144, 145 which could be meticulously utilized depending upon the target site (ATP-competitive/non-ATP-competitive binding or allosteric interaction) for GSK-3 inhibitors. The judicious use of displacement assays and tissue-based assays would no doubt accelerate the screening of new GSK-3 inhibitors. Higher binding affinity, greater selectivity, minimal reversibility, lower non-specific binding, higher metabolic stability, excellent solubility of the drug in an injectable formulation, and known or defined mode of interaction are the ideal characteristics of the imaging probe.
There are often challenges in synthetic chemistry or radiochemistry to obtain the desired probe once the biochemical and physiological characteristics are optimized. Depending on the imaging modality, the synthetic chemistry requirements may vary considerably. For example, optical probes are challenged to maximize photon yield while minimizing photobleaching. PET probes are challenged by their shorter half-life and dependence on automation to reduce radiation exposure. For a PET imaging agent to succeed, a reliable and efficient method of synthesis that is translatable across radiochemistry facilities must be established. Furthermore, the synthesis method must be amenable to current Good Manufacturing Practice (cGMP) methods with a comprehensive quality assurance program if the probe is to be applied in human subjects.
GSK-3 Imaging Probe Developments
The disparate levels of GSK-3 in various pathophysiological conditions offer the potential for it to be exploited as an imaging biomarker for diagnosis and therapy monitoring. Recently, radiolabeled imaging probes for PET have been developed based on known GSK-3 inhibitors (Table 16). Hick et al.94, 146 and Vasdev et al.95 reported two 11C-labeled compounds (98, 99) based on AR-A014418 (IC50 = 104 nM) can act as PET probes for GSK-3β. These compounds were successfully radiolabeled; however, their ex vivo biodistribution showed no appreciable brain uptake in conscious rodents.95 Wang et al.98 developed the 11C-radiochemistry to label the GSK-3 inhibitor SB-216763 (IC50 = 34 nM for both GSK-3α/β isoforms) (100), but no biological evaluation data were reported. However, recently Li et al.147 conducted a preliminary study using the same compound, [11C]SB-216763, in rodents and non-human primates for PET imaging. Authors reported a high initial uptake in GSK-3 rich regions of the brain (e.g., thalamus, striatum, cortex, and cerebellum) followed by washout. In 2014, Cole et al.97 reported the 11C-labeling of pyrATP-1 (IC50 = 4.9 nM) to form 11C- pyrATP-1 (101) that can serve as a PET probe of GSK-3β for imaging in neurodegenerative diseases. Although autoradiography using this compound in ex vivo rat brain slices showed specific binding in the cortex, caudate putamen, and nucleus accumbens but there was very little uptake in the brain after in vivo administration in rodents and in non-human primates. Brain uptake of 11C-pyrATP-1 was moderately enhanced when rats were pre-treated with a known P-glycoprotein (Pgp) blocker cyclosporine A, suggesting that the radioligand was subjected to back-flux from the brain through Pgp transporter.
Table 16.
PET Imaging Probes for GSK-3β

Abbreviation: Nd, not determined.
*The number written inside the parenthesis corresponds to the number/code given to the compound in the original publication.
Recently, Mossine et al.148, 149 reported radiosynthesis of three PET probes (102-104) for potential GSK-3 imaging of CNS. Two of these molecules (102, 103) are analogs of 5-imino-1, 2, 4-thiadiazole (ITDZ) class of GSK-3 inhibitors reported by Perez et al.150 Neither IC50 value nor imaging data of any of the new analogs (102-104) were reported, but biological evaluation studies are ongoing.148, 149
In a preliminary communication, Mossine et al.151 labelled tideglusib (105) via [11C] CO2 fixation and cyclization as [11C]tideglusib but did not present imaging or biodistribution data. Tideglusib is a well-known non-ATP competitive and irreversible inhibitor of GSK-3β as described previously.99-101 Liang et al.152 discovered and labeled a new GSK-3α/β inhibitor N-(3-(1H-1,2,4-triazol-1-yl)propyl)-5-(3-chloro-4-methoxyphenyl)oxazole-4-carboxamide ( PF-367) having IC50 values of 1.1 nM, 2.1 nM and 9.0 nM as measured via three different enzymatic assays. PET probe 106 showed equal affinity for both GSK-3α and β, while exhibiting >450- fold selectivity for GSK-3 in a panel of >400 neuroreceptors, enzymes and kinases.152 The imaging potential of labeled [11C]PF-367 (106) was investigated in non-human primates and showed fairly uniform brain distribution with a peak uptake of 2 SUV units followed by a fast clearance152.
More effort is likely forthcoming in the development of brain-penetrant PET probes for GSK-3. In addition to the challenge of avoiding the potential effects of Pgp transport, the radioligand should selectively bind to GSK-3 in its non-phosphorylated active state rather than its phosphorylated inactive state. As previously mentioned, most GSK-3 inhibitors from which a radioligand would be derived were identified from ATP-competitive binding assays performed while the kinase was in its active state. This methodology provides excellent structural stability of the kinase protein for identification of high-affinity inhibitors. However, the homology of the ATP-binding site across a broad range of kinases can result in poor specificity of the inhibitor for the target kinase.153 The corresponding imaging radioligand may likewise suffer poor specificity. For this reason, we believe that an alternative approach may be needed to achieve ligand/inhibitor constructs with sufficient affinity and selectivity to succeed as GSK-3 imaging agents. Clearly, a thorough understanding of the signaling pathways and factors governing GSK-3 expression and its phosphorylation state in various pathological conditions will be instrumental in the meaningful interpretation of GSK-3 radioligand imaging.
Perspectives and Conclusions
GSK-3 is involved in a large number of key cellular processes and exhibits dysregulation in a wide variety of disease states. Accordingly, modulation of GSK-3 activity has been deemed an important approach for therapy and imaging. As shown in this limited review, a very large number of GSK-3 inhibitors have been synthesized. However, as reflected in the slow progress of these inhibitors toward clinical translation, a number of challenges remain. Adverse effects caused by off-target activity of GSK-3 inhibitors were determined after the screening of compounds that bind to the ATP-competitive binding site conserved across a broad range of kinases. Therefore, it would be extremely advantageous to design and develop GSK-3 inhibitors that can selectively target individual pathways and differentiate between the phosphorylated and non-phosphorylated forms of GSK-3. In this regard, parallel development of a therapeutic molecule with a closely related imaging biomarker may allow early evaluation of drug candidates based on noninvasive imaging measurements of the target-to-background ratios. Imaging can also be instrumental in defining the brain uptake patterns of GSK inhibitors intended for brain-related disorders, which would allow for the early dismissal of candidates that show poor brain penetration or retention. Alternative approaches to ligand design may be needed to improve GSK-3 targeting specificity for both therapy and imaging purposes.
Acknowledgments
The authors thank the Department of Radiology at Mayo Clinic in Rochester, Minnesota, for their financial support and Mr Jeffrey M. McEarchern for proofreading the manuscript.
Abbreviations
- GSK-3
glycogen synthase kinase-3
- nM
nanomolar
- ATP
adenosine triphosphate
- GS
glycogen synthase
- PI-3 kinase
phosphatidylinositol-4, 5-bisphosphate 3-kinase
- PKB
protein kinase B
- BD
bipolar disorder
- AD
Alzheimer's disease
- Wnt pathway
a pathway that transfers signals from the outside of the cell to the inside of the cell through cell surface receptors
- β-catenin
cadherin association protein
- KDa
kilodalton
- IC50
the half maximal inhibitory concentration
- SAR
structure activity relationship
- HBD
hydrogen bond donor
- HBA
hydrogen bond acceptor
- PBMC
peripheral blood mononuclear cells
- AIDS
acquired immunodeficiency syndrome
- Pgp
P-glycoprotein
- PET
positron emission tomography
- MDR-MDCK assay
a permeability assay using Madin-Darby canine kidney cells with the MDR1 gene
- CDK1
cyclin-dependent kinase 1
- ERK2
extracellular signal-regulated kinase 2, also known as mitogen activated protein kinase
- IL-8
interleukin 8
- ZDF
Zucker diabetic fatty
- PIM1
proto-oncogene serine/threonine-protein kinase
- PAK1
serine/threonine-protein kinase 1
- DAPK1
death-associated protein kinase 1
- MLCK
myosin light-chain kinase
- FLT4
fms-related tyrosine kinase 4
- CK2
casein kinase-II
- CLK1
dual specificity protein kinase
- PD
Parkinson's disease
- HD
Huntington's disease
- CNS
central nervous system
- TMED
threshold minimum effective dose
- TDZD
Thiadiazolidinones
- PKA
protein kinase A
- PKC
protein kinase C
- PSP
progressive supranuclear palsy
- MMSE
mini-mental status examination, cGMP, current Good Manufacturing Practice
- and ITDZ
5-imino-1, 2, 4-thiadiazole.
Biographies
Mukesh K. Pandey, PhD, received his BSc (H) and MSc in chemistry from Banaras Hindu University. He completed his MPhil and PhD in natural product and synthetic organic chemistry at the University of Delhi. After receiving his doctorate, he joined the University of Massachusetts at Lowell for his post-doctoral training on the design and synthesis of natural product-based polymeric material for targeted drug delivery. In 2009, he moved to Harvard Medical School for radiochemistry and molecular imaging training as a research fellow with Dr Timothy DeGrado. In 2012, he joined the Molecular Imaging Research program at Mayo Clinic in Rochester, Minnesota, as an Assistant Professor of Radiology. His research interests include the design, synthesis, and evaluation of novel molecular PET imaging probes for neurodegenerative diseases.
Timothy R. DeGrado, PhD, received his BS in nuclear engineering from the University of California-Santa Barbara in 1982. He received his MS (1985) and PhD (1988) degrees in medical physics from the University of Wisconsin-Madison, followed by post-doctoral research fellowships in Jülich, Germany (1988), and the University of Michigan-Ann Arbor (2001). He is currently the Director of Molecular Imaging Research at Mayo Clinic in Rochester, Minnesota. His specific interests include molecular imaging probe development for multiple imaging modalities, radioisotope production, radiochemistry, and radiopharmacology. His research has focused on receptor ligands and metabolizable small molecule radiotracers for cancer, neuronal imaging, and cardiovascular imaging.
References
- 1.Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–86. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Summers SA, Kao AW, Kohn AD, Backus GS, Roth RA, Pessin JE. et al. The role of glycogen synthase kinase 3beta in insulin-stimulated glucose metabolism. J Biol Chem. 1999;274:17934–40. doi: 10.1074/jbc.274.25.17934. [DOI] [PubMed] [Google Scholar]
- 3.Lochhead PA, Coghlan M, Rice SQ, Sutherland C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001;50:937–46. doi: 10.2337/diabetes.50.5.937. [DOI] [PubMed] [Google Scholar]
- 4.Ciaraldi TP, Oh DK, Christiansen L, Nikoulina SE, Kong APS, Baxi S. et al. Tissue-specific expression and regulation of GSK-3 in human skeletal muscle and adipose tissue. Am J Physiol-Endoc M. 2006;291:E891–E8. doi: 10.1152/ajpendo.00176.2006. [DOI] [PubMed] [Google Scholar]
- 5.Nikoulina SE, Carter L, Plodowski R, Chinnapongse S, Henry RR. Effects of modulation of GSK-3 alpha protein expression on glucose metabolism in human skeletal muscle cell cultures. Diabetes. 2001;50:A275–A6. [Google Scholar]
- 6.Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–71. doi: 10.2337/diabetes.49.2.263. [DOI] [PubMed] [Google Scholar]
- 7.Watkins CC, Sawa A, Pomper MG. Glia and immune cell signaling in bipolar disorder: insights from neuropharmacology and molecular imaging to clinical application. Transl Psychiatry. 2014;4:e350. doi: 10.1038/tp.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurer U, Preiss F, Brauns-Schubert P, Schlicher L, Charvet C. GSK-3-at the crossroads of cell death and survival. J Cell Sci. 2014;127:1369–78. doi: 10.1242/jcs.138057. [DOI] [PubMed] [Google Scholar]
- 9.Montori-Grau M, Tarrats N, Osorio-Conles O, Orozco A, Serrano-Marco L, Vazquez-Carrera M. et al. Glucose dependence of glycogen synthase activity regulation by GSK3 and MEK/ERK inhibitors and angiotensin-(1-7) action on these pathways in cultured human myotubes. Cell Signal. 2013;25:1318–27. doi: 10.1016/j.cellsig.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Singh V, Lin R, Yang J, Cha B, Sarker R, Tse CM. et al. AKT and GSK-3 are necessary for direct ezrin binding to NHE3 as part of a C-terminal stimulatory complex: role of a novel Ser-rich NHE3 C-terminal motif in NHE3 activity and trafficking. J Biol Chem. 2014;289:5449–61. doi: 10.1074/jbc.M113.521336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song J, McColl J, Camp E, Kennerley N, Mok GF, McCormick D. et al. Smad1 transcription factor integrates BMP2 and Wnt3a signals in migrating cardiac progenitor cells. Proc Natl Acad Sci USA. 2014;111:7337–42. doi: 10.1073/pnas.1321764111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yucel G, Oro AE. Cell migration: GSK3beta steers the cytoskeleton's tip. Cell. 2011;144:319–21. doi: 10.1016/j.cell.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuemmerle J. Endogenous IGF-I promotes survival of human intestinal smooth muscle cells by Akt-dependent inhibition of GSK-3 beta activity. Mol Biol Cell. 2002;13:165A–165A. [Google Scholar]
- 14.Kuemmerle JF. Endogenous IGF-I protects human intestinal smooth muscle cells from apoptosis by regulation of GSK-3 beta activity. Am J Physiol-Gastr L. 2005;288:G101–G10. doi: 10.1152/ajpgi.00032.2004. [DOI] [PubMed] [Google Scholar]
- 15.Kuemmerle JF. Endogenous IGF-I inhibits GSK-3 beta activity and promotes cell survival. Gastroenterology. 2002;122:A380–380A. [Google Scholar]
- 16.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 17.Embi N, Rylatt DB, Cohen P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem. 1980;107:519–27. [PubMed] [Google Scholar]
- 18.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug discovery. 2004;3:479–87. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 19.Woodgett JR, Cohen P. Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase-3 and casein kinase-II (glycogen synthase kinase-5) Biochim Biophys Acta. 1984;788:339–47. doi: 10.1016/0167-4838(84)90047-5. [DOI] [PubMed] [Google Scholar]
- 20.Peat AJ, Boucheron JA, Dickerson SH, Garrido D, Mills W, Peckham J. et al. Novel pyrazolopyrimidine derivatives as GSK-3 inhibitors. Bioorg Med Chem Lett. 2004;14:2121–25. doi: 10.1016/j.bmcl.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 21.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–26. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 22.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J. 1994;303:21–6. doi: 10.1042/bj3030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–51. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valvezan AJ, Klein PS. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front Mol Neurosci. 2012;5:1. doi: 10.3389/fnmol.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Liu M, Cai Z, Wang G, Li X. Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 2010;12:741–52. doi: 10.1111/j.1399-5618.2010.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beurel E, Mines MA, Song L, Jope RS. Glycogen synthase kinase-3 levels and phosphorylation undergo large fluctuations in mouse brain during development. Bipolar disord. 2012;14:822–30. doi: 10.1111/bdi.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li DW, Liu ZQ, Chen W, Yao M, Li GR. Association of glycogen synthase kinase-3beta with Parkinson's disease (review) Mol Med Rep. 2014;9:2043–50. doi: 10.3892/mmr.2014.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Llorens-Maritin M, Jurado J, Hernandez F, Avila J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7:46. doi: 10.3389/fnmol.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Llorens-Martin M, Blazquez-Llorca L, Benavides-Piccione R, Rabano A, Hernandez F, Avila J. et al. Selective alterations of neurons and circuits related to early memory loss in Alzheimer's disease. Front Neuroanat. 2014;8:38. doi: 10.3389/fnana.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llorens-Martin M, Fuster-Matanzo A, Teixeira CM, Jurado-Arjona J, Ulloa F, deFelipe J. et al. Alzheimer disease-like cellular phenotype of newborn granule neurons can be reversed in GSK-3 beta-overexpressing mice. Mol Psychiatr. 2013;18:395–95. doi: 10.1038/mp.2013.27. [DOI] [PubMed] [Google Scholar]
- 33.Llorens-Martin M, Fuster-Matanzo A, Teixeira CM, Jurado-Arjona J, Ulloa F, deFelipe J. et al. GSK-3 beta overexpression causes reversible alterations on postsynaptic densities and dendritic morphology of hippocampal granule neurons in vivo. Mol Psychiatr. 2013;18:451–60. doi: 10.1038/mp.2013.4. [DOI] [PubMed] [Google Scholar]
- 34.Lim NK, Hung LW, Pang TY, McLean CA, Liddell JR, Hilton JB. et al. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington's disease affected brain. Hum Mol Genet. 2014;23:4051–63. doi: 10.1093/hmg/ddu119. [DOI] [PubMed] [Google Scholar]
- 35.Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J. et al. HIV-1 Tat promotes Kaposi's sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3beta signaling pathway. Plos One. 2013;8:e53145. doi: 10.1371/journal.pone.0053145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharm. 2007;2:93–6. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 37.Garcia I, Fall Y, Gomez G. Using Topological Indices to Predict Anti-Alzheimer and Anti-Parasitic GSK-3 Inhibitors by Multi-Target QSAR in Silico Screening. Molecules. 2010;15:5408–22. doi: 10.3390/molecules15085408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia I, Fall Y, Gomez G, Gonzalez-Diaz H. First computational chemistry multi-target model for anti-Alzheimer, anti-parasitic, anti-fungi, and anti-bacterial activity of GSK-3 inhibitors in vitro, in vivo, and in different cellular lines. Mol Divers. 2011;15:561–7. doi: 10.1007/s11030-010-9280-3. [DOI] [PubMed] [Google Scholar]
- 39.Beurel E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front Mol Neurosci. 2011;4:18. doi: 10.3389/fnmol.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beurel E, Jope RS. Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neurosci. 2010;169:1063–70. doi: 10.1016/j.neuroscience.2010.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. 2011;12:607–15. doi: 10.1038/ni.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan M, Ma S, Huang Q, Hu K, Song B, Li M. GSK-3alpha/beta-mediated phosphorylation of CRMP-2 regulates activity-dependent dendritic growth. J Neurochem. 2013;125:685–97. doi: 10.1111/jnc.12230. [DOI] [PubMed] [Google Scholar]
- 43.Wisniewska MB. Physiological role of beta-catenin/TCF signaling in neurons of the adult brain. Neurochem Res. 2013;38:1144–55. doi: 10.1007/s11064-013-0980-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denham M, Bye C, Leung J, Conley BJ, Thompson LH, Dottori M. Glycogen synthase kinase 3beta and activin/nodal inhibition in human embryonic stem cells induces a pre-neuroepithelial state that is required for specification to a floor plate cell lineage. Stem Cells. 2012;30:2400–11. doi: 10.1002/stem.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fung TK, Gandillet A, So CW. Selective treatment of mixed-lineage leukemia leukemic stem cells through targeting glycogen synthase kinase 3 and the canonical Wnt/beta-catenin pathway. Curr Opin Hematol. 2012;19:280–6. doi: 10.1097/MOH.0b013e3283545615. [DOI] [PubMed] [Google Scholar]
- 46.Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM. et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/beta-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA. 2014;111:5349–54. doi: 10.1073/pnas.1317731111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen G, Jiang QQ, You ZH, Yao J, Mou LP, Lin X. et al. Regulation of GSK-3 beta in the proliferation and apoptosis of human thyrocytes investigated using a GSK-3 beta-targeting RNAi adenovirus expression vector: involvement the Wnt/beta-catenin pathway. Mol Biol Rep. 2010;37:2773–9. doi: 10.1007/s11033-009-9819-5. [DOI] [PubMed] [Google Scholar]
- 48.McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL. et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881–911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD. et al. Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65:5792–801. doi: 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- 50.Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA. et al. The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res. 2007;67:7756–64. doi: 10.1158/0008-5472.CAN-06-4665. [DOI] [PubMed] [Google Scholar]
- 51.Ma K, Yang LM, Chen HZ, Lu Y. Activation of muscarinic receptors inhibits glutamate-induced GSK-3 beta overactivation in PC12 cells. Acta Pharmacol Sin. 2013;34:886–92. doi: 10.1038/aps.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong J, Peng J, Zhang H, Mondesire WH, Jian W, Mills GB. et al. Role of glycogen synthase kinase 3beta in rapamycin-mediated cell cycle regulation and chemosensitivity. Res. 2005;65:1961–72. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL. et al. Adiponectin modulates the glycogen synthase kinase-3beta/beta-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res. 2006;66:11462–70. doi: 10.1158/0008-5472.CAN-06-1969. [DOI] [PubMed] [Google Scholar]
- 54.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. The EMBO J. 1990;9:2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- 56.ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat Struct Biol. 2001;8:593–6. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- 57.Woodgett JR. Judging a protein by more than its name: GSK-3. Sci. STKE. 2001;100:re12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- 58.Thomas GM, Frame S, Goedert M, Nathke I, Polakis P, Cohen P. A GSK3-binding peptide from FRAT1 selectively inhibits the GSK3-catalysed phosphorylation of axin and beta-catenin. FEBS letters. 1999;458:247–51. doi: 10.1016/s0014-5793(99)01161-8. [DOI] [PubMed] [Google Scholar]
- 59.Bijur GN, Jope RS. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–9. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- 60.Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3 beta. J Biol Chem. 2001;276:37436–42. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S. et al. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci USA. 1996;93:2719–23. doi: 10.1073/pnas.93.7.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–45. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 65.Sutherland C. What Are the bona fide GSK3 Substrates? Int J Alzheimers Dis. 2011;2011:505607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peat AJ, Garrido D, Boucheron JA, Schweiker SL, Dickerson SH, Wilson JR. et al. Novel GSK-3 inhibitors with improved cellular activity. Bioorg Med Chem Lett. 2004;14:2127–30. doi: 10.1016/j.bmcl.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 67.Smalley TL, Peat AJ, Boucheron JA, Dickerson S, Garrido D, Preugschat F. et al. Synthesis and evaluation of novel heterocyclic inhibitors of GSK-3. Bioorg Med Chem Lett. 2006;16:2091–4. doi: 10.1016/j.bmcl.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 68.Shin D, Lee SC, Heo YS, Lee WY, Cho YS, Kim YE. et al. Design and synthesis of 7-hydroxy-1H-benzoimidazole derivatives as novel inhibitors of glycogen synthase kinase-3beta. Bioorg Med Chem Lett. 2007;17:5686–9. doi: 10.1016/j.bmcl.2007.07.056. [DOI] [PubMed] [Google Scholar]
- 69.Coffman K, Brodney M, Cook J, Lanyon L, Pandit J, Sakya S. et al. 6-amino-4-(pyrimidin-4-yl)pyridones: novel glycogen synthase kinase-3beta inhibitors. Bioorg Med Chem Lett. 2011;21:1429–33. doi: 10.1016/j.bmcl.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 70.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1:435–49. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nuss JM, Harrison SD, Ring DB, Boyce RS, Johnson K, Pfister KB, Preparation of aminopyrimidines, -pyridines as glycogen synthase kinase 3 inhibitors. Patent 2002; WO 0220495 A2 20020314: pp 268.
- 72.Nuss JM, Harrison SD, Ring DB, Boyce RS, Brown SP, Goff D, Johnson K, Pfister KB, Ramurthy S, Renhowe PA, Seely L, Subramanian S, Wagman AS, Zhou XA. Preparation of aminopyrimidines and -pyridines as glycogen synthase kinase 3 inhibitors. Patent; 1999. WO 9965897 A1 19991223: pp.262. [Google Scholar]
- 73.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr Pharm Design. 2004;10:1105–37. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 74.Zhang HC, White KB, Ye H, McComsey DF, Derian CK, Addo MF. et al. Macrocyclic bisindolylmaleimides as inhibitors of protein kinase C and glycogen synthase kinase-3. Bioorg Med Chem Lett. 2003;13:3049–53. doi: 10.1016/s0960-894x(03)00644-9. [DOI] [PubMed] [Google Scholar]
- 75.Engler TA, Malhotra S, Burkholder TP, Henry JR, Mendel D, Porter WJ. et al. The development of potent and selective bisarylmaleimide GSK3 inhibitors. Bioorg Med Chem Lett. 2005;15:899–903. doi: 10.1016/j.bmcl.2004.12.063. [DOI] [PubMed] [Google Scholar]
- 76.Engler TA, Henry JR, Malhotra S, Cunningham B, Furness K, Brozinick J. et al. Substituted 3-imidazo[1,2-a]pyridin-3-yl- 4-(1,2,3,4-tetrahydro-[1,4]diazepino-[6,7,1-hi]indol-7-yl)pyrrole-2,5-diones as highly selective and potent inhibitors of glycogen synthase kinase-3. J Med Chem. 2004;47:3934–7. doi: 10.1021/jm049768a. [DOI] [PubMed] [Google Scholar]
- 77.Feng L, Geisselbrecht Y, Blanck S, Wilbuer A, Atilla-Gokcumen GE, Filippakopoulos P. et al. Structurally sophisticated octahedral metal complexes as highly selective protein kinase inhibitors. J Am Chem Soc. 2011;133:5976–86. doi: 10.1021/ja1112996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yngve U, Soderman P, Svensson M, Rosqvist S, Arvidsson PI. Imidazopyridine-based inhibitors of glycogen synthase kinase 3: synthesis and evaluation of amide isostere replacements of the carboxamide scaffold. Chem Biodiversity. 2012;9:2442–52. doi: 10.1002/cbdv.201200308. [DOI] [PubMed] [Google Scholar]
- 79.Saitoh M, Kunitomo J, Kimura E, Hayase Y, Kobayashi H, Uchiyama N. et al. Design, synthesis and structure-activity relationships of 1,3,4-oxadiazole derivatives as novel inhibitors of glycogen synthase kinase-3beta. Bioorg Med Chem. 2009;17:2017–29. doi: 10.1016/j.bmc.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 80.Berg S, Bergh M, Hellberg S, Hogdin K, Lo-Alfredsson Y, Soderman P. et al. Discovery of novel potent and highly selective glycogen synthase kinase-3beta (GSK3beta) inhibitors for Alzheimer's disease: design, synthesis, and characterization of pyrazines. J Med Chem. 2012;55:9107–19. doi: 10.1021/jm201724m. [DOI] [PubMed] [Google Scholar]
- 81.Perez DI, Palomo V, Perez C, Gil C, Dans PD, Luque FJ. et al. Switching reversibility to irreversibility in glycogen synthase kinase 3 inhibitors: clues for specific design of new compounds. J Med Chem. 2011;54:4042–56. doi: 10.1021/jm1016279. [DOI] [PubMed] [Google Scholar]
- 82.Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3 beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer's disease. J Med Chem. 2002;45:1292–9. doi: 10.1021/jm011020u. [DOI] [PubMed] [Google Scholar]
- 83.Gebhardt R, Lerche KS, Gotschel F, Gunther R, Kolander J, Teich L. et al. 4-Aminoethylamino-emodin-a novel potent inhibitor of GSK-3beta acts as an insulin-sensitizer avoiding downstream effects of activated beta-catenin. J Cell Mol Med. 2010;14:1276–93. doi: 10.1111/j.1582-4934.2009.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palomo V, Perez DI, Perez C, Morales-Garcia JA, Soteras I, Alonso-Gil S. et al. 5-imino-1,2,4-thiadiazoles: first small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J Med Chem. 2012;55:1645–61. doi: 10.1021/jm201463v. [DOI] [PubMed] [Google Scholar]
- 85.Conde S, Perez DI, Martinez A, Perez C, Moreno FJ. Thienyl and phenyl alpha-halomethyl ketones: new inhibitors of glycogen synthase kinase (GSK-3beta) from a library of compound searching. J Med Chem. 2003;46:4631–3. doi: 10.1021/jm034108b. [DOI] [PubMed] [Google Scholar]
- 86.Alonso D, Martinez A. Marine Compounds as a New Source for Glycogen Synthase Kinase 3 Inhibitors. In: A. Martinez, A. Castro, M. Medina., editors. Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors: Drug Discovery and Development, Hoboken, NJ, USA. John Wiley & Sons, Inc; 2006. pp. 307–331. (doi: 10.1002/0470052171) [Google Scholar]
- 87.Hu JF, Hamann MT, Hill R, Kelly M. The manzamine alkaloids. Alkaloids Chem Biol. 2003;60:207–85. doi: 10.1016/s0099-9598(03)60004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rao KV, Donia MS, Peng J, Garcia-Palomero E, Alonso D, Martinez A. et al. Manzamine B and E and ircinal A related alkaloids from an Indonesian Acanthostrongylophora sponge and their activity against infectious, tropical parasitic, and Alzheimer's diseases. J Nat Prod. 2006;69:1034–40. doi: 10.1021/np0601399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamann M, Alonso D, Martin-Aparicio E, Fuertes A, Perez-Puerto MJ, Castro A. et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure-activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer's disease. J Nat Prod. 2007;70:1397–405. doi: 10.1021/np060092r. [DOI] [PubMed] [Google Scholar]
- 90.Yousaf M, Hammond NL, Peng J, Wahyuono S, McIntosh KA, Charman WN. et al. New manzamine alkaloids from an Indo-Pacific sponge. Pharmacokinetics, oral availability, and the significant activity of several manzamines against HIV-I, AIDS opportunistic infections, and inflammatory diseases. J Med Chem. 2004;47:3512–7. doi: 10.1021/jm030475b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bidon-Chanal A, Fuertes A, Alonso D, Perez DI, Martinez A, Luque FJ. et al. Evidence for a new binding mode to GSK-3: allosteric regulation by the marine compound palinurin. Eur J Med Chem. 2013;60:479–89. doi: 10.1016/j.ejmech.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 92.Palomo V, Soteras I, Perez DI, Perez C, Gil C, Campillo NE. et al. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J Med Chem. 2011;54:8461–70. doi: 10.1021/jm200996g. [DOI] [PubMed] [Google Scholar]
- 93.Cole E, Shao X, Fawaz M, Scott P. Radiosynthesis of [11C]PyrATP1, a novel radiotracer targeting the ATP-binding site of GSK-3β. J Nucl Med. 2014;55:1110. [Google Scholar]
- 94.Hicks JW, VanBrocklin HF, Wilson AA, Houle S, Vasdev N. Radiolabeled small molecule protein kinase inhibitors for imaging with PET or SPECT. Molecules. 2010;15:8260–78. doi: 10.3390/molecules15118260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vasdev N, Garcia A, Stableford WT, Young AB, Meyer JH, Houle S. et al. Synthesis and ex vivo evaluation of carbon-11 labelled N-(4-methoxybenzyl)-N'-(5-nitro-1,3-thiazol-2-yl)urea ([11C]AR-A014418): a radiolabelled glycogen synthase kinase-3beta specific inhibitor for PET studies. Bioorg Med Chem Lett. 2005;15:5270–3. doi: 10.1016/j.bmcl.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 96.Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S. et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluation. J Clin Invest. 2009;119:1382–95. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cole EL, Shao X, Sherman P, Quesada C, Fawaz MV, Desmond TJ. et al. Synthesis and evaluation of [11C]PyrATP-1, a novel radiotracer for PET imaging of glycogen synthase kinase-3beta (GSK-3beta) Nucl Med Biol. 2014;41:507–12. doi: 10.1016/j.nucmedbio.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang M, Gao M, Miller KD, Sledge GW, Hutchins GD, Zheng QH. The first synthesis of [11C]SB-216763, a new potential PET agent for imaging of glycogen synthase kinase-3 (GSK-3) Bioorg Med Chem Lett. 2011;21:245–9. doi: 10.1016/j.bmcl.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 99.Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV. et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29:470–8. doi: 10.1002/mds.25824. [DOI] [PubMed] [Google Scholar]
- 100.Hoglinger GU, Huppertz HJ, Wagenpfeil S, Andres MV, Belloch V, Leon T. et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord. 2014;29:479–87. doi: 10.1002/mds.25815. [DOI] [PubMed] [Google Scholar]
- 101.del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M. et al. Treatment of Alzheimer's Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J Alzheimers Dis. 2013;33:205–15. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- 102.Zamek-Gliszczynski MJ, Mohutsky MA, Rehmel JL, Ke AB. Investigational small-molecule drug selectively suppresses constitutive CYP2B6 activity at the gene transcription level: physiologically based pharmacokinetic model assessment of clinical drug interaction risk. Drug Metab Dispos. 2014;42:1008–15. doi: 10.1124/dmd.114.057018. [DOI] [PubMed] [Google Scholar]
- 103.Zamek-Gliszczynski MJ, Abraham TL, Alberts JJ, Kulanthaivel P, Jackson KA, Chow KH. et al. Pharmacokinetics, metabolism, and excretion of the glycogen synthase kinase-3 inhibitor LY2090314 in rats, dogs, and humans: a case study in rapid clearance by extensive metabolism with low circulating metabolite exposure. Drug Metab Dispos. 2013;41:714–26. doi: 10.1124/dmd.112.048488. [DOI] [PubMed] [Google Scholar]
- 104.Kreisl TN, Kim L, Moore K, Duic P, Kotliarova S, Walling J. et al. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin Cancer Res. 2009;15:3617–23. doi: 10.1158/1078-0432.CCR-08-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nwankwo N, Zhang Z, Wang T, Collins C, Resta L, Ermisch S. et al. Phase I study of enzastaurin and bevacizumab in patients with advanced cancer: safety, efficacy and pharmacokinetics. Invest New Drugs. 2013;31:653–60. doi: 10.1007/s10637-012-9850-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Forlenza OV, Diniz BS, Radanovic M, Santos FS, Talib LL, Gattaz WF. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: randomised controlled trial. Br J Psychiatry. 2011;198:351–6. doi: 10.1192/bjp.bp.110.080044. [DOI] [PubMed] [Google Scholar]
- 107.Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A. et al. Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–31. [PubMed] [Google Scholar]
- 108.Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S. A feasibility and tolerability study of lithium in Alzheimer's disease. Int J Geriatr Psychiatry. 2008;23:704–11. doi: 10.1002/gps.1964. [DOI] [PubMed] [Google Scholar]
- 109.Guo XF, Yang WT, Shi L, Sang YH, Zhu RY, Chen YB. Judging the postoperative lymph node metastasis of esophagus cancer patients with pN0 stage by detecting MTA1 protein expression. Zhonghua Yi Xue Za Zhi. 2012;92:3204–6. [PubMed] [Google Scholar]
- 110.Collaborative pooled analysis of data on C-reactive protein gene variants and coronary disease. judging causality by Mendelian randomisation. Eur J Epidemiol. 2008;23:531–40. doi: 10.1007/s10654-008-9249-z. [DOI] [PubMed] [Google Scholar]
- 111.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–9. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hooft RW, Sander C, Vriend G. Objectively judging the quality of a protein structure from a Ramachandran plot. Comput Appl Biosci. 1997;13:425–30. doi: 10.1093/bioinformatics/13.4.425. [DOI] [PubMed] [Google Scholar]
- 113.Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN. et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005;65:7462–9. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- 114.Freland L, Beaulieu JM. Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front Mol Neurosci. 2012;5:14. doi: 10.3389/fnmol.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.O'Brien WT, Klein PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009;37:1133–8. doi: 10.1042/BST0371133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kaladchibachi SA, Doble B, Anthopoulos N, Woodgett JR, Manoukian AS. Glycogen synthase kinase 3, circadian rhythms, and bipolar disorder: a molecular link in the therapeutic action of lithium. J Circadian Rhythms. 2007;5:3. doi: 10.1186/1740-3391-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kozlovsky N, Nadri C, Agam G. Low GSK-3 beta in schizophrenia as a consequence of neurodevelopmental insult. Eur Neuropsychopharm. 2005;15:1–11. doi: 10.1016/j.euroneuro.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 118.Kozlovsky N, Belmaker RH, Agam G. GSK-3 and the neurodevelopmental hypothesis of schizophrenia. Eur Neuropsychopharm. 2002;12:13–25. doi: 10.1016/s0924-977x(01)00131-6. [DOI] [PubMed] [Google Scholar]
- 119.Kozlovsky N, Belmaker RH, Agam G. Low GSK-3 activity in frontal cortex of schizophrenic patients. Schizophr Res. 2001;52:101–5. doi: 10.1016/s0920-9964(00)00174-2. [DOI] [PubMed] [Google Scholar]
- 120.Kozlovsky N, Belmaker RH, Agam G. Low GSK-3 beta immunoreactivity in postmortem frontal cortex of schizophrenic patients. Am J Psychiat. 2000;157:831–3. doi: 10.1176/appi.ajp.157.5.831. [DOI] [PubMed] [Google Scholar]
- 121.Lipina TV, Wang M, Liu F, Roder JC. Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology. 2012;62:1252–62. doi: 10.1016/j.neuropharm.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 122.Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–9. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sereno L, Coma M, Rodriguez M, Sanchez-Ferrer P, Sanchez MB, Gich I. et al. A novel GSK-3 beta inhibitor reduces Alzheimer's pathology and rescues neuronal loss in vivo. Neurobiol Dis. 2009;35:359–67. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 124.Morales-Garcia JA, Susin C, Alonso-Gil S, Perez DI, Palomo V, Perez C. et al. Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem Neurosci. 2013;4:350–60. doi: 10.1021/cn300182g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.King MK, Pardo M, Cheng Y, Downey K, Jope RS, Beurel E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol Ther. 2014;141:1–12. doi: 10.1016/j.pharmthera.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ougolkov AV, Bone N, Billadeau D, Lee YK, Kay NE. Inhibition of GSK-3 induces apoptosis of CLL cells by abrogating NFkB nuclear activity. Blood. 2006;108:792A–A. [Google Scholar]
- 127.Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- 128.Kawazoe H, Bilim V, Yuuki K, Muto A, Ougolkov AV, Billadeau DD. et al. Gsk-3 Is a Potential Therapeutic Target for the Treatment of Renal Cell Carcinoma (Rcc): Preclinical in Vitro and in Vivo Investigation. J Urology. 2009;181:152–3. [Google Scholar]
- 129.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–95. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–80. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 131.Motawi TM, Bustanji Y, El-Maraghy SA, Taha MO, Al Ghussein MA. Naproxen and cromolyn as new glycogen synthase kinase 3beta inhibitors for amelioration of diabetes and obesity: an investigation by docking simulation and subsequent in vitro/in vivo biochemical evaluation. J Biochem Mol Toxicol. 2013;27:425–36. doi: 10.1002/jbt.21503. [DOI] [PubMed] [Google Scholar]
- 132.Fava GA, Bartolucci G, Rafanelli C, Mangelli L. Cognitive-behavioral management of patients with bipolar disorder who relapsed while on lithium prophylaxis. J Clin Psychiatry. 2001;62:556–9. doi: 10.4088/jcp.v62n07a10. [DOI] [PubMed] [Google Scholar]
- 133.Mur M, Portella MJ, Martinez-Aran A, Pifarre J, Vieta E. Long-term stability of cognitive impairment in bipolar disorder: a 2-year follow-up study of lithium-treated euthymic bipolar patients. J Clin Psychiatry. 2008;69:712–9. [PubMed] [Google Scholar]
- 134.Chen CH, Lin SK. Carbamazepine treatment of bipolar disorder: a retrospective evaluation of naturalistic long-term outcomes. BMC Psychiatry. 2012;12:47. doi: 10.1186/1471-244X-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rukat A, Musisi S, Strohle A, Mundt AP. Prescription Patterns of Psychotropic Medications for the Treatment of Psychotic Disorders in the Largest Mental Health Institutions of Uganda. J Clin Psychopharmcol. 2014;34(5):571–6. doi: 10.1097/JCP.0000000000000166. [DOI] [PubMed] [Google Scholar]
- 136.Jope RS. Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–3. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 137.Nunes PV, Forlenza OV, Gattaz WF. Lithium and risk for Alzheimer's disease in elderly patients with bipolar disorder. Br J Psychiatry. 2007;190:359–60. doi: 10.1192/bjp.bp.106.029868. [DOI] [PubMed] [Google Scholar]
- 138.Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3beta: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25:329–37. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- 139.Wang YG, Liu WP, He XS, Zhou F. Parkinson's Disease-Associated Dj-1 Mutations Increase Abnormal Phosphorylation of Tau Protein through Akt/Gsk-3 beta Pathways. J Mol Neurosci. 2013;51:911–8. doi: 10.1007/s12031-013-0099-0. [DOI] [PubMed] [Google Scholar]
- 140.Cohen P. Guidelines for the effective use of chemical inhibitors of protein function to understand their roles in cell regulation. Biochem J. 2010;425:53–4. doi: 10.1042/BJ20091428. [DOI] [PubMed] [Google Scholar]
- 141.Vainshtein I, Silveria S, Kaul P, Rouhani R, Eglen RM, Wang J. A high-throughput, nonisotopic, competitive binding assay for kinases using nonselective inhibitor probes (ED-NSIP) J Biomol Screen. 2002;7:507–14. doi: 10.1177/1087057102238624. [DOI] [PubMed] [Google Scholar]
- 142.Parker GJ, Law TL, Lenoch FJ, Bolger RE. Development of high throughput screening assays using fluorescence polarization: nuclear receptor-ligand-binding and kinase/phosphatase assays. J Biomol Screen. 2000;5:77–88. doi: 10.1177/108705710000500204. [DOI] [PubMed] [Google Scholar]
- 143.Guarnieri MT, Blagg BS, Zhao R. A high-throughput TNP-ATP displacement assay for screening inhibitors of ATP-binding in bacterial histidine kinases. Assay Drug Dev Technol. 2011;9:174–83. doi: 10.1089/adt.2010.0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Patel S, Doble BW, MacAulay K, Sinclair EM, Drucker DJ, Woodgett JR. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol Cell Biol. 2008;28:6314–28. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Patel S, Macaulay K, Woodgett JR. Tissue-Specific Analysis of Glycogen Synthase Kinase-3 alpha (GSK-3 alpha) in Glucose Metabolism: Effect of Strain Variation. Plos One. 2011;6(1):e15845. doi: 10.1371/journal.pone.0015845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [11C-carbonyl] AR-A014418 by [11C]CO2 fixation. Bioorg Med Chem Lett. 2012;22:2099–101. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 147.Li L, Shao X, Cole EL, Ohnmacht SA, Ferrari V, Hong YT. et al. Synthesis and Initial in Vivo Studies with [11C]SB-216763: The First Radiolabeled Brain Penetrative Inhibitor of GSK-3. ACS Med Chem Lett. 2015;6:548–52. doi: 10.1021/acsmedchemlett.5b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mossine AV, Cole EL, Brooks AF, Li L, Aigbirhio F, Scott PJ. Synthesis of Radiolabeled Glycogen Synthase Kinase 3β Inhibitors for CNS imaging. J Label Compd Radiopharm. 2015;58:S411. [Google Scholar]
- 149.Mossine A, Lie L, Cole E, Brooks A, Aigbirhio F, Scott P. Glycogen Synthase Kinase 3β Inhibitor Scaffolds for PET Radiotracer Development. J Nucl Med. 2015;56:1094. [Google Scholar]
- 150.Perez DI, Pistolozzi M, Palomo V, Redondo M, Fortugno C, Gil C. et al. 5-Imino-1,2-4-thiadiazoles and quinazolines derivatives as glycogen synthase kinase 3 beta (GSK-3 beta) and phosphodiesterase 7 (PDE7) inhibitors: Determination of blood-brain barrier penetration and binding to human serum albumin. Eur J Pharm Sci. 2012;45:677–84. doi: 10.1016/j.ejps.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 151.Mossine A, Scott P, Shao X. Radiosynthesis of [11C]tideglusib via [11C]CO2 fixation. World Molecular Imaging Congress 2015, Honolulu Hawaii; 2015. [Google Scholar]
- 152.Liang S, Chen J, Normandin M, Collier T, Perils R, Holson E, Haggarty S, El Fakhri G, Kurumbail R, Vasdev N. Discovery of [11C]PF-367 for neuroimaging of glycogen synthase kinase 3. J Nucl Med. 2015;56:491. [Google Scholar]
- 153.Smyth LA, Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J Chem Biol. 2009;2:131–51. doi: 10.1007/s12154-009-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]