

Abstract
Ledipasvir (LDV; GS-5885), a component of Harvoni (a fixed-dose combination of LDV with sofosbuvir [SOF]), is approved to treat chronic hepatitis C virus (HCV) infection. Here, we report key preclinical antiviral properties of LDV, including in vitro potency, in vitro resistance profile, and activity in combination with other anti-HCV agents. LDV has picomolar antiviral activity against genotype 1a and genotype 1b replicons with 50% effective concentration (EC50) values of 0.031 nM and 0.004 nM, respectively. LDV is also active against HCV genotypes 4a, 4d, 5a, and 6a with EC50 values of 0.11 to 1.1 nM. LDV has relatively less in vitro antiviral activity against genotypes 2a, 2b, 3a, and 6e, with EC50 values of 16 to 530 nM. In vitro resistance selection with LDV identified the single Y93H and Q30E resistance-associated variants (RAVs) in the NS5A gene; these RAVs were also observed in patients after a 3-day monotherapy treatment. In vitro antiviral combination studies indicate that LDV has additive to moderately synergistic antiviral activity when combined with other classes of HCV direct-acting antiviral (DAA) agents, including NS3/4A protease inhibitors and the nucleotide NS5B polymerase inhibitor SOF. Furthermore, LDV is active against known NS3 protease and NS5B polymerase inhibitor RAVs with EC50 values equivalent to those for the wild type.
INTRODUCTION
Hepatitis C virus (HCV) infection is a significant global health challenge with an estimated 150 million individuals infected worldwide (1, 2). Several direct-acting antiviral (DAA) agents have been approved to treat patients with HCV, including the nucleotide analog NS5B polymerase inhibitor sofosbuvir (Sovaldi; SOF) and the NS5A inhibitor ledipasvir (LDV) (3–5).
The availability of SOF represents a major advance in the treatment of HCV infection as SOF-based regimens are shorter in duration, are better tolerated, and result in higher sustained virologic response (SVR) rates than prior therapies (6). Combinations of DAAs, including a fixed-dose combination of LDV and SOF (Harvoni), obviate administration of pegylated interferon (Peg-IFN) and ribavirin (RBV) in patients with certain genotypes of HCV (7–9). Harvoni is approved in the United States for patients with genotype 1 HCV infection following 8 to 24 weeks of treatment across treatment-naive and treatment-experienced patients and in patients with cirrhosis (4).
LDV inhibits HCV replication by targeting the NS5A protein, which is an essential viral protein that plays roles in both viral RNA replication and the assembly of HCV virions (10, 11). Recently, LDV was shown to inhibit NS5A through direct binding to the NS5A protein, and resistance to LDV was shown to be the result of lower binding affinity to NS5A mutants (12). Targeting NS5A has been validated clinically, as treatment with NS5A inhibitors elicits a rapid decline of HCV viral load (13, 14) and enhanced SVR rates when combined with Peg-IFN and RBV (15) or other DAAs, including SOF (7, 8, 16).
Here, we describe key preclinical antiviral properties of LDV, including in vitro potency against genotype 1 to 6 HCV, antiviral selectivity, in vitro resistance profile, cross-resistance profile, and combination activity.
MATERIALS AND METHODS
Compounds.
LDV, SOF, GS-9451, GS-9669, simeprevir (SMV), daclatasvir (DCV) (17), BILN-2061, efavirenz (EFV), elvitegravir (EVG), tenofovir (TFV), emtricitabine (FTC), rilpivirine (RPV), and raltegravir (RAL) were synthesized by Gilead Sciences (Foster City, CA). 2′-C-methyl-adenosine (2′-C-Me-A) was synthesized by Acme Bioscience (Palo Alto, CA). RBV and alpha interferon (IFN-α) were purchased from Sigma (St. Louis, MO). Atazanavir (ATV) and darunavir (DRV) were obtained from Toronto Research Chemicals (Toronto, Ontario, Canada).
Cell lines and replicon constructs.
The 1C cell line, which is highly permissive for genotype 1a replication, was described previously (18). A bovine viral diarrhea virus (BVDV) replicon clone was established in Huh-7 cells as described previously (19). HEp-2 cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and infected with respiratory syncytial virus (RSV) strain A2 (ABI, Columbia, MD). HeLa cells were obtained from the ATCC and infected with a mixture of three rhinovirus strains (human rhinovirus 1A [HRV1A], HRV16, and HRV14; ATCC, Manassas, VA). Normal human bronchial/tracheal epithelial (NHBE) cells were obtained from Lonza and cultured in bronchial epithelial growth medium (BEGM) supplemented with growth factors (Lonza, Basel, Switzerland). Influenza A and B viruses were obtained from the ATCC. All flavivirus testing was performed at the Institute for Antiviral Research at Utah State University using standard procedures. The AD-38 cell line was derived from HepG2 cells (ATCC) and expresses wild-type hepatitis B virus (HBV) under the control of a tetracycline-inducible promoter (20). MT-4 cells were obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD) and infected with HIV-1 IIIB (ABI).
Unless noted otherwise, all cells and replicon cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX (Invitrogen [Gibco], Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM nonessential amino acids (Invitrogen), and 0.5 mg/ml of G-418 (Invitrogen). HEp-2 cells were maintained in minimum essential medium (MEM) supplemented with 10% FBS. Huh7-Lunet and Lunet-CD81 cells were maintained in DMEM supplemented with 10% FBS only. AD-38 cells were maintained in DMEM–F-12 medium supplemented with 10% FBS, 50 μg/ml penicillin, 50 μg/ml streptomycin, 20 μg/ml gentamicin, and 0.4 mg/ml G-418 (Invitrogen). MT-4 cells were maintained in RPMI 1640 medium supplemented with 100 U/ml penicillin, 10 μg/ml streptomycin, and 10% FBS.
Construction of genotype 1a, 1b, 2a, 2b, 3a, 4a, 4d, 5a, 6a, and 6e HCV subgenomic and NS5A chimeric replicons.
Genotype 1a subgenomic replicon H77/SG-RlucNeo (H77 strain), genotype 1b subgenomic replicon Con1/SG-hRlucNeo (Con-1 strain), and genotype 2a subgenomic replicon JFH-1/RlucNeo2a were described previously (21, 22). Subgenomic replicons GT3a-PI-hRluc, GT4aED43SG, and GT6aRlucNeoSG were established recently (18, 23, 24). Replicons H77, HSG-51, HSG-57, 1a-HGluc-1, 1a-HGluc-2, 1a-Sf9Rluc-1, 1a-Sf9Rluc-2, Con-1, Huh-luc, and SL-3 have previously been described (25, 26).
Plasmids encoding genotype 2a and 2b NS5A chimeric replicons, pRluNeo2a-J6NS5A, pRlucNeo2a-MD2b8-2NS5A, and pRluNeo2a-MD2b-1NS5A, were constructed by cloning genotype 2a (J6 strain) and 2b (MD2b8-2 and MD2b-1 strains) full-length NS5A genes into the above genotype 2a JFH-1 replicon backbone pRlucNeo2a, respectively. DNA fragments which carried the 264-nucleotide sequence for the NS4B C-terminal sequence from the genotype 2a JFH-1 strain, the full-length NS5A gene from the genotype 2a J6 strain or the genotype 2b MD2b8-2 or MD2b-1 strain, and the 122-nucleotide sequence for the NS5B N-terminal sequence of the JFH-1 strain were synthesized by GenScript (Piscataway, NJ).
Plasmid pGT4d Neo FJ 462437 QC, encoding a subgenomic genotype 4d replicon based on the HCV isolate QC382 (GenBank accession no. FJ462437), and plasmid pGT6aSUBID6636SG (GSI6a-1), encoding a subgenomic genotype 6a replicon based on the sequence of an HCV isolate (SUBID 6636), were prepared by DNA synthesis (GenScript). Genotype 5a and 6e NS5A chimeric replicons, pGT1b-PI-Rluc-GT5a-SA13NS5A(9–184) and pGT1b-PI-Rluc-GT6e-D88NS5A(9–184), were constructed by cloning genotype 5a strain SA13 and 6e strain D88 NS5A nucleotide sequences encoding N-terminal amino acids 9 to 184 into genotype 1b Con-1 replicon backbone pGT1b-PI-Rluc (23), respectively (full-length NS5A chimeric replicons were constructed but did not replicate sufficiently for antiviral assays). To construct these NS5A chimeric replicons, DNA fragments were synthesized by GenScript (Piscataway, NJ); these fragments include the restriction site AseI in NS4B to the 24-nucleotide sequence encoding the N-terminal sequence of NS5A from genotype 1b, the NS5A nucleotide sequence coding for amino acids 9 to 184 of genotype 5a strain SA13 or 6e strain D88, and the nucleotide sequence from encoding the amino acid 185 of NS5A to the restriction site BclI in NS5B from genotype 1b.
Generation of replicon RNA for transient transfection.
Genotype 1a replicon plasmids were linearized with HpaI. The genotype 1b replicon and other replicon plasmids based on the genotype 1b backbone were linearized with SpeI. The genotype 2a replicon and other replicon plasmids based on the genotype 2a backbone were linearized with XbaI. Linearized replicon constructs were purified using a PCR purification kit (Qiagen, Valencia, CA). RNA was synthesized with T7 MEGAscript reagents (Ambion, Austin, TX) according to the manufacturer's suggested protocol, and reactions were stopped by digestion with RNase-free DNase. RNA was purified by column purification using an RNeasy kit (Qiagen) according to the manufacturer's instructions.
Resistance selection and genotypic analysis of resistant replicon clones.
Stable replicon cells (2 × 105 of genotype 1a or 1b) were seeded in 10-cm tissue culture dishes. After overnight culture, cells were treated with 0.5 mg/ml G-418 along with several concentrations of LDV. After 3 weeks, surviving colonies were isolated and expanded. Total RNAs were extracted from drug-resistant or wild-type replicon cells using an RNeasy minikit (Qiagen). The cDNA of the entire NS5A coding sequence was synthesized using the MonsterScript 1st-Strand cDNA synthesis kit (Epicentre Biotechnologies, Madison, WI). PCR products were sequenced by Elim Biopharmaceuticals Inc. (Hayward, CA). Changes in amino acid residues were identified by comparing the NS5A protein sequences between drug-selected and parental replicons.
Cross-resistance studies.
PI-hRluc, a bicistronic replicon carrying the Renilla luciferase (Rluc) gene downstream of the poliovirus internal ribosome entry site (IRES) and the genotype 1b (Con-1 strain) HCV nonstructural genes (NS3 to NS5B) downstream of the encephalomyocarditis virus (EMCV) IRES, was used for transient-transfection studies (27). NS3 and NS5B mutations were introduced into a plasmid encoding the PI-Rluc replicon using a QuikChange II XL mutagenesis kit, according to the manufacturer's instructions (Stratagene, San Diego, CA). Mutations were confirmed by DNA sequencing. Replicon RNAs were transcribed in vitro from replicon-encoding plasmids using a MEGAscript kit (Ambion). In vitro-transcribed replicon RNAs were transfected into 1C cells as described previously (21, 24).
Antiviral combination assays.
HCV and HIV inhibitor combination assays were performed in either HCV GT1a replicons or MT-4 cells. Serial dilutions were performed in 384-well polypropylene plates (Greiner Bio-One, Monroe, NC) using a Biomek FX workstation. For combination studies, test compound A was serially diluted at a 1:3 ratio in columns 3 to 20 of the first 384-well plate to form a horizontal 10-point dose response. Test compound B was plated in three different concentrations in the second 384-well plate together with a dimethyl sulfoxide (DMSO) control. The combination of the first and second serial dilution plates resulted in the third serial dilution plate, which contained the combination of compounds A and B. For each individual drug, the 50% effective concentration (EC50) was selected as the midpoint for the concentration range tested. All serial dilutions were performed in four replicates per compound within the same 384-well plate. One hundred percent DMSO was added into columns 1 and 2 of each serial dilution 384-well plate. For HCV replicon assays, an HCV protease inhibitor (PI), ITMN-191, was added into column 23 at 100 μM as a control for 100% inhibition of HCV replication while puromycin at 10 mM was added into column 24 as a control for 100% cytotoxicity. For anti-HIV MT-4 assays, zidovudine (AZT) was added into column 23 at 2.5 μM as a control of 100% inhibition on HIV while puromycin at 10 mM was added into column 24 as a control for 100% cytotoxicity.
EC50 and 50% cytotoxic concentration (CC50) were determined as described below in stable replicon antiviral assays. Data were analyzed using the MacSynergy II program developed by Prichard and Shipman (28–30). Accordingly, combination effects are defined by the produced volume of surface deviations (volumes are expressed as μM concentration times μM concentration times percentage, or μM2%): strong synergy, >100 μM2%; moderate synergy, >50 and ≤100 μM2%; minor synergy, >25 and ≤50 μM2%; additivity, ≤25 and >−25 μM2%; minor antagonism, ≤−25 and >−50 μM2%; moderate antagonism, ≤−50 and >−100 μM2%; strong antagonism, ≤−100 μM2%.
Stable HCV replicon antiviral assays in 384-well format.
Stable replicon cell lines were assayed in 384-well plates (Greiner Bio-One, Monroe, NC; cell culture treated). The HCV replicon assay is a multiplex assay, assessing both cytotoxicity and antiviral activity in the same well. CC50 assays were performed first. The medium in the 384-well cell culture plate was aspirated, and wells were washed four times with 100 μl phosphate-buffered saline (PBS; with Ca2+ and Mg2+), using a BioTek ELX405 plate washer. Fifty microliters of a solution containing 400 nM calcein AM (Anaspec, Fremont, CA; catalog no. 25200-056) in PBS was added to each well of the plates with a BioTek μFlow workstation. Plates were incubated for 30 min at room temperature before the fluorescence signal (excitation, 490 nm; emission, 520 nm) was measured with a PerkinElmer Envision plate reader (PerkinElmer, Waltham, MA).
EC50 assays were performed next. The calcein-PBS solution in 384-well cell culture plates was aspirated with a BioTek ELX405 plate washer. Twenty microliters of Dual-Glo luciferase buffer (Promega) was added to each well of the plates with a BioTek μFlow workstation. Plates were incubated for 10 min at room temperature. Twenty microliters of a solution containing a 1:100 mixture of Dual-Glo Stop & Glo substrate (Promega) and Dual-Glo Stop & Glo buffer (Promega) was added to each well with a BioTek μFlow workstation. Plates were incubated at room temperature for 10 min before the luminescence signals were measured with a PerkinElmer Envision plate reader.
HCV transient-transfection replicon antiviral assays in 384-well and 96-well format.
In vitro-transcribed replicon RNAs were transfected into 1C cells as described previously (21, 24). Cells were subsequently transferred into prewarmed culture medium and seeded into 384-well plates at 2 × 103 cells per well in a total volume of 90 μl of DMEM culture medium. Low-replicating replicons were seeded into 96-well plates at 2 × 104 cells per well in a total volume of 200 μl of DMEM culture medium, to improve detection of luciferase signals. After cell attachment was allowed for 4 h, 384-well plates were dosed with serially diluted compound as described previously (22). Ninety-six-well-plate cells were dosed with compound serially 3-fold diluted and dispensed using the HP D300 digital dispenser (Hewlett-Packard Company, Corvallis, OR) at a final concentration of 0.5% DMSO. Plates were incubated at 37°C for 3 days, after which culture medium was removed and cells were lysed and assayed for Rluc activity using a commercial Rluc assay (Promega) and a PerkinElmer Envision plate reader for 384-well plates or a Victor instrument for 96-well plates as described previously (22).
Data analysis.
Fifty percent effective concentrations (EC50 values) against HCV replicons were calculated by nonlinear regression as described previously (22).
RESULTS
Antiviral activity in cell-based assays.
The antiviral activity of LDV was determined using a panel of HCV replicons, representing prevalent NS5A sequences for genotypes 1 to 6 in standard 3-day cell-based replicon assays. LDV has picomolar antiviral activity against genotype 1a (H77 isolate) and genotype 1b (Con-1 isolate) replicons with EC50 values of 0.031 nM and 0.004 nM, respectively (Table 1). In addition, LDV showed similar antiviral activity in other genotype 1 HCV replicon assays, including those using either different HCV isolates or different cellular backgrounds (Table 2).
TABLE 1.
LDV antiviral activity against HCV of genotypes 1 to 6
| Genotype | HCV replicon strain | EC50 (nM)a |
|---|---|---|
| 1ab | H77 | 0.031 |
| 1bb | Con-1 | 0.004 |
| 2ab,c | JFH-1 (L31 in NS5A) | 21 |
| 2ad,e | J6 (M31 in NS5A) | 249 |
| 2bd,e | MD2b8-2 (L31 in NS5A) | 16 |
| 2bd,e | MD2b-1 (M31 in NS5A) | 530 |
| 3ad | S52 | 168 |
| 4ab | ED43 | 0.39 |
| 4db | QC382 | 0.29 |
| 5ad,e | SA13 | 0.15 |
| 6ab | HK6a consensus | 1.1 |
| 6ab | GSI6a-1 | 0.11 |
| 6ed,e | D88 | 264 |
Values represent the geometric means from at least three independent experiments.
Stable subgenomic replicon cell lines assayed in a high-throughput screening (HTS) 384-well format.
Transiently transfected JFH-1 (L31 in NS5A) yielded an EC50 of 6.8 nM.
Transiently transfected replicons in 1C cells assayed in either 96-well (2a, 2b, and 3a) or 384-well (5a and 6e) format.
NS5A chimeric replicons encoding either full-length NS5A (2a and 2b) or NS5A amino acids 9 to 184 (5a and 6e).
TABLE 2.
LDV exhibits consistent activity against different HCV genotype 1 replicons
| Genotype test system (replicona) | EC50 (nM) of agentc: |
||
|---|---|---|---|
| LDV | BILN-2061b | 2′-C-Me-Ab | |
| GT1a | |||
| 1a-RlucP | 0.031 ± 0.014 | 5.1 ± 2.6 | 189 ± 54 |
| H77 | 0.053 ± 0.013 | 17 ± 5.0 | 716 ± 281 |
| HSG-51a | 0.019 ± 0.003 | 12 ± 2.7 | 497 ± 45 |
| HSG-57a | 0.012 ± 0.004 | 13 ± 6.9 | 544 ± 284 |
| 1a-HGluc-1 | 0.010 ± 0.005 | 10 ± 1.1 | 414 ± 65 |
| 1a-HGluc-2 | 0.013 ± 0.002 | 14 ± 1.0 | 156 ± 27 |
| 1a-Sf9Rluc-1 | 0.010 ± 0.001 | 4.9 ± 0.8 | 221 ± 55 |
| 1a-Sf9Rluc-2 | 0.012 ± 0.001 | 7.6 ± 0.2 | 170 ± 19 |
| GT1b | |||
| 1b-Rluc-2 | 0.0037 ± 0.0004 | 2.3 ± 0.6 | 411 ± 157 |
| Con-1 | 0.0041 ± 0.0008 | 3.1 ± 1.1 | 605 ± 196 |
| Huh-luc | 0.0029 ± 0.0003 | 0.76 ± 0.05 | 479 ± 46 |
| SL-3a | 0.003 ± 0.001 | 1.5 ± 0.5 | 531 ± 80 |
EC50 values were determined based on NS3 protease assay (21).
BILN-2061 and 2′-C-Me-A were used as controls for the replicon assays.
Values represent the arithmetic means ± standard deviations from two or more independent experiments using 96- or 384-well assays.
LDV has various levels of antiviral activity against genotypes 2 to 6, with EC50 values ranging from 0.11 nM to 530 nM (Table 1). LDV has an EC50 of 21 nM against the genotype 2a JFH-1 replicon, which has leucine at amino acid position 31 (L31) in NS5A, but has 12-fold less activity (EC50, 249 nM) against the 2a J6 HCV strain, which has methionine at position 31 (M31). Similarly, LDV has differential activities against genotype 2b replicons that have L31 or M31 in NS5A, with EC50 values of 16 nM (2b MD2b8-2, L31) and 530 nM (2b MD2b-1, M31), respectively. LDV is active against genotypes 4a, 4d, 5a, and 6a, with EC50 values of 0.39 nM, 0.29 nM, 0.15 nM, and 0.11 to 1.1 nM, respectively. LDV has relatively less in vitro antiviral activity against genotypes 3a and 6e, with EC50 values of 168 nM and 264 nM, respectively.
Antiviral selectivity in cell-based assays.
The antiviral activity of LDV was tested against viruses other than HCV in cell-based assay systems. These viruses include five closely related viruses (bovine viral diarrhea virus [BVDV], West Nile virus, yellow fever virus, dengue virus, and Banzi virus), as well as six unrelated viruses, including respiratory syncytial virus (RSV), human rhinovirus, influenza A virus, influenza B virus, hepatitis B virus (HBV), and HIV. LDV showed active antiviral activity against HCV with an EC50 of 0.004 nM (Table 3). In contrast, EC50 values against the 11 other viruses were >10,000 nM, indicating that LDV is highly selective for HCV compared with other tested viruses.
TABLE 3.
LDV is a specific inhibitor of HCV
| Virus | Description | Host cell line | EC50 (nM)b | CC50 (nM)b |
|---|---|---|---|---|
| HCV GT1b replicon | Hepacivirus (positive ssRNAc) | Huh-7 | 0.004 | >44,400 |
| BVDV replicon | Pestivirus (positive ssRNA) | Huh-7 | 19,300 | >50,000 |
| West Nile virus Kern 2002 | Flavivirus (positive ssRNA) | Vero | >100,000 | >100,000 |
| Yellow fever virus 17D | Flavivirus (positive ssRNA) | Vero | >100,000 | >100,000 |
| Dengue virus type 2 | Flavivirus (positive ssRNA) | Vero | >41,000 | 41,000 |
| Banzi virus | Flavivirus (positive ssRNA) | Vero | >100,000 | >100,000 |
| RSV | Paramyxovirus (negative ssRNA) | HEp-2 | >10,000 | >10,000 |
| Human rhinovirusa | Enterovirus (positive ssRNA) | HeLa | >50,000 | >50,000 |
| Influenza A virus PC/1/73 | Orthomyxovirus (negative ssRNA) | NHBE | >100,000 | >100,000 |
| Influenza B virus LEE/40 | Orthomyxovirus (negative ssRNA) | NHBE | >100,000 | >100,000 |
| HBV | Hepadnavirus (DNA reverse transcribing) | AD-38 | >10,000 | >10,000 |
| HIV | Lentivirus (RNA reverse transcribing) | MT-4 | >10,000 | 10,100 |
Infectious mixture of HRV1A, HRV16, and HRV14.
Values represent the arithmetic means from two or more independent experiments.
ssRNA, single-stranded RNA.
In vitro resistance selection and analyses.
To define the in vitro resistance profile of LDV, drug-resistant colonies were selected and then isolated (or pooled) after treating stable genotype 1a or 1b replicon cells with LDV for 3 weeks. Genotypic analyses of resistant replicon RNA and subsequent phenotypic analyses of mutations emerging in the NS5A gene identified Y93H as the primary resistance-associated variant (RAV) to LDV in genotypes 1a and 1b, with Q30E also observed in genotype 1a (Table 4). Y93H conferred 3,309- and 1,319-fold resistance to LDV in transient replicon assays of genotypes 1a and 1b, respectively (Fig. 1). However, LDV remained a relatively active inhibitor of genotype 1b replicons encoding this Y93H mutation (EC50 of 5.3 nM compared to the EC50 of 103 nM in genotype 1a). Q30E in genotype 1a conferred 952-fold resistance to LDV. Both Y93H and Q30E conferred cross-resistance to daclatasvir (DCV) (Fig. 1).
TABLE 4.
Genotypic changes in NS5A in LDV in vitro resistance selectionb
| HCV replicon | LDV concn (nM) for selection | No. of clones picked and sequenced | NS5A mutation(s) observed |
|---|---|---|---|
| GT1a | 40 | 10 | Q30E (n = 4), Y93H (n = 6) |
| 20 | 6 | Q30E (n = 5), Y93H (n = 1) | |
| 10 | Pool | Q30E or Y93H | |
| GT1b | 1.25 | 10 | Y93H (n = 10)a |
| 0.625 | 5 | Y93H (n =5)a | |
| 0.313 | Pool | Y93H |
Q54H was identified in two clones in combination with Y93H. The mutant replicon did not replicate.
Mutations detected in only a single clone are not listed.
FIG 1.
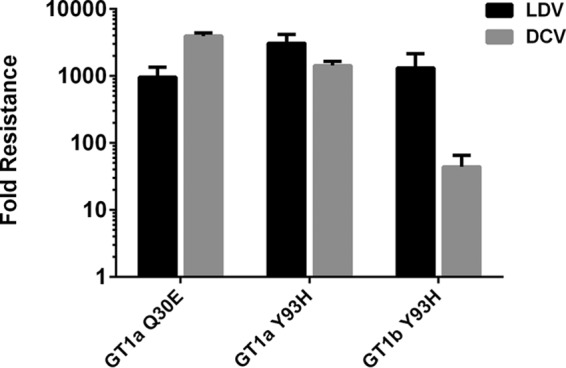
Phenotypic analysis of LDV RAVs selected in vitro. RAVs emerging to LDV during in vitro resistance selection assays were evaluated by introducing them into wild-type genotype 1a or 1b replicons followed by transient transfection. The antiviral activities of LDV and daclatasvir (DCV) against these RAVs were determined. Fold resistance was calculated as the ratio of RAV EC50 to wild-type EC50. All values represent an average from at least two independent experiments with standard deviations.
Cross-resistance evaluation.
To investigate whether known RAVs of other classes of HCV DAAs confer cross-resistance to LDV, transient transfection was used to assay the LDV susceptibility of genotype 1b HCV replicons carrying mutations for resistance to NS3/4A protease inhibitors or several classes of nucleoside and nonnucleoside NS5B polymerase inhibitors. LDV was fully active (<2-fold change in EC50 versus wild type) against all tested RAVs selected by NS3/4A protease inhibitors (V36M, T54A, R155K, A156T, and D168V), active-site NS5B inhibitors (S282T), nonnucleoside NS5B thumb site II inhibitors (M423T and M419T), palm site III inhibitors (M414T), and palm site III/IV inhibitors (Y448H and C316Y) (Fig. 2). Wild-type EC50 values and mutant EC50 shifts for the control NS3 protease inhibitor (BILN-2061) and nucleoside NS5B polymerase inhibitor (2′-C-Me-A) agreed with historical results (data not shown). As expected, LDV showed reduced activity (25- to 327-fold) against NS5A RAVs selected in genotype 1a patients during 3-day monotherapy with LDV in a phase 1 study (13, 14).
FIG 2.
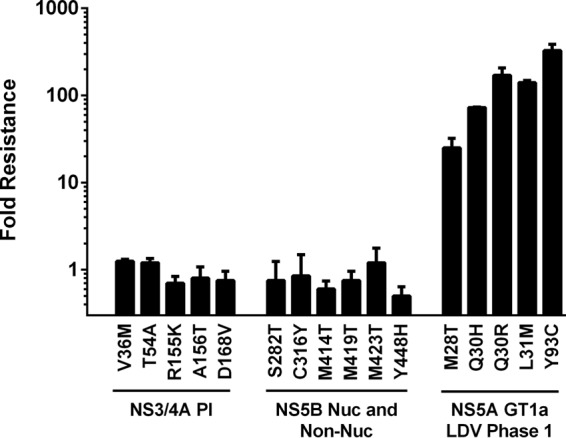
Activity of LDV against other classes of DAA RAVs. The indicated RAVs were evaluated by introducing them into wild-type genotype 1a or 1b replicons followed by transient transfection. Fold resistance was calculated as the ratio of RAV EC50 to wild-type EC50. Values represent the means from two or more independent experiments. LDV is fully active against NS3 protease inhibitor RAVs and NS5B nucleoside and NS5B nonnucleoside inhibitor RAVs. LDV is less effective against NS5A RAVs selected in genotype 1a patients during 3-day monotherapy in a phase 1 study.
Combination activities.
LDV is or will be used in combination with other classes of anti-HCV agents for the treatment of chronic HCV infection. Therefore, we assessed antiviral drug interactions with IFN-α, ribavirin, and NS3 protease inhibitors as well as nucleotide and nonnucleoside NS5B polymerase inhibitors. Additive to moderately synergistic in vitro antiviral activities were observed when LDV was combined with other HCV inhibitors (Table 5). LDV had minor antiviral synergy when combined with IFN-α and moderate antiviral synergy when combined with RBV. Combinations of LDV with other DAAs, including the NS3/4A protease inhibitors GS-9451 and simeprevir (SMV) and the NS5A inhibitor DCV, as well as nucleotide (SOF) or nonnucleoside (GS-9669) NS5B polymerase inhibitors, resulted in additive to minor synergistic antiviral activity. Importantly, neither significant antagonism nor additive cytotoxicity were observed when LDV was combined with any other inhibitor.
TABLE 5.
LDV has additive or synergistic antiviral activity when combined with other classes of HCV inhibitorsa
| Drug tested | Drug class | Synergy vol (μM2%) | Antagonism vol (μM2%) | Interaction |
|---|---|---|---|---|
| IFN-α | Indirect | 32.0 ± 1.4 | 0.0 ± 0.0 | Minor synergy |
| RBV | Direct/indirect | 60.8 ± 0.5 | −0.5 ± 0.1 | Moderate synergy |
| GS-9451 | NS3/4A PI | 6.5 ± 4.0 | −2.3 ± 2.9 | Additive |
| SMV | NS3/4A PI | 3.7 ± 3.8 | −11.5 ± 10.2 | Additive |
| GS-9669 | NS5B Non-Nuc | 34 ± 19 | 0 ± 0 | Minor synergy |
| DCV | NS5A inhibitor | 4.3 ± 6.7 | −11.5 ± 5.1 | Additive |
| SOF | NS5B Nuc | 3.3 ± 4.2 | −7.7 ± 13.3 | Additive |
Antiviral combination assays were performed in GT1a replicon cells. Values represent the geometric means from at least two independent experiments in quadruplicate. Abbreviations: PI, protease inhibitor; Non-Nuc, nonnucleos(t)ide; Nuc, nucleos(t)ide.
HCV and HIV coinfection is a common comorbidity worldwide. Fifteen to 30% of HIV-infected patients are also infected with HCV, and coinfected patients have a significantly higher rate of HCV disease progression, resulting in high mortality rates due to liver disease in this population (31). As such, any treatment for the cure of HCV infection would ideally be compatible with antiretroviral medications utilized for treatment of HIV, and in vitro combination studies were performed combining LDV with two common HIV inhibitors of various classes. The HIV nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) TFV and FTC, nonnucleoside reverse transcriptase inhibitors (NNRTIs) EFV and RPV, protease inhibitors (PIs) ATV and DRV, and integrase strand transfer inhibitors (INSTIs) EVG and RAL were tested in combination with LDV. As seen in Tables S1 and S2 in the supplemental material, LDV EC50 values were similar regardless of the presence of any of the HIV inhibitors at the concentrations tested. The EC50 values for all of the HIV inhibitors tested were similar regardless of the presence of LDV at the concentrations tested. These data indicate that there is no antagonistic effect between LDV and these HIV inhibitors in vitro. Furthermore, no additive cell toxicity was observed in these experiments.
DISCUSSION
LDV has picomolar and selective antiviral activity in multiple HCV genotype 1a and 1b replicon cell lines, with EC50 values of 0.031 nM and 0.004 nM, respectively (Table 1), and has low cellular toxicity (CC50, ≥10,000 nM [Table 3]). This potency, combined with the long plasma half-life of LDV in patients (13), results in significant antiviral activity in patients with genotype 1a and 1b HCV infection at low doses during 3 days of monotherapy (median maximal reduction in HCV RNA of >3 log10 IU/ml from baseline with 3-mg once-a-day [QD] dosing) (13). LDV is also active against genotypes 4a, 4d, 5a, and 6a, with EC50 values of 0.39 nM, 0.29 nM, 0.15 nM, and 0.11 to1.1 nM, respectively. Based on the systemic exposure achieved at the 90-mg QD dose, LDV thus would show antiviral activity in patients with genotype 4a, 4d, 5a, or 6a HCV infection. Combination treatment of LDV with SOF indeed results in high SVR rates at 12 weeks posttreatment (SVR12) in patients with chronic genotype 4, 5, and 6 HCV infection (9, 32, 33). Despite the comparatively lower in vitro activity of LDV against genotypes 2a, 2b, 3a, and 6e (EC50, 16 to 530 nM), high rates of SVR12 (26 of 26 patients) were achieved in patients with HCV genotype 3 infection who received 12 weeks of LDV-SOF plus RBV in a small phase 2 study (33).
Resistance-associated variants to LDV were selected in both genotype 1a and 1b replicon cells. Genotypic and phenotypic analyses of the NS5A gene in these resistant replicon cells identified the single mutation of Y93H or Q30E that confers high levels of resistance to LDV (Fig. 1). A number of other mutations (data not shown) were identified in combination with Y93H, but all appeared at very low frequency and did not confer significant resistance by themselves alone. This in vitro resistance profile of LDV is consistent with the emergence of Y93H in NS5A as the predominant RAV observed in genotype 1b HCV patients after a 3-day monotherapy treatment at 10-mg QD dosing (13). In patients with genotype 1a HCV infection, a different set of NS5A RAVs (M28T, Q30H, Q30R, L31M, and Y93C) were observed at different doses (1 mg to 30 mg); Y93H was selected in genotype 1a patients treated with higher doses (30 mg and 90 mg) of LDV, which correlates with the higher level of resistance conferred by Y93H. In consistency, low-level RAVs such as M28T and L31M (25- and 140-fold shift) were not isolated in vitro likely because the LDV concentrations used in the resistance selection studies were higher than the levels at which those RAVs can survive. Selection of these RAVs in the NS5A gene also supports the idea that LDV interferes with HCV replication by inhibiting NS5A function, which is further validated by a recent demonstration of direct binding of LDV to NS5A (12). LDV has no significant activity against NS3/4A serine protease, NS3 helicase, NS5B polymerase, or HCV IRES in biochemical assays (data not shown). LDV is also highly specific for HCV and does not have significant antiviral activity against a wide range of related (BVDV, West Nile virus, and dengue virus) or unrelated (HIV, HBV, RSV, influenza viruses, and HRVs) viruses (Table 3).
In vitro antiviral combination experiments indicate that LDV has additive to moderately synergistic antiviral activity when combined with IFN-α, RBV, and other DAAs, including the NS3/4A protease inhibitor, and nucleotide NS5B polymerase inhibitor SOF (Table 5). There were no antagonistic effects or combinational cytotoxicity observed when LDV was combined with any of the tested compounds. These results support the idea that LDV can be used in combination with different classes of DAAs in HCV patients to effectively suppress HCV RNA replication. Evaluation of the cross-resistance profile of LDV with known RAVs from several classes of HCV inhibitors demonstrated that LDV remained fully active against all tested NS3/4A protease inhibitor RAVs and all tested NS5B polymerase inhibitor RAVs, including the nucleotide NS5B inhibitor RAV S282T (Fig. 2). Conversely, it has been shown that NS5A inhibitor RAVs are fully susceptible to NS3/4A protease inhibitors and nucleotide NS5B inhibitors, such as SOF (26, 34). LDV also showed no antagonistic interactions with a range of HIV inhibitors when tested in combination in in vitro HCV or HIV antiviral assays.
In summary, LDV active antiviral activity against multiple HCV genotypes and its orthogonal resistance profile and additive to synergistic antiviral activity when combined with other classes of HCV inhibitors support its use in combination with SOF and other DAA agents for the treatment of chronic HCV infection.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the entire Gilead LDV discovery and development teams. Special thanks to Becky Norquist for providing medical writing services to Gilead Sciences.
Funding Statement
All authors were employed by Gilead Sciences during the duration of the study. No other sources of funding were used.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02524-15.
REFERENCES
- 1.Negro F, Alberti A. 2011. The global health burden of hepatitis C virus infection. Liver Int 31(Suppl 2):S1–S3. doi: 10.1111/j.1478-3231.2011.02537.x. [DOI] [PubMed] [Google Scholar]
- 2.Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect 17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 3.Bourliere M, Oules V, Ansaldi C, Adhoute X, Castellani P. 2014. Sofosbuvir as backbone of interferon free treatments. Dig Liver Dis 46(Suppl 5):S212–S220. doi: 10.1016/j.dld.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 4.Gilead Sciences Inc. 2014. Harvoni (ledipasvir and sofosbuvir) tablets, for oral use. U.S. prescribing information. Gilead Sciences Inc, Foster City, CA. [Google Scholar]
- 5.Link JO, Taylor JG, Xu L, Mitchell M, Guo H, Liu H, Kato D, Kirschberg T, Sun J, Squires N, Parrish J, Keller T, Yang ZY, Yang C, Matles M, Wang Y, Wang K, Cheng G, Tian Y, Mogalian E, Mondou E, Cornpropst M, Perry J, Desai MC. 2014. Discovery of ledipasvir (GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection. J Med Chem 57:2033–2046. doi: 10.1021/jm401499g. [DOI] [PubMed] [Google Scholar]
- 6.Younossi ZM, Stepanova M, Marcellin P, Afdhal N, Kowdley KV, Zeuzem S, Hunt SL. 2015. Treatment with ledipasvir and sofosbuvir improves patient-reported outcomes: results from the ION-1, -2, and -3 clinical trials. Hepatology 61:1798–1808. doi: 10.1002/hep.27724. [DOI] [PubMed] [Google Scholar]
- 7.Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, Nahass R, Ghalib R, Gitlin N, Herring R, Lalezari J, Younes ZH, Pockros PJ, Di Bisceglie AM, Arora S, Subramanian GM, Zhu Y, Dvory-Sobol H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Sulkowski M, Kwo P. 2014. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 370:1483–1493. doi: 10.1056/NEJMoa1316366. [DOI] [PubMed] [Google Scholar]
- 8.Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, Romero-Gomez M, Zarski JP, Agarwal K, Buggisch P, Foster GR, Brau N, Buti M, Jacobson IM, Subramanian GM, Ding X, Mo H, Yang JC, Pang PS, Symonds WT, McHutchison JG, Muir AJ, Mangia A, Marcellin P. 2014. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 370:1889–1898. doi: 10.1056/NEJMoa1402454. [DOI] [PubMed] [Google Scholar]
- 9.Abergel A, Loustaud-Ratti V, Metivier S, Jiang D, Kersey K, Knox SJ, Pang PS, Samuel D, Asselah T. 2015. Ledipasvir/sofosbuvir treatment results in high SVR rates in patients with chronic genotype 4 and 5 HCV infection. J Hepatol 62:S219–S220. doi: 10.1016/S0168-8278(15)30070-2. [DOI] [Google Scholar]
- 10.Kim S, Welsch C, Yi M, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J Virol 85:6645–6656. doi: 10.1128/JVI.02156-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes M, Griffin S, Harris M. 2009. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles. J Gen Virol 90:1329–1334. doi: 10.1099/vir.0.009332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon HJ, Xing W, Chan K, Niedziela-Majka A, Brendza KM, Kirschberg T, Kato D, Link JO, Cheng G, Liu X, Sakowicz R. 2015. Direct binding of ledipasvir to HCV NS5A: mechanism of resistance to an HCV antiviral agent. PLoS One 10:e0122844. doi: 10.1371/journal.pone.0122844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawitz EJ, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A, Cheng G, Link JO, Wong KA, Mo H, McHutchison JG, Brainard DM. 2012. A phase 1, randomized, placebo-controlled, 3-day, dose-ranging study of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J Hepatol 57:24–31. doi: 10.1016/j.jhep.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 14.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hezode C, Hirschfield GM, Ghesquiere W, Sievert W, Rodriguez-Torres M, Shafran SD, Thuluvath PJ, Tatum HA, Waked I, Esmat G, Lawitz EJ, Rustgi VK, Pol S, Weis N, Pockros PJ, Bourliere M, Serfaty L, Vierling JM, Fried MW, Weiland O, Brunetto MR, Everson GT, Zeuzem S, Kwo PY, Sulkowski M, Brau N, Hernandez D, McPhee F, Wind-Rotolo M, Liu Z, Noviello S, Hughes EA, Yin PD, Schnittman S. 2015. Daclatasvir plus peginterferon alfa and ribavirin for treatment-naive chronic hepatitis C genotype 1 or 4 infection: a randomised study. Gut 64:948–956. doi: 10.1136/gutjnl-2014-307498. [DOI] [PubMed] [Google Scholar]
- 16.Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, Symonds WT, McHutchison JG, Membreno FE. 2014. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 383:515–523. doi: 10.1016/S0140-6736(13)62121-2. [DOI] [PubMed] [Google Scholar]
- 17.Bristol-Myers Squibb Company, Kim S, Gao Q, Yang F. 12 February 2009. Crystalline form of methyl ((1S)-1-(((2S)-2-(5-(4′-(2-((2S)-1-((2S)-2-((methoxycarbonyl)amino)-3-methylbutanoyl)-2-pyrrolidinyl)-1H-imidazol-5-yl)-4-biphenylyl)-1H-imidazol-2-yl)-1-pyrrolidinyl)carbonyl)-2-methylpropyl)carbamate dihydrochloride salt. World Intellectual Property Organization; International publication number WO 2009/020828 A1-12. [Google Scholar]
- 18.Peng B, Yu M, Xu S, Lee YJ, Tian Y, Yang H, Chan K, Mo H, McHutchison J, Delaney W IV, Cheng G. 2013. Development of robust hepatitis C virus genotype 4 subgenomic replicons. Gastroenterology 144:59–61. doi: 10.1053/j.gastro.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 19.Horscroft N, Bellows D, Ansari I, Lai VC, Dempsey S, Liang D, Donis R, Zhong W, Hong Z. 2005. Establishment of a subgenomic replicon for bovine viral diarrhea virus in Huh-7 cells and modulation of interferon-regulated factor 3-mediated antiviral response. J Virol 79:2788–2796. doi: 10.1128/JVI.79.5.2788-2796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladner S, Otto M, Barker C, Zaifert K, Wang G, Guo J, Seeger C, King R. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng G, Chan K, Yang H, Corsa A, Pokrovskii M, Paulson M, Bahador G, Zhong W, Delaney W IV. 2011. Selection of clinically relevant protease inhibitor-resistant viruses using the genotype 2a hepatitis C virus infection system. Antimicrob Agents Chemother 55:2197–2205. doi: 10.1128/AAC.01382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng JY, Cheng G, Perry J, Barauskas O, Xu Y, Fenaux M, Eng S, Tirunagari N, Peng B, Yu M, Tian Y, Lee YJ, Stepan G, Lagpacan LL, Jin D, Hung M, Ku KS, Han B, Kitrinos K, Perron M, Birkus G, Wong KA, Zhong W, Kim CU, Carey A, Cho A, Ray AS. 2014. Inhibition of hepatitis C virus replication by GS-6620, a potent C-nucleoside monophosphate prodrug. Antimicrob Agents Chemother 58:1930–1942. doi: 10.1128/AAC.02351-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu M, Corsa AC, Xu S, Peng B, Gong R, Lee YJ, Chan K, Mo H, Delaney W IV, Cheng G. 2013. In vitro efficacy of approved and experimental antivirals against novel genotype 3 hepatitis C virus subgenomic replicons. Antiviral Res 100:439–445. doi: 10.1016/j.antiviral.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Yu M, Peng B, Chan K, Gong R, Yang H, Delaney W IV, Cheng G. 2014. Robust and persistent replication of genotype 6a hepatitis C virus replicon in cell culture. Antimicrob Agents Chemother 58:2638–2646. doi: 10.1128/AAC.01780-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson M, Yang H, Sun SC, Peng B, Tian Y, Pagratis N, Greenstein AE, Delaney WE IV. 2010. Novel hepatitis C virus reporter replicon cell lines enable efficient antiviral screening against genotype 1a. Antimicrob Agents Chemother 54:3099–3106. doi: 10.1128/AAC.00289-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H, Robinson M, Corsa AC, Peng B, Cheng G, Tian Y, Wang Y, Pakdaman R, Shen M, Qi X, Mo H, Tay C, Krawczyk S, Sheng XC, Kim CU, Yang C, Delaney WE IV. 2014. Preclinical characterization of the novel hepatitis C virus NS3 protease inhibitor GS-9451. Antimicrob Agents Chemother 58:647–653. doi: 10.1128/AAC.00487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friebe P, Lohmann V, Krieger N, Bartenschlager R. 2001. Sequences in the 5′ nontranslated region of hepatitis C virus required for RNA replication. J Virol 75:12047–12057. doi: 10.1128/JVI.75.24.12047-12057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prichard MN, Aseltine KR, Shipman C Jr. 1993. MacSynergy II, version 1.0. University of Michigan, Ann Arbor, MI. [Google Scholar]
- 29.Prichard MN, Shipman C Jr. 1996. Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir Ther 1:9–20. [PubMed] [Google Scholar]
- 30.Prichard MN, Prichard LE, Shipman C Jr. 1993. Strategic design and three-dimensional analysis of antiviral drug combinations. Antimicrob Agents Chemother 37:540–545. doi: 10.1128/AAC.37.3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naggie S, Sulkowski MS. 2012. Management of patients coinfected with HCV and HIV: a close look at the role for direct-acting antivirals. Gastroenterology 142:1324–1334. doi: 10.1053/j.gastro.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohli A, Kapoor R, Sims Z, Nelson A, Sidharthan S, Lam B, Silk R, Kotb C, Gross C, Teferi G, Sugarman K, Pang PS, Osinusi A, Polis MA, Rustgi V, Masur H, Kottilil S. 2015. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept, single-centre, open-label phase 2a cohort study. Lancet Infect Dis 15:1049–1054. doi: 10.1016/S1473-3099(15)00157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gane EJ, Hyland RH, An D, Svarovskaia E, Pang PS, Brainard D, Stedman CA. 2015. Efficacy of ledipasvir and sofosbuvir, with or without ribavirin, for 12 weeks in patients with HCV genotype 3 or 6 infection. Gastroenterology 149:1454–1461.e1. doi: 10.1053/j.gastro.2015.07.063. [DOI] [PubMed] [Google Scholar]
- 34.Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, Keilman M, Zhu Y, Lan S, Otto MJ, Furman PA. 2012. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother 56:3359–3368. doi: 10.1128/AAC.00054-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.