

Abstract
Streptococcus agalactiae (group B Streptococcus [GBS]) is a leading cause of sepsis in neonates. The rate of invasive GBS disease in nonpregnant adults also continues to climb. Aminoglycosides alone have little or no effect on GBS, but synergistic killing with penicillin has been shown in vitro. High-level gentamicin resistance (HLGR) in GBS isolates, however, leads to the loss of a synergistic effect. We therefore performed a multicenter study to determine the frequency of HLGR GBS isolates and to elucidate the molecular mechanisms leading to gentamicin resistance. From eight centers in four countries, 1,128 invasive and colonizing GBS isolates were pooled and investigated for the presence of HLGR. We identified two strains that displayed HLGR (BSU1203 and BSU452), both of which carried the aacA-aphD gene, typically conferring HLGR. However, only one strain (BSU1203) also carried the previously described chromosomal gentamicin resistance transposon designated Tn3706. For the other strain (BSU452), plasmid purification and subsequent DNA sequencing resulted in the detection of plasmid pIP501 carrying a remnant of a Tn3 family transposon. Its ability to confer HLGR was proven by transfer into an Enterococcus faecalis isolate. Conversely, loss of HLGR was documented after curing both GBS BSU452 and the transformed E. faecalis strain from the plasmid. This is the first report showing plasmid-mediated HLGR in GBS. Thus, in our clinical GBS isolates, HLGR is mediated both chromosomally and extrachromosomally.
INTRODUCTION
Streptococcus agalactiae, alternatively designated group B Streptococcus (GBS), is a leading cause of morbidity and mortality in neonates and pregnant women. Recommendations for diagnosing maternal GBS colonization and administering intrapartum antimicrobial prophylaxis have led to a significant decrease in these infections (1). The rate of invasive GBS disease in nonpregnant adults, however, continues to climb (2). Elderly persons and those with underlying diseases—two expanding segments of the population—are at increased risk (3). Treatment concepts for invasive GBS infections in nonpregnant adults have not been established. Clinical isolates of GBS are susceptible to penicillin, the antimicrobial agent of choice for treating invasive diseases. Several publications advocate the addition of an aminoglycoside to penicillin or ampicillin for infective endocarditis (4) and periprosthetic joint infections (5), although aminoglycosides have ototoxic and nephrotoxic side effects, in particular in the elderly. Aminoglycosides alone have little or no effect on GBS, but synergistic killing with penicillin has been shown in vitro (6). In case of the presence of high-level gentamicin resistance (HLGR) in a bacterial isolate, there is a lack of a synergistic effect.
While HLGR in Enterococcus spp. is frequently found (7), to the best of our knowledge, only two HLGR GBS strains have been previously reported (8, 9). Most diagnostic laboratories do not test routinely for HLGR in GBS. Thus, little is known about the frequency of HLGR GBS, the mechanisms of acquiring HLGR, and the potential to spread genetic elements associated with HLGR.
The aim of this study was to estimate the frequency of HLGR GBS isolates (i) in systematically and continuously collected GBS isolates from colonized pregnant and nonpregnant women and (ii) in GBS isolates pooled in a collection that stems from various selected patient populations. Upon detection of HLGR isolates, we elaborated the molecular mechanism conferring this resistance.
MATERIALS AND METHODS
GBS isolates.
The study included 1,128 GBS isolates. Of these, 464 (41%) were pooled from various GBS collections (Table 1). These isolates stem from various centers and were previously investigated in other contexts (10–15). The other 664 (59%) GBS isolates were prospectively collected and screened for the presence of HLGR. The origin of GBS isolation, the association with diseases or colonization, and the sampling period are presented in Table 1.
TABLE 1.
Pooled collection of isolates investigated for the presence of high-level gentamicin resistance
| Referencea | No. of GBS isolatesa | Disease/case definition | Origin of GBS isolation | Study type | Collection period | Geographic origin(s) | HLGR |
|---|---|---|---|---|---|---|---|
| 10 | 75 | No disease/colonization | Vaginal and rectal swabs from pregnant and nonpregnant women | Cross-sectional study | 2001–2003 | Aachen and Munich, Germany | 0 |
| 12 | 60 | EODb with invasive neonatal GBS infections | Isolation of GBS from blood or CSFg and other sterile body fluids | Part of the prospective active surveillance study | 2001–2003 | Freiburg, Germany | 0 |
| 13 | 50c | Suspicion of EOD without proven invasive GBS disease | GBS isolates from nonsterile sites | Part of the prospective active surveillance study | 2001–2003 | Freiburg, Germany | 0 |
| 14 | 30 | Patients with cystic fibrosis | Respiratory samples | Collection of isolatesd | 2002–2008 | Münster, Germany | 1 |
| 11 | 150 | No disease/colonization | Rectovaginal specimens from pregnant and nonpregnant women | Part of the national surveillance study | 2005–2009 | Lisbon, Portugal | 0 |
| 97 | No disease/colonization | Vaginal swabs from pregnant and nonpregnant women | —e | 2009 | Ulm, Germany | 0 | |
| 15 | 99 | No disease/colonization | Vaginal swabs from pregnant and nonpregnant women | Cross-sectional study | 2010 | Ismailia, Egypt | 0 |
| 36 | 364 | No disease/colonization | Vaginal swabs from pregnant women | Cross-sectional study | 2009–2010 | Bern, Switzerland | 1 |
| 203 | Invasive GBS infections | Isolation of GBS from blood, CSF and other sterile body fluids | —f | 1998–2013 | Bern, Switzerland | 0 | |
| Total | 1,128 | 2 |
GBS isolates investigated and published in a context other than the presence of HLGR. The number of GBS isolates investigated for HLGR may vary from the number in the source publication for technical reasons.
EOD, early-onset disease.
For this study, only GBS isolates with serotype III were available.
GBS isolation occurred during a routine visit or during a visit due to an exacerbation of clinical symptoms.
Prospective collection during routine diagnostic microbiology laboratory analysis. GBS isolates were investigated for this study.
Collection of invasive GBS isolates (all age groups) during routine diagnostic microbiology laboratory analysis. GBS isolates were investigated for this study.
CSF, cerebrospinal fluid.
Definition and identification of HLGR in GBS.
No definition of HLGR in GBS has been published. According to the recommended screening tests for the detection of HLGR in Enterococcus spp., the resistant isolates have an MIC of ≥500 mg/liter (16). In addition, HLGR isolates with an MIC >500 mg/liter have been reported (17, 18). Therefore, HLGR in GBS was defined when the gentamicin MIC determined by Etest was ≥512 mg/liter. All MIC determinations were confirmed with ≥3 measurements.
Two different methods for the identification of HLGR in GBS were applied. Five hundred sixty-one isolates (49.7%) were plated on Mueller-Hinton agar supplemented with 256 mg/liter of gentamicin. Subsequently, the MIC of growing GBS colonies was determined by Etest. For 567 (50.3%) isolates, the MIC was primarily determined by Etest without a prior HLGR screening test.
Bacterial strains.
The strains used in this study are presented in Table 2. The plasmid-free recipient used in the mating experiments was Enterococcus faecalis (BSU386), a clinical blood culture isolate without HLGR.
TABLE 2.
Bacterial strains and their corresponding genetic elements conferring HLGR
| Species | Strain | Description | MIC of gentamicin (mg/liter) | aac(6′)-Ie-aph(2″) -Ia gene | Transposon |
|---|---|---|---|---|---|
| GBS | BSU1203 | Wild-type strain | ≥1,024 | Yes | Tn3706 |
| GBS | BSU452 | Wild-type strain | 512 | Yes | Tn3-like |
| E. faecalis | BSU386 | Wild-type strain | 12 | No | No |
| E. faecalis | BSU580 | BSU386 + pIP501BSU452 | ≥1,024 | Yes | Tn3-like |
| E. faecalis | BSU720 | BSU580 cured | 12 | No | No |
| GBS | BSU729 | BSU452 cured | 24 | No | No |
Genetic basis of HLGR.
Standard recombinant DNA techniques were used for nucleic acid preparation and analysis. Plasmid DNA was isolated and purified using the QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. PCR was performed with Taq polymerase according to the manufacturer's protocol (Roche Diagnostics, Mannheim, Germany), with 35 cycles of amplification steps consisting of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C. PCR products were sequenced on an ABI PRISM 310 genetic analyzer, using the ABI PRISM BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems, Weiterstadt, Germany). To identify the HLGR resistance gene, we performed multiplex PCR as described by Vakulenko et al. (19). For the detection of Tn3706-specific nucleotide sequences, we used PCR with primers O1, O2, and O3 as described by Horaud et al. (20). The primers used for this study are presented in Table 3. Detection of open reading frames (ORFs) was carried out by using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Sequence comparison was performed by using the BLAST system (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
TABLE 3.
Primers used for PCR and DNA sequencing
| Primer | Sequence (5′-3′) | Target gene |
|---|---|---|
| Inverse primer HLGR1 | CTTCATCTTCCCAAGGCTCTG | aac(6′)- aph(2″) |
| Inverse primer HLGR2 | GCCAGAACATGAATTACACGAGG | aac(6′)- aph(2″) |
| 369 Vakulenko PCR | CAGGAATTTATCGAAAATGGTAGAAAAG | |
| 369 Vakulenko PCR | CACAATCGACTAAAGAGTACCAATC | |
| 348 Vakulenko PCR | CAGAGCCTTGGGAAGATGAAG | |
| 348 Vakulenko PCR | CCTCGTGTAATTCATGTTCTGGC | |
| Primer O1 | GGACCTACATGATGAATGGA | |
| Primer O2 | CCTTTACAGAATATTCAATAATGC | |
| Primer O3 | GTATAG CAATATGCAAATCC | |
| pIP501-for | TCGCTCAATCACTACCAAGC | |
| pIP501-rev | CTTGAACGAGTAAAGCCCTT |
Transfer and mobilization of the genetic element conferring HLGR.
To investigate the potential transfer of resistance, we transformed E. faecalis (BSU386) with plasmid DNA, as described previously (21). Curing the transformed E. faecalis strain and GBS BSU452 with HLGR was achieved as follows. The strains were exposed in an overnight culture to serial 2-fold dilutions of ciprofloxacin in Todd-Hewitt broth plus 0.5% yeast extract and Luria-Bertani medium. Bacterial cultures containing the highest subinhibitory concentrations of ciprofloxacin (i.e., 0.625 mg/liter for GBS BSU452 and 10 mg/liter for E. faecalis BSU580) were plated on antibiotic-free tryptic soy agar blood plates and grown overnight at 37°C. Single colonies were then tested for loss of resistance to gentamicin by subculture onto Mueller-Hinton agar plates containing 256 mg/liter of gentamicin. The MIC for gentamicin was then determined by Etest.
Nucleotide sequence accession number.
Nucleotide sequences were submitted to GenBank under accession number KP698941.
RESULTS
GBS isolates with HLGR.
Among 1,128 GBS isolates, 2 (0.17%) strains with HLGR were identified. One strain (BSU1203; MIC, >1,024 mg/liter) was obtained from a 35-year-old Swiss woman during prenatal screening. The second HLGR GBS strain (BSU452; MIC, 512 mg/liter) was isolated in a respiratory specimen from a 26-year-old man with cystic fibrosis.
PCR detection of genes conferring HLGR on GBS.
To investigate the resistance determinants in HLGR GBS strains, we performed PCRs that were specific for the aac(6′)-Ie-aph(2″)-Ia gene and the flanking IS256 element (19). The aac(6′)-Ie-aph(2″)-Ia gene was readily detected in both strains, yielding the expected product of 348 bp (19).
Detection of a previously known transposon and a novel element.
A PCR with a primer set annealing on the structural gentamicin resistance gene and the IS256 sequence, located downstream of this gene, showed the expected 369-bp product for strain GBS BSU1203 (19), indicating the presence of the previously described chromosomal transposon Tn3706. For further characterization of the resistance determinant, PCRs that were specific for insertion sequence elements of Tn3706 were performed as published previously (20). These PCRs were positive with GBS strain BSU1203 and matched those previously described for HLGR GBS strain B128 (20), confirming the presence of Tn3706. In GBS strain BSU452, the aac(6′)-Ie-aph(2″)-Ia gene was found, but none of the PCRs specific for the transposon structures of Tn5281, Tn4001, or Tn3706 (20) yielded a product, suggesting the presence of a novel HLGR determinant in this strain.
Characterization of a novel mobile genetic element conferring HLGR on GBS.
To identify the genetic structure of GBS strain BSU452 carrying the aac(6′)-Ie-aph(2″)-Ia gene, we performed an inverse PCR on a plasmid preparation of BSU452. Primers annealing to the gentamicin resistance gene (Table 3) and directed toward DNA regions upstream and downstream of aac(6′)-Ie-aph(2″)-Ia yielded a PCR product about 2 kb in length, which was completely sequenced. Nucleotide comparison with the GenBank database revealed a 100% identity of nucleotides 1 to 253 and 721 to 2029 with plasmid pTEF1 of E. faecalis strain V583 (GenBank accession number AE016833.1). Several ORFs were identified on the 2-kb PCR product, and comparison of the deduced amino acids with the GenBank database revealed a DNA resolvase fragment and one copy of an insertion sequence element with high homology to IS1216. This structure displayed high similarities to a Tn3 family transposon.
Transfer and mobilization of the resistance determinant in association with HLGR.
The HLGR genes and the flanking DNA sequences in GBS BSU1203 matched those previously identified in GBS B128, and because thorough molecular analyses on the acquisition of HLGR have been published for that strain (8, 20, 22), further investigations focused on GBS BSU452. Tn3 family transposons are typically located on plasmids. To investigate if this is the case in strain GBS BSU452 and to characterize the potential of spreading HLGR to other isolates, we transformed the gentamicin-susceptible E. faecalis strain BSU386 with the plasmid preparation obtained from GBS BSU452. Positive clones (i.e., designated E. faecalis BSU580, which carries the mobile element of BSU452) were obtained upon plating the transformed strain onto the HLGR screening agar, as described above. A subsequent gentamicin evaluation revealed an increase in MIC from 12 mg/liter to ≥1,024 mg/liter (Table 2). To ensure that the increased MIC was due to the uptake of the plasmid DNA, plasmid preparations were subjected to gel electrophoresis (Fig. 1), which showed the presence of large plasmids in the HLGR strains BSU452 and BSU580. Further confirmation of the successful transfer was achieved by PCR showing the presence of the aac(6′)-Ie-aph(2″)-Ia gene and a lack of any flanking IS256 sequences in E. faecalis strain BSU580. To confirm that the newly detected mobile genetic element is indeed located on a plasmid, we attempted to cure GBS BSU452 and the transformed E. faecalis BSU580 from the plasmid. This was successfully achieved by growing the strains in subinhibitory conditions of ciprofloxacin, as described by Eliopoulos et al. (23). Under these conditions, clones of both GBS BSU452 and the transformed E. faecalis strain BSU580 lost their elevated resistance to gentamicin. The MICs decreased in GBS strain BSU729 (i.e., strain BSU452 after plasmid curing) from 512 mg/liter to 24 mg/liter and in E. faecalis BSU720 (i.e., strain BSU580 after plasmid curing) from ≥1,024 mg/liter to 12 mg/liter (Table 2). In addition, the lack of a plasmid in the cured strains could be demonstrated by gel electrophoresis (Fig. 1). Plasmid loss in the presence of subinhibitory ciprofloxacin occurred at a frequency of about 0.02% (1 in 4,500 colonies in GBS and 1 in 6,000 colonies of E. faecalis). One of the most commonly found plasmids in GBS and enterococci is pIP501. To determine if the detected plasmid is pIP501, the plasmid preparation obtained from GBS BSU 452 was subjected to PCR as detailed elsewhere (24). PCR products were sequenced and the presence of pIP501 in GBS BSU452 was confirmed.
FIG 1.
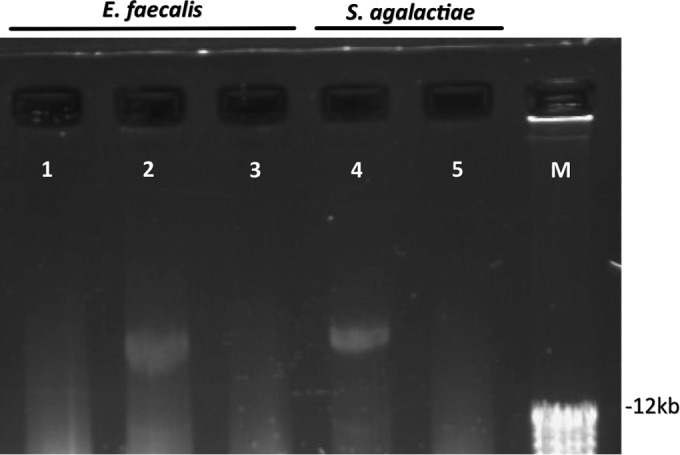
Plasmid preparations of GBS and E. faecalis strains. Shown are plasmid preparations of E. faecalis and GBS strains separated by agarose gel electrophoresis (0.8% gel). Lanes: 1, E. faecalis strain BSU386 (wild-type strain without HLGR); 2, E. faecalis strain BSU580 (wild-type strain BSU386 after transformation with plasmid preparation from S. agalactiae strain BSU452, displaying HLGR); 3, E. faecalis strain BSU720 (E. faecalis strain BSU580 after plasmid curing and loss of HLGR); 4, S. agalactiae strain BSU452 (patient isolate displaying HLGR); 5, S. agalactiae strain BSU729 (S. agalactiae strain BSU452 after plasmid curing and loss of HLGR). M, molecular size marker.
DISCUSSION
In contrast to screens for Enterococcus spp., GBS surveillance schemes usually do not include gentamicin susceptibility testing, and screening for HLGR is often omitted in clinical laboratories. The prevalence of HLGR in GBS is, therefore, unknown. Previously, two HLGR GBS strains were reported. One of them (B128) was isolated from an infected leg wound in 1987 (8) and the other from a 49-year-old woman with a urinary tract infection, as published in 2002 (9). We identified two further colonizing strains in a collection of more than 1,000 isolates (0.17%). This proportion is in contrast to that in a previously published Argentinian study (18). Among 141 strains, the authors found 13.5% HLGR GBS. Although no firm epidemiological conclusion about the frequency of HLGR isolates can be made on the basis of these two studies, it should be noted that up to 20% to 35% of women are colonized with GBS (25) and that the absolute number of HLGR GBS isolates may be much higher than previously estimated (26). Thus, for a patient receiving penicillin-gentamicin combination therapy for invasive GBS infection, and considering the potential side effects of aminoglycosides, the presence of HLGR is of significant clinical relevance.
Investigations on the genetic basis for HLGR in E. faecalis led to the identification of the aacA-aphD gene (27). It is typically found on the composite transposon Tn5281. The transposon resembles Tn4001 in Staphylococcus aureus and is characterized by the presence of two IS256 copies flanking the transposon structure. The aacA-aphD gene, later designated aac(6′)-aph(2″), encodes a bifunctional enzyme with an acetyltransferase and a phosphotransferase function. The enzyme catalyzes inactivation of the vast majority of aminoglycosides, with the exception of streptomycin. In most Enterococcus spp. with HLGR, the transposon harboring the aac(6′)-aph(2″) gene is found on a plasmid (28). Truncated forms of Tn4001 are typically located on plasmid DNA (29). Intact Tn4001 transposons can also be located on chromosomal DNA. In the previously described HLGR GBS strain B128, the aacA-aphD gene was found on a Tn4001 derivative (designated Tn3706), located on chromosomal DNA (8). In one of our strains (BSU1203), the finding of transposon Tn3706 conferring HLGR is in agreement with the previously published findings about HLGR GBS strain B128 (8, 20, 22). Horaud et al. (20) described that its transposition from E. faecalis occurred on GBS plasmid pIP501. However, after conjugative transfer between GBS strains, the hybrid replicons pIP501::Tn3706 were found to be structurally unstable. This observation indicated that streptococcal pIP501-like plasmids do not constitute appropriate delivery vectors for the dissemination of Tn3706 among GBS, and therefore, HLGR is found relatively rarely among GBS isolates (20). Although these arguments speak against a high potential for spread, the persistence of HLGR in GBS strain B128 and BSU1203 indicates that Tn3706 can be stably integrated into the chromosome.
In GBS BSU452, we identified a different mobile genetic element. The genes surrounding the aac(6′)-aph(2″) gene did not display the structures of transposon Tn4001 or of any of the closely related derivatives or its truncated forms. We detected plasmid pIP501, which is a conjugative plasmid that often carries multiresistance genes. It has previously been described for S. agalactiae in association with HLGR and belongs to the Inc18 group of plasmids (24). Tn3 family transposons are commonly associated with Inc18 plasmids and often confer antibiotic resistance on Enterococcus spp. (30). They are, however, typically associated with glycopeptide and macrolide resistance (31) and not HLGR. Investigators have previously reported the presence of an IS1216 transposase on Tn3-like remnants (32), as we found in our GBS BSU452 strain; however, IS1216 is typically associated with tetracycline resistance in streptococcal species (33). To the best of our knowledge, the detection of the aacA-aphD gene on a Tn3-like transposon and the presence of IS216 in association with HLGR constitute novel findings. They have been reported neither for enterococci nor for GBS.
The resistance determinant in GBS BSU452 shows close homologies to parts of the enterococcal resistance plasmid pTEF1 of E. faecalis strain V583 (34), suggesting that it may have been transferred through horizontal gene transfer. This is, however, speculative for GBS strain BSU452, since the presence of an HLGR Tn3-like transposon in GBS has not been previously described. Nevertheless, horizontal gene transfer of resistance genes from Enterococcus spp. to other Gram-positive bacteria by mobile genetic elements is a well-described mechanism in the spread of antibiotic resistance (30, 31). Horizontal gene transfer has recently been suggested for the acquisition of vancomycin resistance genes in GBS (35). GBS strain BSU452 was isolated from the sputum of a cystic fibrosis patient, but there was no evidence of enterococcal colonization. Considering that patients with cystic fibrosis are often treated with antibiotics (including aminoglycosides), and their microbiome in the respiratory tract is different from that of untreated healthy patients, it is possible that horizontal gene transfer to GBS originated from the selected flora. However, this hypothesis cannot be proven in our case and remains speculation. Though a plasmid-borne HLGR has high potential for further spread in a GBS population, the likelihood of this happening cannot be predicted yet. In this study, we demonstrated that pIP501, including the HLGR determinant of GBS452, could easily be transferred to E. faecalis. Thus, it is conceivable that transfer to other GBS isolates is also possible, especially in view of the fact that pIP501 is a broad-host-range plasmid, well established in GBS and enterococci.
In conclusion, the overall frequency of HLGR GBS in our large collection of isolates was low. Molecular investigations revealed a transposon located on the chromosome, as previously described for a single isolate (8, 20, 22), and a Tn3 family transposon conferring HLGR in association with pIP501. These findings point toward a new dimension of potential spread of HLGR within GBS.
ACKNOWLEDGMENTS
We are indebted to Alessandra Carattoli for valuable comments and critical review of the manuscript.
This work was supported in part by the Velux Foundation, Zurich, Switzerland (project no. 724 to P.S.).
We have no conflicts of interest to declare.
REFERENCES
- 1.Schrag SJ, Zell ER, Lynfield R, Roome A, Arnold KE, Craig AS, Harrison LH, Reingold A, Stefonek K, Smith G, Gamble M, Schuchat A. 2002. A population-based comparison of strategies to prevent early-onset group B streptococcal disease in neonates. N Engl J Med 347:233–239. doi: 10.1056/NEJMoa020205. [DOI] [PubMed] [Google Scholar]
- 2.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, Craig AS, Schaffner W, Zansky SM, Gershman K, Stefonek KR, Albanese BA, Zell ER, Schuchat A, Schrag SJ. 2008. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA 299:2056–2065. doi: 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 3.Skoff TH, Farley MM, Petit S, Craig AS, Schaffner W, Gershman K, Harrison LH, Lynfield R, Mohle-Boetani J, Zansky S, Albanese BA, Stefonek K, Zell ER, Jackson D, Thompson T, Schrag SJ. 2009. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990–2007. Clin Infect Dis 49:85–92. doi: 10.1086/599369. [DOI] [PubMed] [Google Scholar]
- 4.Westling K, Aufwerber E, Ekdahl C, Friman G, Gardlund B, Julander I, Olaison L, Olesund C, Rundstrom H, Snygg-Martin U, Thalme A, Werner M, Hogevik H. 2007. Swedish guidelines for diagnosis and treatment of infective endocarditis. Scand J Infect Dis 39:929–946. doi: 10.1080/00365540701534517. [DOI] [PubMed] [Google Scholar]
- 5.Zimmerli W, Trampuz A, Ochsner PE. 2004. Prosthetic-joint infections. N Engl J Med 351:1645–1654. doi: 10.1056/NEJMra040181. [DOI] [PubMed] [Google Scholar]
- 6.Baker CN, Thornsberry C, Facklam RR. 1981. Synergism, killing kinetics, and antimicrobial susceptibility of group A and B streptococci. Antimicrob Agents Chemother 19:716–725. doi: 10.1128/AAC.19.5.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schouten MA, Voss A, Hoogkamp-Korstanje JA. 1999. Antimicrobial susceptibility patterns of enterococci causing infections in Europe. The European VRE Study Group. Antimicrob Agents Chemother 43:2542–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buu-Hoï A, Le Bouguenec C, Horaud T. 1990. High-level chromosomal gentamicin resistance in Streptococcus agalactiae (group B). Antimicrob Agents Chemother 34:985–988. doi: 10.1128/AAC.34.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liddy H, Holliman R. 2002. Group B Streptococcus highly resistant to gentamicin. J Antimicrob Chemother 50:142–143. doi: 10.1093/jac/dkf090. [DOI] [PubMed] [Google Scholar]
- 10.Brimil N, Barthell E, Heindrichs U, Kuhn M, Lutticken R, Spellerberg B. 2006. Epidemiology of Streptococcus agalactiae colonization in Germany. Int J Med Microbiol 296:39–44. doi: 10.1016/j.ijmm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Florindo C, Damiao V, Silvestre I, Farinha C, Rodrigues F, Nogueira F, Martins-Pereira F, Castro R, Borrego MJ, Santos-Sanches I. 2014. Epidemiological surveillance of colonising group B Streptococcus epidemiology in the Lisbon and Tagus Valley regions, Portugal (2005 to 2012): emergence of a new epidemic type IV/clonal complex 17 clone. Euro Surveill 19(23):pii=20825. http://dx.doi.org/10.2807/1560-7917.ES2014.19.23.20825. [DOI] [PubMed] [Google Scholar]
- 12.Fluegge K, Siedler A, Heinrich B, Schulte-Moenting J, Moennig MJ, Bartels DB, Dammann O, von Kries R, Berner R, German Pediatric Surveillance Unit Study Group. 2006. Incidence and clinical presentation of invasive neonatal group B streptococcal infections in Germany. Pediatrics 117:e1139–e1145. doi: 10.1542/peds.2005-2481. [DOI] [PubMed] [Google Scholar]
- 13.Fluegge K, Wons J, Spellerberg B, Swoboda S, Siedler A, Hufnagel M, Berner R. 2011. Genetic differences between invasive and noninvasive neonatal group B streptococcal isolates. Pediatr Infect Dis J 30:1027–1031. doi: 10.1097/INF.0b013e31822a2a1f. [DOI] [PubMed] [Google Scholar]
- 14.Eickel V, Kahl B, Reinisch B, Dubbers A, Kuster P, Brandt C, Spellerberg B. 2009. Emergence of respiratory Streptococcus agalactiae isolates in cystic fibrosis patients. PLoS One 4:e4650. doi: 10.1371/journal.pone.0004650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shabayek S, Abdalla S, Abouzeid AM. 2014. Serotype and surface protein gene distribution of colonizing group B streptococcus in women in Egypt. Epidemiol Infect 142:208–210. doi: 10.1017/S0950268813000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing: twenty-fourth information supplement M100-s24. Screening test for detection of high-level aminoglycoside resistance (HLAR) in Enterococcus species. CLSI, Wayne, PA. [Google Scholar]
- 17.Weinbren MJ, Johnson AP, Woodford N. 2000. Defining high-level gentamicin resistance in enterococci. J Antimicrob Chemother 45:404–405. doi: 10.1093/jac/45.3.404. [DOI] [PubMed] [Google Scholar]
- 18.Villar HE, Jugo MB. 2013. Emergence of high-level resistance to gentamicin and streptomycin in Streptococcus agalactiae in Buenos Aires, Argentina. Rev Esp Quimioter 26:112–115. (In Spanish.) [PubMed] [Google Scholar]
- 19.Vakulenko SB, Donabedian SM, Voskresenskiy AM, Zervos MJ, Lerner SA, Chow JW. 2003. Multiplex PCR for detection of aminoglycoside resistance genes in enterococci. Antimicrob Agents Chemother 47:1423–1426. doi: 10.1128/AAC.47.4.1423-1426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horaud T, de Cespedes G, Trieu-Cuot P. 1996. Chromosomal gentamicin resistance transposon Tn3706 in Streptococcus agalactiae B128. Antimicrob Agents Chemother 40:1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friesenegger A, Fiedler S, Devriese LA, Wirth R. 1991. Genetic transformation of various species of Enterococcus by electroporation. FEMS Microbiol Lett 63:323–327. [DOI] [PubMed] [Google Scholar]
- 22.Kaufhold A, Podbielski A, Horaud T, Ferrieri P. 1992. Identical genes confer high-level resistance to gentamicin upon Enterococcus faecalis, Enterococcus faecium, and Streptococcus agalactiae. Antimicrob Agents Chemother 36:1215–1218. doi: 10.1128/AAC.36.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliopoulos GM, Wennersten C, Zighelboim-Daum S, Reiszner E, Goldmann D, Moellering RC Jr. 1988. High-level resistance to gentamicin in clinical isolates of Streptococcus (Enterococcus) faecium. Antimicrob Agents Chemother 32:1528–1532. doi: 10.1128/AAC.32.10.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brantl S, Nuez B, Behnke D. 1992. In vitro and in vivo analysis of transcription within the replication region of plasmid pIP501. Mol Gen Genet 234:105–112. [DOI] [PubMed] [Google Scholar]
- 25.Barcaite E, Bartusevicius A, Tameliene R, Kliucinskas M, Maleckiene L, Nadisauskiene R. 2008. Prevalence of maternal group B streptococcal colonisation in European countries. Acta Obstet Gynecol Scand 87:260–271. doi: 10.1080/00016340801908759. [DOI] [PubMed] [Google Scholar]
- 26.Murdoch DR, Reller LB. 2001. Antimicrobial susceptibilities of group B streptococci isolated from patients with invasive disease: 10-year perspective. Antimicrob Agents Chemother 45:3623–3624. doi: 10.1128/AAC.45.12.3623-3624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferretti JJ, Gilmore KS, Courvalin P. 1986. Nucleotide sequence analysis of the gene specifying the bifunctional 6′-aminoglycoside acetyltransferase 2″-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J Bacteriol 167:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horodniceanu T, Bougueleret L, El-Solh N, Bieth G, Delbos F. 1979. High-level, plasmid-borne resistance to gentamicin in Streptococcus faecalis subsp. zymogenes. Antimicrob Agents Chemother 16:686–689. doi: 10.1128/AAC.16.5.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casetta A, Hoi AB, de Cespedes G, Horaud T. 1998. Diversity of structures carrying the high-level gentamicin resistance gene (aac6-aph2) in Enterococcus faecalis strains isolated in France. Antimicrob Agents Chemother 42:2889–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmer KL, Kos VN, Gilmore MS. 2010. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr Opin Microbiol 13:632–639. doi: 10.1016/j.mib.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hegstad K, Mikalsen T, Coque TM, Werner G, Sundsfjord A. 2010. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin Microbiol Infect 16:541–554. doi: 10.1111/j.1469-0691.2010.03226.x. [DOI] [PubMed] [Google Scholar]
- 32.Tanous C, Chambellon E, Sepulchre AM, Yvon M. 2005. The gene encoding the glutamate dehydrogenase in Lactococcus lactis is part of a remnant Tn3 transposon carried by a large plasmid. J Bacteriol 187:5019–5022. doi: 10.1128/JB.187.14.5019-5022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novais C, Freitas AR, Silveira E, Baquero F, Peixe L, Roberts AP, Coque TM. 2012. Different genetic supports for the tet(S) gene in enterococci. Antimicrob Agents Chemother 56:6014–6018. doi: 10.1128/AAC.00758-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. doi: 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 35.Park C, Nichols M, Schrag SJ. 2014. Two cases of invasive vancomycin-resistant group B streptococcus infection. N Engl J Med 370:885–886. doi: 10.1056/NEJMc1308504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fröhlicher S, Reichen-Fahrni G, Muller M, Surbek D, Droz S, Spellerberg B, Sendi P. 2014. Serotype distribution and antimicrobial susceptibility of group B streptococci in pregnant women: results from a Swiss tertiary centre. Swiss Med Wkly 144:w13935. [DOI] [PubMed] [Google Scholar]