

Abstract
Levofloxacin is commonly used in critically ill patients for which existing data suggest nonstandard dosing regimens should be used. The objective of this study was to compare the population pharmacokinetics of levofloxacin in critically ill and in non-critically ill patients. Adult patients with a clinical indication for levofloxacin were eligible for participation in this prospective pharmacokinetic study. Patients were given 500 mg or 750 mg daily by intravenous administration with up to 11 blood samples taken on day 1 or 2 of therapy. Plasma samples were analyzed and population pharmacokinetic analysis was undertaken using Pmetrics. Thirty-five patients (18 critically ill) were included. The mean (standard deviation [SD]) age, weight, and Cockcroft-Gault creatinine clearance for the critically ill and for the non-critically ill patients were 60.3 (16.4) and 72.0 (11.6) years, 78.5 (14.8) and 70.9 (15.8) kg, and 71.9 (65.8) and 68.2 (30.1) ml/min, respectively. A two-compartment linear model best described the data. Increasing creatinine clearance was the only covariate associated with increasing drug clearance. The presence of critical illness did not significantly affect any pharmacokinetic parameter. The mean (SD) parameter estimates were as follows: clearance, 8.66 (3.85) liters/h; volume of the central compartment (Vc), 41.5 (24.5) liters; intercompartmental clearance constants from central to peripheral, 2.58 (3.51) liters/h; and peripheral to central compartments, 0.90 (0.58) liters/h. Monte Carlo dosing simulations demonstrated that achievement of therapeutic exposures was dependent on renal function, pathogen, and MIC. Critical illness appears to have no independent effect on levofloxacin pharmacokinetics that cannot be explained by altered renal function.
INTRODUCTION
Levofloxacin is a fluoroquinolone antibiotic with broad-spectrum activity that is used to treat Gram-positive, Gram-negative, and atypical microorganisms, including Streptococcus pneumoniae, Haemophilus influenzae, and Pseudomonas aeruginosa (1). As many of these bacteria are known to cause lower respiratory tract infections, levofloxacin is a common empirical choice in the treatment of severe pneumonia.
Like other fluoroquinolones, the pharmacokinetic/pharmacodynamic (PK/PD) indices that best describe levofloxacin antibacterial activity are the area under the concentration-time curve to MIC ratio (AUC0–24/MIC) and the peak concentration to MIC ratio (Cmax/MIC) (2). Previous data have shown that levofloxacin is well distributed at the target site of infection for pneumonia (i.e., epithelial lining fluid) (3). The excellent oral bioavailability of levofloxacin (≥99%) means that comparable exposure to the intravenous regimen may be achieved after oral administration (4, 5).
Recent data suggest that conventional levofloxacin doses derived from the general population may need to be altered to attain PD targets in critically ill patients with severe pneumonia (3). Previous work has shown that creatinine clearance (CrCL) is an important descriptor of altered levofloxacin clearance (CL) (6); however, it remains unclear whether critical illness itself contributes to mandatory nonstandard dosing of levofloxacin among these patients.
The objective of this study was to compare the population pharmacokinetics of levofloxacin in critically ill and non-critically ill patients. We then sought to apply Monte Carlo dosing simulations to propose robust dosing regimens for these patients.
MATERIALS AND METHODS
Setting.
This was an observational pharmacokinetic study using convenient sampling at a tertiary referral hospital. Ethical approval was obtained from the local institutional Ethics Committee. Informed consent was only required from the non-critically ill patients. An ethics waiver was granted for the critically ill patients because blood sampling was performed as part of the local therapeutic drug monitoring program.
Study population.
The inclusion criteria for this study were an age of ≥18 years and a serum creatinine concentration of <1.5 mg/dl. Critically ill and non-critically ill patients were eligible for participation. Exclusion criteria included a history of allergy to levofloxacin and the presence of any form of renal replacement therapy.
Study protocol.
Five hundred milligrams or 750 mg levofloxacin (Levoxacin; GlaxoSmithKline) was administered intravenously (i.v.) every 24 h as a 60-min infusion as part of the patient's prescribed therapy. Blood samples to determine plasma levofloxacin concentrations were taken either on day 1 of therapy at 0, 1, 1.5, 2, 3, 5, 7, 9, and 13 h or on day 2 at 0, 1, 1.25, 1.5, 2, 3, 5, 7, 9, 12, and 24 h.
Additional clinical and demographic data were collected, including sex, weight, age, height, sepsis organ failure assessment (SOFA), Cockcroft-Gault creatinine clearance, serum albumin concentration, serum bilirubin concentration, and indication for therapy.
Sample handling, storage, and measurement.
Blood samples were immediately placed on ice and were centrifuged within 60 min at 3,000 rpm for 10 min, and then if necessary they were stored at −80°C until assay. A high-performance liquid chromatography (HPLC)-UV assay was used to measure levofloxacin concentrations in plasma (7). Intra- and interassay coefficients of variation (CV) were always <10%. The lower limit of detection was 0.1 mg/liter.
Population pharmacokinetic modeling.
One- and two-compartment models were developed with the nonparametric adaptive grid (NPAG) algorithm within the Pmetrics package for R (Los Angeles, CA) (8, 9). Clearance from the central compartment and intercompartmental distribution (two-compartment model) into the peripheral compartment were modeled as first-order processes using differential equations. Estimates of assay error were included in the modeling process. Area under the concentration-time curve from 0 to 24 h (AUC0–24) was calculated using Pmetrics. Demographic and clinical characteristics that were considered biologically plausible for affecting levofloxacin pharmacokinetics were tested for inclusion as covariates. If inclusion of the covariate resulted in a statistically significant improvement in the log likelihood (P < 0.05) and/or an improvement in the goodness-of-fit plots, it was supported for inclusion.
Model diagnostics.
Goodness of fit was assessed by regression with an observed-predicted plot, coefficients of determination, and log-likelihood values. Predictive performance evaluation was based on mean prediction error (bias) and on the mean bias-adjusted squared prediction error (imprecision) of the population and of the individual prediction models. A visual predictive check (VPC) was also performed by simulating 1,000 patients to evaluate the predictive performance of the final model. Visual checks were performed by overlaying the observed data points with the 95% confidence intervals of the simulated 5th, 25th, 50th, 75th, and 95th percentile curves.
Probability of target attainment.
Monte-Carlo simulations (n = 1,000) were employed using Pmetrics to determine the probability of target attainment (PTA) of achieving an AUC/MIC of ≥80 for various MICs (0.064 to 8 mg/liter) during the first 24 h of treatment for patients with creatinine clearances of 30, 70, 130, and 200 ml/min for doses of 500 mg by i.v. daily, 750 mg daily, 1,000 mg daily, and 500 mg every 12 h.
Fractional target attainment calculation.
MIC data of S. pneumoniae, H. influenzae, methicillin-susceptible Staphylococcus aureus, and P. aeruginosa from the EUCAST database (www.eucast.org) were used to determine fractional target attainment. The fractional target attainment identifies the likely success of treatment by comparing the pharmacodynamic exposure (i.e., PTA) against the MIC distribution. The fractional target attainment was calculated using an AUC/MIC of ≥80. The PTA for achieving an AUC/MIC of ≥80 was calculated for the Monte Carlo simulations (n = 1,000) at various doses, including 500 mg i.v. every 24 h, 750 mg i.v. every 24 h, 1,000 mg i.v. every 24 h, and 500 mg i.v. every 12 h during the first 24 h of therapy. An a priori dosing regimen was considered successful if the fractional target attainment was >85%.
Statistical analysis.
Continuous data are presented as the mean (SD) or median (interquartile range [IQR]). Categorical data are presented as counts (%). Correlation was assessed by means of a scatter graph and the Pearson correlation coefficient (r). Differences in levofloxacin pharmacokinetic parameters between critically ill and non-critically ill patients were analyzed using Student's t test. A P value of <0.05 was considered statistically significant, and all analyses were performed using SPSS version 21 (Chicago, IL, USA).
RESULTS
Demographic data.
Thirty-five patients were recruited into the study, 18 of which were critically ill. Most patients received therapy as empirical therapy for community-acquired pneumonia, and others received treatment as directed therapy for aspiration against culture-positive S. pneumoniae. Demographic and comparison data between critically ill and non-critically ill patients are presented in Table 1. None of the patients were on any concomitant drug therapy that interacted with the pharmacokinetics of levofloxacin, and there were no reported adverse events associated with levofloxacin therapy in any of the study patients.
TABLE 1.
Demographic data and comparison between critically ill and non-critically ill patientsa
| Characteristic | Critically ill | Non-critically ill | P value |
|---|---|---|---|
| Total no. | 18 | 17 | |
| Male/Female | 12/6 | 10/7 | 0.63 |
| Age (yr) | 61 ± 17 | 70 ± 13 | 0.07 |
| Weight (kg) | 79 ± 15 | 71 ± 15 | 0.13 |
| Creatinine clearance (ml/min) | 70 ± 67 | 70 ± 32 | 0.97 |
| SAPS IIb | 43 (35–50) | ||
| SOFAc | 6 (5–8) |
Data are presented as mean ± SD or median (IQR).
Available for critically ill patients only. SAPS II, simplified acute physiology score.
Available for critically ill patients only. SOFA, sequential organ failure assessment.
Pharmacokinetic model building.
The time course of 329 total plasma concentrations of levofloxacin was best described by a two-compartment linear model. This model included a zero order input of the drug into the central compartment for i.v. administration. The only covariate that improved the fit of the model for levofloxacin clearance was creatinine clearance, which was statistically significant (P = 0.017). The final model was described as follows: Levofloxacin CL = TVCL * (CrCL/70)0.75. In this model, CL is clearance, TVCL is the typical value of clearance, and CrCL is creatinine clearance.
The population pharmacokinetic parameter estimates from the final covariate model are shown in Table 2. There were no statistically significant differences between intensive care unit (ICU) and non-ICU patients for levofloxacin clearance (CL; P = 0.39), volume of the central compartment (Vc; P = 0.15), rate constant for drug distribution from the central to the peripheral compartment (kcp; P = 0.67), or rate constant for drug distribution from the peripheral to the central compartment (kpc; P = 0.98). The diagnostic plots to confirm the goodness of fit of the model were considered acceptable and are shown in Fig. 1. The final covariate model was then used for dosing simulations.
TABLE 2.
Parameter estimates for levofloxacin from the final covariate 2-compartment population pharmacokinetic model
| Parametera | Mean | SD | Coefficient of variation (%) | Median |
|---|---|---|---|---|
| Clearance (liters/h) | 8.66 | 3.85 | 44.5 | 7.49 |
| Vcentral (liters) | 41.5 | 24.5 | 59.0 | 36.1 |
| kcp (h−1) | 2.58 | 3.51 | 136.0 | 1.30 |
| kpc (h−1) | 0.90 | 0.58 | 64.1 | 0.75 |
Vcentral, volume of distribution of the central compartment; kcp, rate constant for drug distribution from the central to the peripheral compartment; kpc, rate constant for drug distribution from the peripheral to the central compartment.
FIG 1.

(a) Diagnostic plots for the final covariate model. Observed versus population predicted concentrations (left) and individual predicted concentrations (right) in plasma are shown. (b) Visual predictive check for the final covariate model where the y axis output is levofloxacin concentration. Data are presented in milligrams per liter.
Dosing simulations.
The Monte Carlo simulations and PTAs for various levofloxacin doses for patients with creatinine clearances of 30, 70, 130, and 200 ml/min are described in Fig. 2. These simulations generally describe that PTA is lower for patients with higher creatinine clearances. Increasing doses results in increased PTA, with an increased dosing frequency having little effect.
FIG 2.

Monte Carlo simulations and the probability of target attainment in plasma for various levofloxacin doses for creatinine clearances of 30 ml/min, 70 ml/min,130 ml/min, and 200 ml/min.
Fractional target attainment.
The fractional target attainments for the simulated PTAs for a range of levofloxacin doses, dose frequencies, and creatinine clearances against susceptible MIC distributions (MIC, ≤1.0 mg/liter) for S. pneumoniae, S. aureus, H. influenzae, and P. aeruginosa are shown in Table 3. These data show that acceptable achievement of PD targets occurs for all doses against the MIC distribution H. influenzae but not for any of the other organisms. S. aureus tended to have fractional target attainments between 70% and 80%, and P. aeruginosa and S. pneumoniae had lower attainments with S. pneumoniae in particular having very low fractional target attainments.
TABLE 3.
Fractional target attainment for the various levofloxacin doses for patients with creatinine clearances of 30, 70, 130, and 200 ml/min for an S. pneumoniae, H influenzae, P. aeruginosa, and methicillin-susceptible S. aureus MIC distributiona
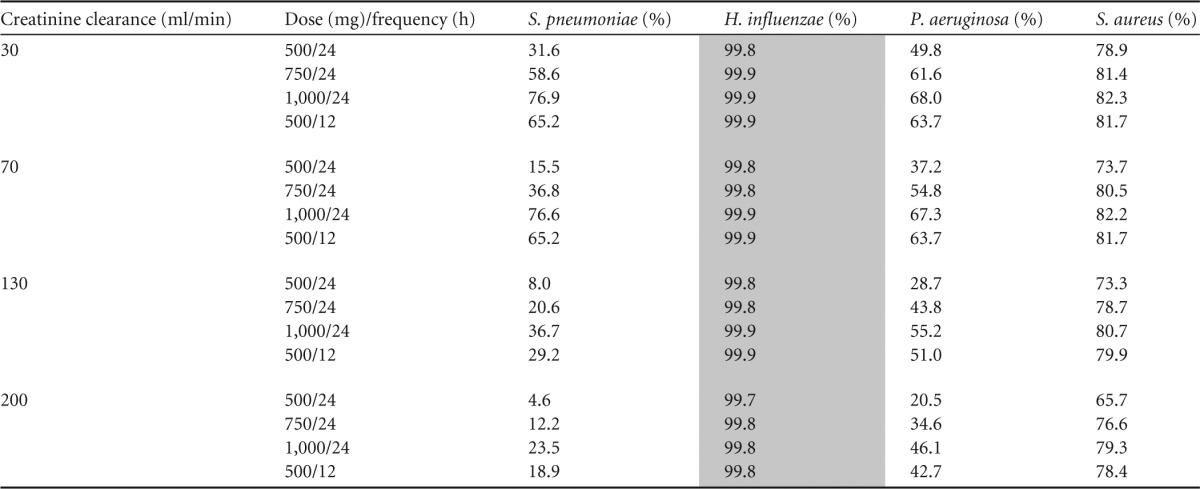
Doses achieving the a priori target of PTA against at least 85% of isolates are shaded gray. PTA, probability of target attainment (AUC/MIC ratio of 80).
DISCUSSION
This study found no significant effect of critical illness on the pharmacokinetics of levofloxacin. Creatinine clearance was the only covariate that influenced levofloxacin clearance as has also been noted in previous population pharmacokinetic studies in various patient groups. Preston et al. concluded that creatinine clearance, age, and race were covariates for their population pharmacokinetic model, with creatinine clearance explaining most of the variability in levofloxacin clearance (6). Additionally, earlier studies found decreasing levofloxacin clearance was associated with lower creatinine clearance, mainly in elderly patients (10, 11), while Kiser et al. (12) showed that renal function was a superior predictor of levofloxacin clearance than the percentage of total body surface area burn injury in a cohort of critically ill burn patients.
The AUC0–24/MIC targets for levofloxacin have been proposed by various groups to be different for Gram-positive (≥50) and for Gram-negative (≥87) pathogens, respectively (2, 13), with other authors suggesting that a target of ≥100 should be used (14). Using the available data, we applied an AUC0–24/MIC target of 80 in this study, and our Monte Carlo dosing simulations showed that achievement of PK/PD targets was not high for MIC distributions of key Gram-negative and Gram-positive pathogens. Twice daily dosing did little to abate the falling fractional target attainment in the face of increasing creatinine clearances in our study, a result that was not observed in previous investigations of the pharmacokinetics of levofloxacin in pneumonia.
In a 2003 study, Pea and colleagues found that levofloxacin dosed at 500 mg i.v. every 12 h met and exceeded Cmax/MIC and AUC0–24/MIC targets in critically ill patients with a mean creatinine clearance of 142 ml/min (7). These conflicting findings, however, were not due to variability in the pharmacokinetics of the drug between studies but were rather a function of the higher susceptibilities of bacterial isolates noted in the 2003 study. The authors of that study duly observed that for an MIC of 2 mg/liter, which is currently listed as the clinical breakpoint for key pathogens causing pneumonia (15), AUC0–24 values would most likely be unattainable in critically ill patients with normal renal function (7). This conclusion closely reflects the findings of this study.
Directly in contrast to Pea and colleagues, Boselli et al. (3) found that PK/PD target attainments in serum and epithelial lining fluid can be achieved for pathogens with MICs of ≤1 mg/liter and of >1 mg/liter, respectively. This was possibly attributed to increases in the volume of distribution leading to a longer elimination half-life, thereby favorably influencing the AUC0–24 values of the drug. The authors hypothesized that large interindividual variability meant that renal function may not be the only pharmacokinetic variable influencing levofloxacin clearance, a conclusion that cannot be confirmed based on the findings of our study.
It should be noted that limitations do exist when interpreting our study results. First, there may be multiple factors influencing the pharmacokinetics of levofloxacin in the critically ill such as severity of illness, site of infection, and fluid status. Due to the small sample size, however, it was not possible to control for these variables. Second, the small sample size also prevented the testing of PK/PD target attainment with patient outcomes. Third, as with all pharmacokinetic studies based on serum drug concentrations, there was an inability to report on levofloxacin concentrations at the site of infection. Previous work has been able to show comparable concentrations of this drug at target sites, such as in epithelial lining fluid (3) and in blood, but due to our study patients having diagnoses other than pneumonia, we cannot extrapolate these findings to ascertain if PK/PD targets were or were not achieved at infection sites outside the lungs. Fourth, our study creatinine clearance was estimated via the Cockcroft-Gault equation rather than by measuring urine creatinine clearance during the sampling period, which is often considered a more accurate estimate of glomerular filtration rate.
In conclusion, critical illness does not appear to be a significant variable in altering levofloxacin clearance per se, with altered renal function being the determinant factor. Importantly, based on the current MIC distribution data, fractional target attainment for key pathogens is poorly achievable even with higher levofloxacin doses or with the use of dosing every 12 h. Larger studies investigating the influence of additional variables, such as infection site and fluid status, are suggested to determine if any other aspect of critical illness impacts the dosing requirements for levofloxacin.
Funding Statement
Jason A. Roberts is funded by a Career Development Fellowship from the National Health and Medical Research Council of Australia (APP1048652).
REFERENCES
- 1.Fish DN, Chow AT. 1997. The clinical pharmacokinetics of levofloxacin. Clin Pharmacokinet 32:101–119. doi: 10.2165/00003088-199732020-00002. [DOI] [PubMed] [Google Scholar]
- 2.Preston SL, Drusano GL, Berman AL, Fowler CL, Chow AT, Dornseif B, Reichl V, Natarajan J, Corrado M. 1998. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA 279:125–129. doi: 10.1001/jama.279.2.125. [DOI] [PubMed] [Google Scholar]
- 3.Boselli E, Breilh D, Rimmele T, Djabarouti S, Saux MC, Chassard D, Allaouchiche B. 2005. Pharmacokinetics and intrapulmonary diffusion of levofloxacin in critically ill patients with severe community-acquired pneumonia. Crit Care Med 33:104–109. doi: 10.1097/01.CCM.0000150265.42067.4C. [DOI] [PubMed] [Google Scholar]
- 4.Furlanut M, Brollo L, Lugatti E, Di Qual E, Dolcet F, Talmassons G, Pea F. 2003. Pharmacokinetic aspects of levofloxacin 500 mg once daily during sequential intravenous/oral therapy in patients with lower respiratory tract infections. J Antimicrob Chemother 51:101–106. doi: 10.1093/jac/dkg035. [DOI] [PubMed] [Google Scholar]
- 5.Rebuck JA, Fish DN, Abraham E. 2002. Pharmacokinetics of intravenous and oral levofloxacin in critically ill adults in a medical intensive care unit. Pharmacotherapy 22:1216–1225. doi: 10.1592/phco.22.15.1216.33484. [DOI] [PubMed] [Google Scholar]
- 6.Preston SL, Drusano GL, Berman AL, Fowler CL, Chow AT, Dornseif B, Reichl V, Natarajan J, Wong FA, Corrado M. 1998. Levofloxacin population pharmacokinetics and creation of a demographic model for prediction of individual drug clearance in patients with serious community-acquired infection. Antimicrob Agents Chemother 42:1098–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pea F, Di Qual E, Cusenza A, Brollo L, Baldassarre M, Furlanut M. 2003. Pharmacokinetics and pharmacodynamics of intravenous levofloxacin in patients with early-onset ventilator-associated pneumonia. Clin Pharmacokinet 42:589–598. doi: 10.2165/00003088-200342060-00008. [DOI] [PubMed] [Google Scholar]
- 8.Tatarinova T, Neely M, Bartroff J, van Guilder M, Yamada W, Bayard D, Jelliffe R, Leary R, Chubatiuk A, Schumitzky A. 2013. Two general methods for population pharmacokinetic modeling: non-parametric adaptive grid and non-parametric Bayesian. J Pharmacokinet Pharmacodyn 40:189–199. doi: 10.1007/s10928-013-9302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanigawara Y, Nomura H, Kagimoto N, Okumura K, Hori R. 1995. Premarketing population pharmacokinetic study of levofloxacin in normal subjects and patients with infectious diseases. Biol Pharm Bull 18:315–320. doi: 10.1248/bpb.18.315. [DOI] [PubMed] [Google Scholar]
- 11.Chien SC, Chow AT, Natarajan J, Williams RR, Wong FA, Rogge MC, Nayak RK. 1997. Absence of age and gender effects on the pharmacokinetics of a single 500-milligram oral dose of levofloxacin in healthy subjects. Antimicrob Agents Chemother 41:1562–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiser TH, Hoody DW, Obritsch MD, Wegzyn CO, Bauling PC, Fish DN. 2006. Levofloxacin pharmacokinetics and pharmacodynamics in patients with severe burn injury. Antimicrob Agents Chemother 50:1937–1945. doi: 10.1128/AAC.01466-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drusano GL, Preston SL, Fowler C, Corrado M, Weisinger B, Kahn J. 2004. Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in patients with nosocomial pneumonia. J Infect Dis 189:1590–1597. doi: 10.1086/383320. [DOI] [PubMed] [Google Scholar]
- 14.Odenholt I, Cars O. 2006. Pharmacodynamics of moxifloxacin and levofloxacin against Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae and Escherichia coli: simulation of human plasma concentrations after intravenous dosage in an in vitro kinetic model. J Antimicrob Chemother 58:960–965. doi: 10.1093/jac/dkl356. [DOI] [PubMed] [Google Scholar]
- 15.European Committee on Antimicrobial Susceptibility Testing. 2015. Breakpoint tables for interpretation of MICs and zone diameters. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_5.0_Breakpoint_Table_01.pdf.