

Abstract
We are in a crisis of bacterial resistance. For economic reasons, most pharmaceutical companies are abandoning antimicrobial discovery efforts, while, in health care itself, infection control and antibiotic stewardship programs have generally failed to prevent the spread of drug-resistant bacteria. At this point, what can be done? The first step has been taken. Governments and international bodies have declared there is a worldwide crisis in antibiotic drug resistance. As discovery efforts begin anew, what more can be done to protect newly developing agents and improve the use of new drugs to suppress resistance emergence? A neglected path has been the use of recent knowledge regarding antibiotic dosing as single agents and in combination to minimize resistance emergence, while also providing sufficient early bacterial kill. In this review, we look at the data for resistance suppression. Approaches include increasing the intensity of therapy to suppress resistant subpopulations; developing concepts of clinical breakpoints to include issues surrounding suppression of resistance; and paying attention to the duration of therapy, which is another important issue for resistance suppression. New understanding of optimizing combination therapy is of interest for difficult-to-treat pathogens like Pseudomonas aeruginosa, Acinetobacter spp., and multidrug-resistant (MDR) Enterobacteriaceae. These lessons need to be applied to our old drugs as well to preserve them and to be put into national and international antibiotic resistance strategies. As importantly, from a regulatory perspective, new chemical entities should have a resistance suppression plan at the time of regulatory review. In this way, we can make the best of our current situation and improve future prospects.
WHAT ARE THE MECHANISMS OF RESISTANCE AND DO THEY INTERACT?
This paper does not have as its aim a comprehensive review of all bacterial resistance mechanisms. Here, we examine the major resistance mechanisms for different drug classes and how they may interact.
Perhaps the most underappreciated resistance mechanism is the efflux pump. As demonstrated previously (1–3), the first mechanism called forth is often upregulation of efflux pumps. Indeed, in Pseudomonas aeruginosa, there are multiple examples of pumps whose expression has been subjected to antibiotic pressure so as to find the one that is most efficient in combating the threat (1).
This phenomenon has also been seen in other bacteria in infection models. It was clearly seen in a Streptococcus pneumoniae study where fluoroquinolones were employed for therapy in a murine thigh model (4). The drug ciprofloxacin is substantially more hydrophilic than the agent levofloxacin. Efflux pumps like pmrA have a preference for hydrophilic substrates. In the challenge experiments, no levofloxacin-resistant isolates were found. Ciprofloxacin allowed recovery of such organisms. The organisms that were less susceptible to ciprofloxacin had a minimally increased levofloxacin MIC (0.6 to 0.8 mg/liter [performed in arithmetic dilution]). However, the mutational frequency of resistance to levofloxacin for this isolate changed by 4 orders of magnitude (<10−8.5 to 10−4.5). Subsequent experiments were able to select for levofloxacin resistance. The original clones had, with one exception, no mutations in gyrA or gyrB or in parC or parE. These isolates were responsive to reserpine and in the presence of this drug had their ciprofloxacin MIC values diminish significantly. In one isolate only, there was a target site mutation found (parC: Ser79Tyr) in combination with efflux pump-mediated resistance. This strongly implies (but does not prove) that the first response in the organism is pump upregulation, followed by continuing rounds of replication resulting in target site mutation. It should be noted that efflux pump expression can be reversibly inducible or can be stably expressed through mutation.
A later in vitro evaluation in the hollow-fiber infection model (HFIM) (5), followed by profiling of two of these isolates through analysis of ethidium bromide uptake and efflux (ethidium bromide is a known substrate of pmrA), demonstrated that uptake was much more rapid in the wild-type isolate than in the mutant isolate (but without a change in the target site) and responded to the presence of reserpine (pump blockade markedly speeded uptake). Likewise, efflux was substantially more rapid in the mutant isolate than in the wild type, again strongly implying that the change in MIC was due to an efflux pump. Members of A. P. MacGowan's laboratory elegantly demonstrated this in a paper examining both moxifloxacin and levofloxacin (6).
Another example of this phenomenon can be found in the evaluation of levofloxacin against Bacillus anthracis in the HFIM (7). This organism had a resistance mutation frequency of less than −8.7 log10 CFU/ml. As the bacterial inoculum was slightly below 107/ml, and as the system volume was 10 ml, there is a relatively low likelihood that there were resistant isolates in the population a priori. An exposure-range experiment employing animal pharmacokinetics demonstrated that the values of the area under the concentration-time curves over 24 h in the steady state divided by the MIC (AUC/MIC ratios) of 150, 200, and 300 did not cause major cell kill but did allow amplification of resistant subpopulations. There was a time dependency, with an AUC/MIC ratio of 150 allowing resistance at day 3, while AUC/MIC values of 200 and 300 allowed resistance to be measured at day 6, with the AUC/MIC arm of 300 amplifying significantly more slowly. Later isolates demonstrated target site mutations (data not shown). Pasipanodya and Gumbo (8) have recently postulated a similar chain of events in Mycobacterium tuberculosis.
One of the issues often ignored is the change in MIC wrought by the resistance mechanism. For example, efflux pump upregulation frequently generates an MIC increase of 4-fold to 8-fold. Stable derepression of an ampC β-lactamase generally also results in a 4-fold-to-8-fold MIC increase. Target site mutation for fluoroquinolones mediates a 4-fold-to-8-fold change. However, rpoB mutation for rifamycins generally mediates an MIC change of >32-fold. The importance of the step size change is that lower values can often be counterselected by proper dosing of antimicrobials. As the step size gets larger, the probability of counterselection of amplification of less-susceptible populations with single-agent therapy goes down substantially.
Another factor often not considered is the drug being employed. Certain agents, such as quinolones, induce error-prone replication (9). Drugs that cause generation of reactive oxygen and nitrogen species are often associated with random mutations (10). Those β-lactams binding primarily to pbp3 also (somewhat unexpectedly) induce error-prone replication (11). Once error-prone replication is induced or increased mutation rates occur, it is a matter of the number of rounds of bacterial replication until a MIC-increasing mutation is generated, as the site of the mutation is random. Many of these mutations are lethal. But with enough rounds of replication, a mutation will be found that is MIC-changing and survivable. This brings up two further issues. The first is that while many mutations result in degraded biofitness for the organism, continuing rounds of replication provide the opportunity for compensatory mutations to improve relative biofitness, as shown in Andersson's laboratory (12).
The other and, arguably, more important issue is that of the interaction of mechanisms. For agents like fluoroquinolones, induction of efflux pumps generates relatively low-level resistance. As fluoroquinolones induce error-prone replication, this provides enough extra rounds of bacterial replication to markedly increase the likelihood of obtaining a target site mutation as well. Generally, these two resistance mechanisms together provide an 8-fold-to-16-fold MIC change (13).
Another example of mechanism interaction is the combination of β-lactamase induction or stable derepression with loss of a porin channel (14). β-Lactamases display Michaelis-Menten-like kinetics (i.e., they are saturable). If penetration into the periplasm is rapid enough, the enzyme can be saturated and “excess” drug can bind to the β-lactam protein binding target. By eliminating the porin channel, the rate of penetration into the periplasm decreases, making it harder to saturate the system. This is why there is a marked increase in the MICs of carbapenems in Pseudomonas aeruginosa when an isolate has stable derepression of its ampC enzyme (stable derepression alone has limited impact on MIC for carbapenems) combined with oprD downregulation (major carbapenem porin channel).
Given the number of resistance mechanisms and their interactions, it becomes obvious that in some instances it will be nearly impossible to identify a drug dose that will produce exposures that will counterselect amplification of a less-susceptible population in an acceptable proportion of patients.
WHAT ABOUT COMBINATION THERAPY?
There are a number of reasons to employ combination chemotherapy. One may broaden the spectrum of coverage. If the choice of agents is salutary, synergy may be attained. Perhaps the most important use of combination chemotherapy is in attempts to suppress emergence of resistance.
Combination therapy has a long history. Here, we look at more-modern evaluations where statistical evaluation of the interaction has been performed. The first issue is that two agents generate more adverse drug reactions than a single agent. Consequently, it is important to realize an advantage from the combination. Such an advantage is most likely when there is a synergistic or, at least, an additive interaction, so that the kill rate is improved. Having a synergistic (or additive) interaction also provides the highest likelihood of achieving resistance suppression.
Even the best single-agent therapy administered at maximal doses and at an optimal schedule may not provide robust counterselection of amplification of less-susceptible populations. As an example (15), meropenem had a resistance-suppression target identified in a murine pneumonia model with P. aeruginosa as the challenge pathogen. When bridging to humans was performed using a maximal clinically approved dose regimen (2 g administered intravenously [i.v.] every 8 h as a 3-h infusion), the resistance-suppression target was attained 64.5% of the time (expectation taken over the MIC distribution) and target attainment by MIC fell below 80% at an MIC of 1 mg/liter. This suggests that single-agent therapy with meropenem may not be adequate for Pseudomonas resistance suppression. Consequently, combination therapy may be warranted in this situation.
Many drug interactions are not synergistic. In Table 1, some studies are listed by pathogen and agents and the type of interaction. Below, we examine this issue for Gram-negative pathogens, with an emphasis on Pseudomonas aeruginosa and also for Mycobacterium tuberculosis.
TABLE 1.
Combination chemotherapy, type of drug interaction by pathogen, and suppression of resistancea
| Species and drug combination | Experimental system | Interaction | Reference or result |
|---|---|---|---|
| Pseudomonas aeruginosa | |||
| Meropenem + tobramycin | In vitro plate assay | Additive | 16 |
| Meropenem + levofloxacin | HFIM | Synergistic | 17 |
| Meropenem + levofloxacin | Murine pneumonia | Synergistic | 18 |
| Cefepime + tobramycin | HFIM | Additive | 19 |
| Meropenem + tobramycin | Murine pneumonia | Additive | 20 |
| Ceftolozane + tazobactam | HFIM | Potentiating | 21 |
| Klebsiella pneumoniae/Enterobacter cloacae/Escherichia coli | |||
| Ceftaroline + avibactam | HFIM | Potentiating | 22 |
| Ceftolozane + tazobactam | HFIM | Potentiating | 23 |
| Mycobacterium tuberculosis | |||
| Moxifloxacin + rifampin | HFIM | Antagonistic | 24 |
| Moxifloxacin + rifampin | HFIM | Antagonistic | 25 |
| Linezolid + rifampin | HFIM | Additive | 26 |
HFIM, hollow-fiber infection model. “Interaction” refers to the drug interaction for bacterial cell kill.
PSEUDOMONAS AERUGINOSA
Surprisingly, the effects of meropenem plus tobramycin were additive but not synergistic for bacterial cell kill both in an in vitro 96-well plate checkerboard drug interaction assay and in a murine Pseudomonas pneumonia model (16, 20). In both evaluations, however, the drug combination was able to suppress amplification of less-susceptible clones.
Another evaluation was performed for the combination of meropenem plus levofloxacin in the HFIM (17). The rationale for this combination is counterintuitive at first glance because the two agents share a resistance mechanism (overexpression of the mexAB efflux pump). In the evaluation, both wild-type Pseudomonas aeruginosa PAO1 and its isogenic daughter isolate overexpressing mexAB genes were evaluated. In both, the drug combination was synergistic for bacterial cell kill and mediated complete suppression of resistance amplification. This may have been because the pump has two entry points (periplasm for meropenem and cytoplasm for levofloxacin) and because the combination may saturate the pump, as pumps also display Michaelis-Menten-like kinetics and can be saturated. The combination of β-lactams plus levofloxacin was actually subjected to trials in patients with hospital-acquired pneumonia (27). In 17 patients with P. aeruginosa pneumonia, there were no instances of resistance emergence, although in a trial of maximal-dose ciprofloxacin (400 mg i.v. every 8 h), the rate of resistance emergence during therapy was 33% for P. aeruginosa (28).
We have very recently evaluated this combination in a murine model of Pseudomonas aeruginosa pneumonia, using our novel combination therapy mathematical model (18). As in the in vitro evaluation, we found that these agents were synergistic for cell kill and also that they prevented resistance amplification.
An evaluation of cefepime with or without tobramycin was performed in the HFIM (19). While the 3′ side chain of cefepime improves stability with respect to hydrolysis by ampC β-lactamases (decreases physiologic affinity for the enzyme), this can be overcome by the use of a very large bacterial burden.
Figure 1 demonstrates that a bacterial burden of 108 CFU/ml in the HFIM (10-ml volume) resulted in regimen failure even with large exposures to either cefepime (up to 3 g every 8 h) or tobramycin (up to 10 mg/kg of body weight/day) when given singly. All combination regimens produced 6 to 8 log10 (CFU/ml) bacterial load declines without resistance emergence (latter data not shown). In another experiment, addition of a β-lactamase inhibitor also prevented regimen failure. The mechanism of the major increase in bacterial cell kill and resistance suppression was then sought. In Fig. 2, a Western blot is shown from a culture of P. aeruginosa exposed to cefepime alone or to tobramycin alone or to the combination of the two. The antibody was directed against the ampC β-lactamase of Pseudomonas. Comparison of the h 2 control with administration of cefepime alone, tobramycin alone, and the combination demonstrates that cefepime showed some induction of β-lactamase production whereas tobramycin virtually shut it off. The combination showed more production than tobramycin alone but significantly lower production than cefepime alone. This demonstrates that inhibition of β-lactamase production is the mechanism behind the salutary effect of the combination with respect to bacterial cell kill and resistance suppression. These findings were recapitulated at 5 h of exposure and also using quantitative PCR as a second endpoint at both time points.
FIG 1.

(A and B) For cefepime (CFPM) and tobramycin (Tobra or TOB) as monotherapy against P. aeruginosa in the hollow-fiber infection model, regimen failure was seen even with supraphysiologic dosing. WT or wt, wild type. Q8h, every 8 h. (C) Combination chemotherapy drove regimen success.
FIG 2.
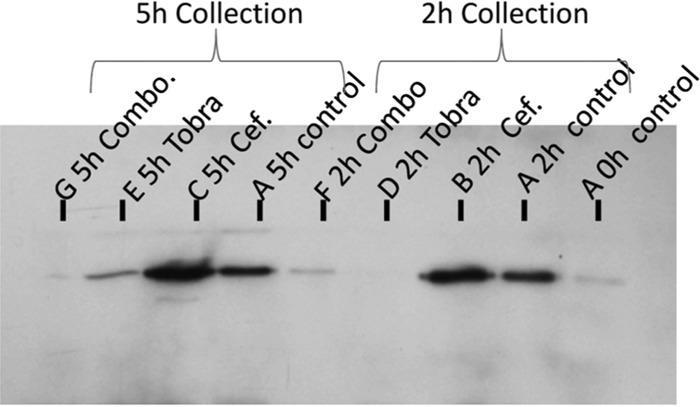
Western blotting for the amount of expressed AmpC β-lactamase in control cultures as well as in cultures exposed to cefepime (Cef.), tobramycin (Tobra), and the combination (Combo.) of cefepime plus tobramycin over time.
Very recently (21), VanScoy et al. looked at the use of ceftolozane plus tazobactam against two isolates of P. aeruginosa, an ATCC strain and a clinical isolate. Resistance emergence was not seen for the ATCC isolate in a 10-day study, but an “inverted U” plot for resistance suppression was demonstrated for the clinical isolate (as seen before; 29). Resistance suppression was attained for this isolate at a ceftolozane/tazobactam dose of 2 g/1 g every 8 h.
KLEBSIELLA PNEUMONIAE, ENTEROBACTER CLOACAE, ESCHERICHIA COLI
Evaluations of β-lactam/β-lactamase inhibitor combinations are slightly different from those in the circumstance where two fully active agents are combined. Most β-lactamase inhibitors are not microbiologically active. A second drug that potentiates the activity of the first without activity of its own has an interaction that is referred to as potentiation. Avibactam (NXL-104) was evaluated in combination with ceftaroline. A strain of K. pneumoniae carrying a Kpc-2 carbapenemase along with a SHV-27 extended-spectrum β-lactamase (ESBL) and a TEM-1 enzyme was evaluated (22). Other organisms evaluated included a K. pneumoniae isolate with a CTX-M15 ESBL and an E. cloacae isolate with a chromosomal ampC β-lactamase. Dose fractionation experiments demonstrated that time > threshold was the dynamically linked index parameter both for bacterial cell kill and for suppression of amplification of less-susceptible populations for the agents in combination.
Vanscoy et al. (23) studied ceftolozane plus tazobactam against a set of isogenic organisms with different amounts of expression of a CTX-M15 β-lactamase. As shown previously, they found that the linked dynamic index parameter was time > threshold and that resistance suppression followed an inverted U plot (23, 29).
MYCOBACTERIUM TUBERCULOSIS
The benefits of combination chemotherapy have been clearly demonstrated in M. tuberculosis, particularly for resistance suppression (30). Ironically, while synergistic or additive combinations are most desirable, antagonistic combinations can sometimes be successful at resistance suppression. Oftentimes, the cost attendant on this is the decrement of the rate of kill of the bacterial burden. The combination of rifampin plus moxifloxacin was evaluated in the HFIM (24). Log-phase growth organisms as well as nonreplicative persister (NRP) phenotype organisms were evaluated. The drug interaction was difficult to evaluate in log-phase organisms because of the confounding of rate of kill by resistance emergence. However, in the NRP population, there was no resistance emergence. Here, the antagonism between moxifloxacin and rifampin with regard to cell kill was demonstrable. With regard to resistance, the combination was able to suppress amplification of resistant populations. That study had the antagonism for cell kill validated in a murine model of tuberculosis (31).
Even though the moxifloxacin/rifampin combination could suppress amplification of less-susceptible organisms in this evaluation, there are circumstances where it may fail. In order to minimize regimen nonadherence (and hence help suppress resistance), directly observed therapy programs have been initiated. In some programs, drugs are administered 5 of 7 (5/7) days, with the patients getting a drug holiday. The moxifloxacin/rifampin combination was evaluated in a HFIM and the impact of the 5/7-day regimen evaluated against a 7/7-day regimen (25). Another important issue is that when a fixed dose of drug is administered to a population of patients, there arises a distribution of exposures in the patients. In this evaluation, the mean exposure as well as plus or minus 1 standard deviation for each drug was evaluated. The experiment was performed in duplicate simultaneously by two different teams. The results are displayed in Fig. 3. In both experiments, there was no resistance emergence when the regimen was administered 7/7 days. In both experiments, the 5/7-day administration group had resistance emerge late in the experiment. However, this occurred only in the case of the exposure that was 1 standard deviation below the mean value for the agents in the combination.
FIG 3.

(A) First trial of moxifloxacin (Moxi) plus rifampin (Rif) in the 7-day/7-day (7/7) versus 5-day/7-day (5/7) regimens. The AUC exposures in the symbol keys are the free AUC 24-h exposures that were infused into that particular hollow-fiber-system experimental arm on the days that the drugs were administered. (B) Second trial of moxifloxacin plus rifampin in the 7-day/7-day versus 5-day/7-day regimens. The AUC exposures in the figure legends are the free AUC 24-h exposures that were infused into that particular hollow-fiber-system experimental arm on the days that the drugs were administered. Resistance occurred only in the 5-day/7-day therapy arms at the lowest exposures.
This leads to the issue of the mechanism by which resistance emerged in the 5/7-day group. In the case of moxifloxacin plus rifampin, there is a major mismatch in the half-life of the drugs. After the first day of the drug holiday, there is little rifampin left, as its half-life and level of clearance are much shorter and higher, respectively, than for moxifloxacin. Fluoroquinolones induce error-prone replication (9). Consequently, in the second day of the drug holiday, there is enough quinolone present to continue induction of error-prone replication and there is little rifampin to suppress resistance. With enough rounds of replication, it is likely that there will be a mutation that generates a less-susceptible clone. Consequently, we need to be careful with regimen design when resistance suppression is a goal. The combination of rifamycin with moxifloxacin has recently been evaluated (REMoxTB trial and moxifloxacin plus high-dose rifapentine; 32, 33) in an attempt to shorten therapy to 4 months. Noninferiority was not achieved in either of the experimental arms of the REMoxTB trial or in the moxifloxacin/high-dose-rifapentine trial for the 4-month durations of therapy. Antagonism is not likely to allow significant shortening of tuberculosis therapy.
Trying to cure M. tuberculosis in a patient within a reasonably short period of time (e.g., 2 months) is a goal of many agencies worldwide. At the heart of the problem is the difficulty of obtaining a rapid decline in bacterial burden combined with suppression of resistance emergence. Determining the way drugs interact with respect to rate of kill and resistance suppression in a statistically rigorous manner may be a way forward with this problem. The regimens chosen should, if possible, have drug interactions that are at least additive and preferably synergistic.
It is important to define what we mean by the terms synergy, additivity, and antagonism. One reasonable approach is to have a definition of “additivity.” In a recent publication (26), the authors employed the definition of Loewe additivity (essentially defining additive interaction as adding a drug to itself, pushing it up the exposure-response surface; that is, the drug exposure for drug 1 is on the x axis and the drug exposure for drug 2 is on the y axis, with the response plotted on the z axis). Under this definition, synergy can be defined as an effect that is statistically greater than additivity and antagonism as an effect that is statistically less than additivity.
Greco et al. derived a fully parametric approach for drug interaction (34). In this formulation, the interaction term “α” determines the interaction. If α is positive and the lower 95% confidence bound does not overlap zero, the interaction represents significant synergy. If the value is negative and the upper 95% confidence bound does not overlap zero, the interaction represents significant antagonism. Any estimate of α where the confidence bound overlaps zero is accorded a definition of additivity.
A new approach (26) expanded this approach by including resistant subpopulations in the evaluations. The approach was also expanded to explicitly handle time-dependent data, so effect endpoints (e.g., total colony counts, resistant colony counts, drug concentrations) can be simultaneously examined over a prolonged period, with intermediate measurements. As an example of the approach, the authors examined the combination of linezolid plus rifampin in a 28-day HFIM experiment. The α values estimated for the fully susceptible population, the linezolid-resistant/rifampin-sensitive population, and the rifampin-resistant/linezolid-sensitive population all were negative (trending to antagonism), but all had 95% confidence bounds that crossed zero and therefore were considered to represent additivity.
Because the approach is fully parametric, it is possible to perform Monte Carlo simulation. When a 1,000-subject simulation was performed and between-patient variability in pharmacokinetics was accounted for, the results provided insight into the problem and a possible way forward to solve it. As shown in Fig. 4, approximately 51% of subjects had eradicated their fully susceptible population by the end of the experiment. Another 32% had eradicated the linezolid- and rifampin-resistant subpopulation. Only 4% achieved total bacterial burden eradication.
FIG 4.

System simulation (1,000-iterate Monte Carlo simulation), susceptible counts (Cts), and subpopulations less susceptible to the study drugs (linezolid [LZD] and rifampin [RIF]) from the Bayesian posterior parameter vectors. The median values and the standard deviations are displayed.
The message is that it is much harder for the regimen to deal with the resistant subpopulations. To obtain M. tuberculosis eradication in a short time period, it may be that the regimen should be completely switched over to a new regimen where there is no overlap of the resistance genotypes generated by the first regimen. As there may be different metabolic populations present in M. tuberculosis and as they change in relative levels of importance over time, the new regimen should also take such an evolution into account.
SUMMARY
One path to preserving our current antimicrobials and extending the lifetimes of new agents as they enter the therapeutic armamentarium is to employ dosing strategies (intensity, duration of therapy) that will help minimize resistance emergence. Understanding that some agents induce error-prone replication allows one to understand the relative risk of getting a target site mutation. Other mechanisms, such as efflux pump upregulation, give more rounds of replication per unit time and increase the likelihood of a target site mutation. Again, this interacts with therapy intensity and duration.
Combination chemotherapy often (but not always) suppresses resistance emergence. The goal is to obtain synergistic or at least additive combinations so that we can obtain resistance suppression while rapidly reducing the bacterial burden. Application of these simple principles will hopefully extend the useful lifetimes of our older agents as well as of new additions to our therapeutic armamentarium.
Resistance to antimicrobial agents will ultimately develop even with good stewardship and proper dosing of regimens alone and in combination. This is because of the numbers of organisms involved, the plethora of resistance mechanisms available to them, and the need, on occasion, to have prolonged therapy. Nonadherence to regimens also plays a role. Nonetheless, to ameliorate the position in which we now find ourselves, we believe it is incumbent upon the regulatory authorities to take under consideration a new requirement for sponsors. Currently, drugs must be safe and effective. New molecular entities, we believe, should be accompanied by a plan for minimizing resistance emergence as part of the regulatory process. For some agents, the dose should help suppress amplification of less-susceptible subpopulations. For drugs where this is not possible (large step size change in MIC with a mutation), exploration of appropriate companion drugs for combination therapy should be implemented. In so doing, we can stave off another crisis.
REFERENCES
- 1.Jumbe N, Louie A, Leary R, Liu W, Deziel MR, Tam VH, Bachhawat R, Freeman C, Kahn JB, Bush K, Dudley MN, Miller MH, Drusano GL. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J Clin Invest 112:275–285. doi: 10.1172/JCI200316814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tam VH, Louie A, Deziel MR, Liu W, Leary R, Drusano GL. 2005. Bacterial-population responses to drug-selective pressure: examination of garenoxacin's effect on Pseudomonas aeruginosa. J Infect Dis 192:420–428. doi: 10.1086/430611. [DOI] [PubMed] [Google Scholar]
- 3.Ruzin A, Visalli MA, Keeney D, Bradford PA. 2005. Influence of transcriptional activator RamA on expression of multidrug efflux pump AcrAB and tigecycline susceptibility in Klebsiella pneumoniae. Antimicrob Agents Chemother 49:1017–1022. doi: 10.1128/AAC.49.3.1017-1022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jumbe NL, Louie A, Miller MH, Liu W, Deziel MR, Tam VH, Bachhawat R, Drusano GL. 2006. Quinolone efflux pumps play a central role in emergence of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 50:310–317. doi: 10.1128/AAC.50.1.310-317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louie A, Brown DL, Liu W, Kulawy RW, Deziel MR, Drusano GL. 2007. In vitro infection model characterizing the effect of efflux pump inhibition on prevention of resistance to levofloxacin and ciprofloxacin in Streptococcus pneumoniae. Antimicrob Agents Chemother 51:3988–4000. doi: 10.1128/AAC.00391-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowker KE, Garvey MI, Noel AR, Tomaselli SG, MacGowan AP. 2013. Comparative antibacterial effects of moxifloxacin and levofloxacin on Streptococcus pneumoniae strains with defined mechanisms of resistance: impact of bacterial inoculum. J Antimicrob Chemother 68:1130–1138. doi: 10.1093/jac/dks344. [DOI] [PubMed] [Google Scholar]
- 7.Deziel M, Heine H, Louie A, Kao M, Byrne WR, Bassett J, Miller L, Bush K, Kelley M, Drusano GL. 2005. Effective antimicrobial regimens for use in humans for therapy of Bacillus anthracis infections and postexposure prophylaxis.Effective antimicrobial regimens for use in humans for therapy of Bacillus anthracis infections and postexposure prophylaxis. Antimicrob Agents Chemother 49:5099–5106. doi: 10.1128/AAC.49.12.5099-5106.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasipanodya JG, Gumbo T. 2011. A new evolutionary and pharmacokinetic-pharmacodynamic scenario for rapid emergence of resistance to single and multiple anti-tuberculosis drugs. Curr Opin Pharmacol 11:457–463. doi: 10.1016/j.coph.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Sullivan DM, Hinds J, Butcher PD, Gillespie SH, McHugh TD. 2008. Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J Antimicrob Chemother 62:1199–1202. doi: 10.1093/jac/dkn387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, Flynn JL, Fortune SM. 2011. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genetics 43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blázquez J, Gómez-Gómez JM, Oliver A, Juan C, Kapur V, Martín S. 2006. PBP3 inhibition elicits adaptive responses in Pseudomonas aeruginosa. Mol Microbiol 62:84–99. doi: 10.1111/j.1365-2958.2006.05366.x. [DOI] [PubMed] [Google Scholar]
- 12.Kugelberg E, Löfmark S, Wretlind B, Andersson DI. 2005. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J Antimicrob Chemother 55:22–30. [DOI] [PubMed] [Google Scholar]
- 13.Singh R, Swick MC, Ledesma KR, Yang Z, Hu M, Zechiedrich L, Tam VH. 2012. Temporal interplay between efflux pumps and target mutations in development of antibiotic resistance in Escherichia coli. Antimicrob Agents Chemother 56:1680–1685. doi: 10.1128/AAC.05693-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vu H, Nikaido H. 1985. Role of beta-lactam hydrolysis in the mechanism of resistance of a beta-lactamase-constitutive Enterobacter cloacae strain to expanded-spectrum beta-lactams. Antimicrob Agents Chemother 27:393–398. doi: 10.1128/AAC.27.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drusano GL, Lodise TP, Melnick D, Liu W, Oliver A, Mena A, Van Scoy B, Louie A. 2011. Meropenem penetration into epithelial lining fluid in mice and humans and delineation of exposure targets. Antimicrob Agents Chemother 55:3406–3412. doi: 10.1128/AAC.01559-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drusano GL, Liu W, Fregeau C, Kulawy R, Louie A. 2009. Differing effects of combination chemotherapy with meropenem and tobramycin on cell kill and suppression of resistance of wild-type Pseudomonas aeruginosa PAO1 and its isogenic MexAB efflux pump-overexpressed mutant. Antimicrob Agents Chemother 53:2266–2273. doi: 10.1128/AAC.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louie A, Grasso C, Bahniuk N, Van Scoy B, Brown DL, Kulawy R, Drusano GL. 2010. The combination of meropenem and levofloxacin is synergistic with respect to both Pseudomonas aeruginosa kill rate and resistance suppression. Antimicrob Agents Chemother 54:2646–2654. doi: 10.1128/AAC.00065-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louie A, Liu W, VanGuilder M, Neely MN, Schumitzky A, Jelliffe R, Fikes S, Kurhanewicz S, Robbins N, Brown D, Baluya D, Drusano GL. 2015. Combination treatment with meropenem plus levofloxacin is synergistic against Pseudomonas aeruginosa infection in a murine model of pneumonia. J Infect Dis 211:1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drusano GL, Bonomo RA, Bahniuk N, Bulitta JB, Vanscoy B, Defiglio H, Fikes S, Brown D, Drawz SM, Kulawy R, Louie A. 2012. Resistance emergence mechanism and mechanism of resistance suppression by tobramycin for cefepime for Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:231–242. doi: 10.1128/AAC.05252-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louie A, Liu W, Fikes S, Brown D, Drusano GL. 2013. Impact of meropenem in combination with tobramycin in a murine model of Pseudomonas aeruginosa pneumonia. Antimicrob Agents Chemother 57:2788–2792. doi: 10.1128/AAC.02624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.VanScoy BD, Mendes RE, Castanheira M, McCauley J, Bhavnani SM, Jones RN, Friedrich LV, Steenbergen JN, Ambrose PG. 2014. Relationship between ceftolozane-tazobactam exposure and selection for Pseudomonas aeruginosa resistance in a hollow-fiber infection model. Antimicrob Agents Chemother 58:6024–6031. doi: 10.1128/AAC.02310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louie A, Castanheira M, Liu W, Grasso C, Jones RN, Williams G, Critchley I, Thye D, Brown D, Vanscoy B, Kulawy R, Drusano GL. 2012. Pharmacodynamics of β-lactamase inhibition by NXL104 in combination with ceftaroline: examining organisms with multiple types of β-lactamases. Antimicrob Agents Chemother 56:258–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanscoy B, Mendes RE, Castanheira M, McCauley J, Bhavnani SM, Forrest A, Jones RN, Okusanya OO, Friedrich LV, Steenbergen J, Ambrose PG. 2013. Relationship between ceftolozane-tazobactam exposure and drug resistance amplification in a hollow-fiber infection model. Antimicrob Agents Chemother 57:4134–4138. doi: 10.1128/AAC.00461-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drusano GL, Sgambati N, Eichas A, Brown DL, Kulawy R, Louie A. 10 August 2010. The combination of rifampin plus moxifloxacin is synergistic for suppression of resistance but antagonistic for cell kill of Mycobacterium tuberculosis as determined in a hollow-fiber infection model. mBio 1:e00139-10. doi: 10.1128/mBio.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drusano GL, Sgambati N, Eichas A, Brown D, Kulawy R, Louie A. 12 July 2011. Effect of administration of moxifloxacin plus rifampin against Mycobacterium tuberculosis for 7 of 7 days versus 5 of 7 days in an in vitro pharmacodynamic system. mBio 2:e00108-11. doi: 10.1128/mBio.00108-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drusano GL, Neely M, Van Guilder M, Schumitzky A, Brown D, Fikes S, Peloquin C, Louie A. 8 July 2014. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PLoS One 9:e101311. doi: 10.1371/journal.pone.0101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West M, Boulanger BR, Fogarty C, Tennenberg A, Wiesinger B, Oross M, Wu SC, Fowler C, Morgan N, Kahn JB. 2003. Levofloxacin compared with imipenem/cilastatin followed by ciprofloxacin in adult patients with nosocomial pneumonia: a multicenter, prospective, randomized, open-label study. Clin Ther 25:485–506. doi: 10.1016/S0149-2918(03)80091-7. [DOI] [PubMed] [Google Scholar]
- 28.Fink MP, Snydman DR, Niederman MS, Leeper KV Jr, Johnson RH, Heard SO, Wunderink RG, Caldwell JW, Schentag JJ, Siami GA, Zameck RL, Haverstock DC, Reinhart HH, Echols RM; the Severe Pneumonia Study Group. 1994. Treatment of severe pneumonia in hospitalized patients: results of a multicenter, randomized, double-blind trial comparing intravenous ciprofloxacin with imipenem-cilastatin. The Severe Pneumonia Study Group. Antimicrob Agents Chemother 38:547–557. doi: 10.1128/AAC.38.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tam VH, Louie A, Deziel MR, Liu W, Drusano GL. 2007. The relationship between quinolone exposures and resistance amplification is characterized by an inverted U: a new paradigm for optimizing pharmacodynamics to counterselect resistance. Antimicrob Agents Chemother 51:744–747. doi: 10.1128/AAC.00334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selkon JB, Devadatta S, Kullarna KG, Mitchison DA, Narayana AS, Nair CN, Ramachandran K. 1964. The emergence of isoniazid-resistant cultures in patients with pulmonary tuberculosis during treatment with isoniazid alone or isoniazid plus PAS. Bull World Health Organ 31:273–294. [PMC free article] [PubMed] [Google Scholar]
- 31.Balasubramanian V, Solapure S, Gaonkar S, Mahesh Kumar KN, Shandil RK, Deshpande A, Kumar N, Vishwas KG, Panduga V, Reddy J, Ganguly S, Louie A, Drusano GL. 2012. Effect of coadministration of moxifloxacin and rifampin on Mycobacterium tuberculosis in a murine aerosol infection model. Antimicrob Agents Chemother 56:3054–3057. doi: 10.1128/AAC.06383-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PP, Nunn AJ; REMoxTB Consortium. 2014. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371:1577–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jindani A, Harrison TS, Nunn AJ, Phillips PP, Churchyard GJ, Charalambous S, Hatherill M, Geldenhuys H, McIlleron HM, Zvada SP, Mungofa S, Shah NA, Zizhou S, Magweta L, Shepherd J, Nyirenda S, van Dijk JH, Clouting HE, Coleman D, Bateson AL, McHugh TD, Butcher PD, Mitchison DA; RIFAQUIN Trial Team. 2014. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N Engl J Med 371:1599–1608. doi: 10.1056/NEJMoa1314210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greco WR, Bravo G, Parsons JC. 1995. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev 47:331–385. [PubMed] [Google Scholar]