

Abstract
Malaria remains a major global health problem, with more than half of the world population at risk of contracting the disease and nearly a million deaths each year. Here, we report the discovery of inhibitors that target multiple stages of malaria parasite growth. To identify these inhibitors, we took advantage of the Tres Cantos Antimalarial Compound Set (TCAMS) small-molecule library, which is comprised of diverse and potent chemical scaffolds with activities against the blood stage of the malaria parasite, and investigated their effects against the elusive liver stage of the malaria parasite using a forward chemical screen. From a screen of nearly 14,000 compounds, we identified and confirmed 103 compounds as dual-stage malaria inhibitors. Interestingly, these compounds show preferential inhibition of parasite growth in liver- versus blood-stage malaria parasite assays, highlighting the drug susceptibility of this parasite form. Mode-of-action studies were completed using genetically modified and drug-resistant Plasmodium parasite strains. While we identified some compound targets as classical antimalarial pathways, such as the mitochondrial electron transport chain through cytochrome bc1 complex inhibition or the folate biosynthesis pathway, most compounds induced parasite death through as yet unknown mechanisms of action. Importantly, the identification of new chemotypes with different modes of action in killing Plasmodium parasites represents a promising opportunity for probing essential and novel molecular processes that remain to be discovered. The chemical scaffolds identified with activity against drug-resistant Plasmodium parasites represent starting points for dual-stage antimalarial development to surmount the threat of malaria parasite drug resistance.
INTRODUCTION
Malaria continues to be a major global health burden in large parts of the world, killing a disproportionate number of pregnant women and children, as well as hindering economic growth in many developing countries (1). Clinical treatments for malaria are failing due to parasite drug resistance, and this demands new therapeutics for disease control. Current guidelines for malaria eradication suggest inhibitors with unique targets and efficacy against multiple parasite forms are necessary. Many pioneering studies have identified potential treatment therapies through systematic searches of diverse small-molecule libraries (2). Unfortunately, active compounds identified in these searches are often later found to have targets that are not therapeutically viable due to the prevalence of parasite strains that have mutated under the selection pressure of currently prescribed antimalarials. These facts highlight the need for an approach that identifies dual-stage malaria inhibitors with novel modes of action, and importantly, compounds with novel modes of action have the potential to reveal new aspects of malaria parasite biology.
A unicellular parasite from the genus Plasmodium causes malaria, and among the five major species that infect people, Plasmodium falciparum is the most deadly. In addition to infecting humans, Plasmodium parasites also have a mosquito host. Within the mosquito, the Plasmodium parasite, taken up from a human blood meal, develops into sporozoites, and this parasite form is released into the human dermis during feeding (3). Once in the dermis, the sporozoite travels in the bloodstream to the liver, and it is there that thousands of merozoites, a blood-infective parasite form, are produced (3, 4). This exoerythrocytic phase is asymptomatic, but once the merozoites infect red blood cells (RBC), the symptomatic cyclical erythrocytic stage begins. Thus, the malaria parasite has distinct life stages, including the vector stage, the liver stage, and the blood stage. The different parasite forms share the same genome, but they morph in size, shape, and metabolism to enable the selective infection of distinct host cells. While many details of this metamorphosis-like process remain unknown, compounds that target these stages are useful probes to advance our understanding of Plasmodium biology. Additionally, compounds that target these processes are useful starting points for drug development, as targeting blood-stage parasites is important for disease treatment and killing liver-stage parasites offers prophylaxis, a necessary step for meeting eradication goals (5).
Current therapies for malaria target a limited number of parasite processes. A significant number of antimalarials function by inhibiting folate biosynthesis, targeting the dihydrofolate reductase, or by disrupting the electron transport chain by inhibiting the cytochrome bc1 complex (reviewed in reference 6). Other essential parasite processes include isoprenoid precursor biosynthesis (reviewed in reference 7) and hemozoin formation (reviewed in reference 8). Unfortunately, resistance to the drugs targeting these pathways has already been reported, and therefore, active compounds that target these processes in phenotypic drug screens may not inhibit Plasmodium parasites in the clinic (reviewed in reference 6). This necessitates the development of new strategies to prioritize screening of active compounds and to identify new targets.
Target identification is challenging in most systems, but the discovery of novel Plasmodium targets is particularly difficult compared to well-studied model systems due to the inability to systemically explore essential genes with genetic tools, such as knockout libraries or RNA interference (9). In malaria, unique drug targets are often identified after a small-molecule disruptor, or chemical probe, is discovered that inhibits parasite growth. Thus, it becomes easier to discover novel targets with an inhibitor in hand that binds to this new target. Such methods identified the parasite's ATPase (10), lysyl-tRNA synthetase (11), prolyl-tRNA synthetase (12), translation elongation factor 2 (13), dihydroorotate dehydrogenase (DHODH) (14), and phosphatidylinositol-4-OH kinase (PI4K) (15) as potential drug targets. Significantly, compounds targeting both DHODH (DMS265) and PI4K (MMV390048) are in clinical development, but there remains a need to identify still more essential processes, as a suite of unique targets presents the best route to disease eradication.
Several large malaria drug discovery campaigns by organizations, including GlaxoSmithKline (GSK) (16), Novartis (17), and St. Jude's Children's Research Hospital (18), have been completed. These ambitious endeavors have identified thousands of chemical starting points for antimalarial development. Selecting only candidates with dual-stage activity and novel targets in cell-based assays from these starting points poses a significant hurdle, especially for GlaxoSmithKline's library of ∼14,000 blood-stage malaria inhibitors. This library is particularly attractive for further exploration due to its large number of chemotypes and potent inhibition of blood-stage malaria parasites. Therefore, we utilized a forward chemical genetic screen to identify compounds within GSK's library that have efficacy against malaria's asymptomatic liver stage, as they have potential uses for prophylaxis, as well as treatment. Active compounds that exhibited dual-stage activity were further filtered for inhibition of previously known malaria targets, such as electron transport, folate biosynthesis, and pyrimidine pathways, and several diverse scaffolds with nanomolar activity were discovered to inhibit malaria parasites by a different mode of action. This work identifies hundreds of small molecules with potent dual-stage activity, but perhaps most importantly, our approach to filter out scaffolds with previously known targets enables the selection of candidates with the greatest potential for novel targets. These candidates may now be harnessed as chemical probes to identify their targets to further our understanding of malaria parasite biology.
MATERIALS AND METHODS
Chemicals and reagents.
HepG2 cells (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen), 10% fetal bovine serum (FBS) (Sigma), and 1% antibiotic-antimycotic (Invitrogen) in a standard tissue culture incubator (37°C; 5% CO2). Plasmodium-infected Anopheles stephensi mosquitoes were purchased from the New York University Langone Medical Center Insectary. Compounds used in this study were from the Tres Cantos antimalarial set, and their purity was confirmed by high-performance liquid chromatography-mass spectrometry (HPLC-MS) analysis to be >95%.
Phenotypic liver-stage malaria screen.
The Tres Cantos antimalarial set was screened for inhibition of the liver-stage Plasmodium berghei ANKA parasite load in HepG2 cells and HepG2 viability in duplicate. The library was provided as 1 mM stocks (50 nl) in dimethyl sulfoxide (DMSO) in compound storage plates (Eppendorf 384-well twin.tec), which were stored at −20°C until use. For assays, 15,000 HepG2 cells/well were added to 384-well microtiter plates. After 18 to 24 h at 37°C, the medium was aspirated, cell medium was added to the compound plates, and then compounds in the cell medium (25 μl) were transferred to the assay plates with a Velocity 11 Bravo liquid handler (Agilent Technologies) to give a final concentration of 1.3 μM. Halofuginone (1 μM) was used as the positive control for parasite inhibition (19), and DMSO was used as the negative control. The parasite form that infects liver cells was obtained by isolation and disruption of salivary glands from previously infected A. stephensi mosquitoes. Approximately 1 h after compound addition, luciferase-expressing P. berghei ANKA parasites (4) were added to the plates at a density of 3,000 to 4,000 parasites/well, the plates were spun for 10 min at 1,000 rpm, and then they were incubated at 37°C for ∼45 h. The final assay volume postinfection was 30 μl. After ∼45 h at 37°C, HepG2 viability was assessed by adding CellTiter-Fluor (Promega) and measuring fluorescence. After the fluorescence measurement, the parasite load in the liver cells was determined by adding Bright-Glo (Promega) and measuring luminescence. The relative fluorescence and luminescence signal intensity of each plate was evaluated with an EnVision system (PerkinElmer). The parasite and HepG2 signal in the presence of compounds was normalized to the negative control (DMSO) and evaluated as the relative percent viability.
Compounds that significantly inhibited the parasite load and had a minimal effect on HepG2 viability were considered active compounds (156 in total). All active compounds were retested at 320 nM and 160 nM in singlicate. Among these compounds, 103 were selected for 50% effective concentration (EC50) analysis based on their activity at 160 nM and structural diversity.
Dose-response curves for compound inhibition of the P. berghei ANKA parasite load in liver cells were generated to acquire EC50s. Compounds (0 to 14 μM) were added to HepG2 cells in a 384-well plate using a Velocity 11 Bravo liquid handler (Agilent Technologies) in duplicate. Plasmodium infection and assay measurements were completed as described above. For dose-response analysis, the parasite signal was normalized to the negative control and reported as the relative percent parasite viability. Data analysis was carried out using GraphPad Prism.
Mode-of-action studies.
Compounds were tested using functional assays to identify inhibitors of the mitochondrial electron transport chain (ETC), as well as folate biosynthesis blockers. Dose-response assays were conducted at concentrations ranging from 20 μM to 0.003 μM. The 50% inhibitory concentration (IC50) calculations were performed with GraFit 5 software.
Lactate dehydrogenase (LDH) activity was used as a surrogate of Plasmodium viability. Predispensed plates with 50 nl of compounds were inoculated with 25 μl of a parasitized RBC suspension (2% hematocrit; 0.25% parasitemia) and incubated for 72 h. LDH activity was monitored in a final volume of 100 μl in 100 mM Tris-HCl, pH 8.0, 0.5% Tween 20, 100 mM lactate, 100 μM 3-acetylpyridine adenine dinucleotide (APAD+), 125 μM nitroblue tetrazolium (NBT), and 1 U/ml diaphorase. Absorbance at 650 nm was used to follow the reaction.
Folate pathway studies were carried out using P. falciparum 3D7A adapted to grow in low levels (100 ng/ml) of folic acid (FA)- and para-aminobenzoic acid (pABA)-depleted RPMI medium until normal 3D7A growth rates were observed (compared with P. falciparum 3D7A in standard RPMI medium). We assessed whether the compounds were targeting the folate pathway by comparing the IC50s in FA/pABA-depleted medium and standard RPMI medium using the normal and adapted strains. Antifolates were identified by the IC50 shift in the growth medium supplemented with folic acid.
Mitochondrial electron transport pathway studies were conducted using the Dd2-attB_yeastDHODH (Dd2_yDHODH) strain and its parental strain, Dd2-attB. Transgenic parasites expressing Saccharomyces cerevisiae DHODH (yDHODH) are resistant to mitochondrial electron transport inhibitors. Addition of proguanil (1 μM) restores the sensitivity of Dd2_yDHODH to electron transport inhibitors, such as atovaquone, but has no effect with specific P. falciparum DHODH (PfDHODH) inhibitors.
RESULTS
Chemical genetic liver-stage screen.
We designed a forward genetic screen to identify small molecules that inhibit the malaria parasite's asymptomatic liver stage (20). The screen recapitulated liver-stage infection using Plasmodium sporozoites to infect HepG2 liver cells in a 384-well microtiter plate, and relative infection was assessed with a luciferase reporter. The assay uses a mouse-infective P. berghei ANKA strain, since it has a higher infection rate than other in vitro human malaria models and facilitates downstream animal studies. The screen was used to test 13,553 compounds that target the P. falciparum intraerythrocytic cycle, aptly named the Tres Cantos Antimalarial Compound Set (TCAMS), from GSK (16). The compound set was tested for inhibition of P. berghei infection of human HepG2 cells and for cytotoxicity in HepG2 cells at 1.3 μM as a primary screen. Screening actives were identified as compounds that decreased the parasite load by 95% compared to DMSO-treated controls but did not inhibit HepG2 cell viability. All active compounds were then retested in the cell-based assay at 160 nM and 320 nM in singlicate. Compounds that were active (80% inhibition) at 160 nM were reacquired, and the 50% inhibitory concentration was determined with dose-response analysis. From this phenotypic liver-stage malaria screen, we identified 103 compounds (0.8% hit rate) that decreased the parasite load in HepG2 cells with an EC50 below 5 μM. Most screening actives are submicromolar inhibitors of liver-stage P. berghei parasites (see Table S1 in the supplemental material). Furthermore, we compared the anti-liver-stage malaria potencies of these compounds to those previously reported in the blood-stage P. falciparum assay (16) and observed a moderate positive correlation (Spearman's correlation; Rs = 0.414) (Fig. 1). This correlation indicates a difference in susceptibility of Plasmodium parasites between liver and blood stages upon treatment with these active compounds, with preferential inhibition of the liver stage.
FIG 1.

Relationship of active-compound potencies between inhibition of Plasmodium liver and blood stages. The compounds are plotted against their P. berghei liver-stage and P. falciparum blood-stage potencies (pXC50 = −logXC50, where XC50 is in molar units and pXC50 is dimensionless; an XC50 value of 1 μM corresponds to a pXC50 value of 6). The calculated Spearman's correlation (RS) is 0.414, with a P value of <0.0001; n = 101.
Chemotype analysis.
To determine the number of chemotypes present within the screening actives, we employed molecular framework and fingerprint cluster analyses (16, 21). The molecular frameworks describe the general core structure of a molecule, whereas the fingerprint analysis describes changes in substituent groups. Thus, fingerprint clustering is useful to separate distinct classes of molecules from within a larger group organized by molecular frameworks. Using the Daylight fingerprint methods with a Tanimoto similarity index of 0.85 (22), we identified 15 chemical clusters and 58 “singletons” (see Table S1 in the supplemental material). Moreover, the combination of molecular framework and fingerprint cluster analyses enables the exploration of the chemical structure space represented by these active compounds (Fig. 2). Thus, the chemical clustering analysis of active compounds against liver-stage P. berghei provided an initial framework to begin addressing the important question of the mode of action.
FIG 2.

Three-dimensional plot of active compounds determined from a liver-stage malaria parasite screen. Compounds are represented by spheres plotted against their assigned molecular framework numbers, chemical fingerprint cluster numbers, and liver-stage P. berghei potencies. Chemical clusters and singletons are indicated by colored and gray spheres, respectively. The structure diagrams are examples of potent compounds from chemical clusters and singletons.
Mechanism-of-action studies.
The chemical clustering of active compounds likely groups molecules with similar mechanisms, thereby providing a starting point to begin investigations of the mechanism of action. To date, the majority of malaria drugs used clinically target only a few parasite processes, including the mitochondrial electron transport chain and pyrimidine biosynthesis and folate pathways (6). Therefore, we first sought to test if active compounds target these known pathways with pathway-specific orthogonal whole-organism screens (23, 24). All available singletons and one or more members from each chemical cluster were evaluated in three different mode-of-action assays.
To investigate whether screening actives act through the inhibition of the Plasmodium mitochondrial ETC, similarly to atovaquone, or via inhibition of the pyrimidine biosynthesis pathway, parasite strains expressing either endogenous (P. falciparum) or yeast (S. cerevisiae) dihydroorotate dehydrogenase were utilized. DHODH is a critical enzyme in the pyrimidine biosynthesis pathway and a validated antimalarial drug target (reviewed in reference 25). It uses ubiquinone as an electron acceptor in the generation of orotate from dihydroorotate, thus coupling ETC with pyrimidine biosynthesis (Fig. 3A). Compounds were tested for differential growth inhibition of a wild-type P. falciparum Dd2 (Dd2-attB) strain versus a transgenic P. falciparum parasite strain expressing the S. cerevisiae DHODH (Dd2-yDHODH). Importantly, this transgenic strain overexpressing the yDHODH utilizes fumarate as an electron acceptor in the generation of orotate for pyrimidine synthesis and thus bypasses the endogenous PfDHODH. Therefore, P. falciparum Dd2-yDHODH is resistant to ETC and/or DHODH inhibitors. We observed that 3 chemical clusters and 8 singletons exhibited reduced activity against the transgenic P. falciparum Dd2-yDHODH strain compared to the parental P. falciparum Dd2-attB strain, as shown by a resistance value of >1-fold (Table 1; see Fig. S1 in the supplemental material). The differential activity observed for these compounds suggests that they inhibit the Plasmodium mitochondrial ETC and/or DHODH.
FIG 3.

In vitro parasite growth curves for mode-of-action studies. (A) The P. falciparum mitochondrial membrane potential, which is driven by the cytochrome bc1 complex- and cytochrome c oxidase-mediated reactions, is coupled with pyrimidine biosynthesis through the PfDHODH. Following the generation of oxidized ubiquinone (Q) via the cytochrome bc1 complex, reduction of ubiquinol (QH2) by PfDHODH occurs during pyrimidine synthesis. The ETC and pyrimidine biosynthesis pathway are sensitive to cytochrome bc1 complex inhibitors, such as atovaquone. Expression of the yDHODH bypasses the ETC and thus rescues pyrimidine synthesis in the presence of cytochrome bc1 inhibitors. Inhibition of a proguanil-sensitive pathway, which contributes to mitochondrial membrane potential, restores sensitivity to cytochrome bc1 inhibitors (adapted from reference26 with permission of the publisher). (B and C) Representative P. falciparum growth curves obtained for TCMDC-135546 (B) and TCMDC-132690 (C). The dose-response curves of wild-type P. falciparum Dd2 (blue), transgenic P. falciparum Dd2-yDHODH (red), P. falciparum Dd2 with 1 μM proguanil (brown), and P. falciparum Dd2-yDHODH with 1 μM proguanil (green) are shown. Addition of proguanil restored sensitivity to cytochrome bc1 complex inhibitors, such as TCMDC-135546, but not to compounds with an unknown mode of action, like TCMDC-132690. The error bars indicate standard deviations.
TABLE 1.
Growth inhibition of parental and transgenic P. falciparum Dd2 parasite strains
| Compound | Chemical cluster no. | IC50 growtha (μM) |
Fold resistance | ||
|---|---|---|---|---|---|
| Dd2-attB | Dd2-scDHODH | Dd2-scDHODH + PG | |||
| TCMDC-123667 | 87 | 0.077 | 7.9 | 0.037 | 102 |
| TCMDC-125258 | 87 | 0.25 | 1.0 | 0.075 | 4 |
| TCMDC-125465 | 87 | 0.14 | 0.69 | 0.057 | 5 |
| TCMDC-135461 | 87 | 0.015 | 1.4 | 0.0040 | 94 |
| TCMDC-135546 | 87 | 0.022 | 0.97 | 0.0060 | 44 |
| TCMDC-135678 | 87 | 0.20 | 4.7 | 0.031 | 23 |
| TCMDC-135795 | 87 | 0.11 | 1.1 | 0.041 | 10 |
| TCMDC-137383 | 87 | 0.12 | 0.77 | 0.049 | 6 |
| TCMDC-137384 | 87 | 0.12 | 4.1 | 0.025 | 35 |
| TCMDC-137395 | 87 | 0.017 | 0.96 | 0.0050 | 57 |
| TCMDC-137410 | 87 | 0.069 | 1.0 | 0.022 | 15 |
| TCMDC-123745 | 130 | 0.029 | 0.69 | 0.010 | 24 |
| TCMDC-137414 | 130 | 0.13 | 3.5 | 0.039 | 27 |
| TCMDC-135796 | 143 | 0.038 | 1.1 | 0.010 | 28 |
| TCMDC-136185 | 143 | 0.25 | 3.9 | 0.093 | 16 |
| TCMDC-136286 | 143 | 1.7 | 11 | 0.26 | 7 |
| TCMDC-136303 | 143 | 0.29 | 9.3 | 0.088 | 32 |
| TCMDC-138214 | 202 | 1.1 | 3.4 | 0.098 | 3 |
| TCMDC-137973 | 420 | 0.12 | 11 | 0.044 | 88 |
| TCMDC-139660 | 1,098 | 0.92 | 4.8 | 0.070 | 5 |
| TCMDC-135289 | 2,199 | 0.76 | 2.8 | 0.12 | 4 |
| TCMDC-136157 | 2,255 | 2.6 | 9.0 | 0.42 | 4 |
| TCMDC-137377 | 2,399 | 0.90 | 4.7 | 0.092 | 5 |
| TCMDC-138010 | 2,483 | 0.40 | 11 | 0.048 | 29 |
| TCMDC-123515 | 2,744 | 1.1 | 10 | 0.11 | 9 |
Reported IC50s for Dd2-attB and Dd2-scDHOD parasite strains are based on dose-response curves. PG, proguanil.
To further narrow down the mechanism of action of these compounds, we tested them for inhibition of the Plasmodium ETC or DHODH using cell-based assays. Compounds that target the ETC likely act on the cytochrome bc1 complex, a validated drug target that plays an essential role throughout the parasite's life cycle by regenerating ubiquinone, the cofactor of DHODH (Fig. 3A). To specifically address the targeting of the mitochondrial ETC via inhibition of the cytochrome bc1 complex, we assessed the susceptibility of P. falciparum Dd2-yDHODH transgenic parasites to screening actives with and without cotreatment with proguanil, a dihydrofolate reductase inhibitor. It was previously shown that proguanil restores the sensitivity of transgenic parasites, P. falciparum Dd2-yDHODH, to cytochrome bc1 complex inhibitors (24, 26), thereby providing a method to identify compounds that target this enzyme complex (Fig. 3B). However, compounds for which the parasite's susceptibility is not restored by proguanil are designated compounds having an as yet unknown mode of action (Fig. 3C). It is likely that these compounds target the parasite's DHODH, but this prediction awaits verification with enzymatic assays. Therefore, proguanil (1 μM) was coadministered with screening actives to P. falciparum Dd2-yDHODH cultures to generate dose-response curves of parasite inhibition. The supplementation of transgenic P. falciparum Dd2-yDHODH cultures with proguanil did not alter the EC50s of screening actives that retained potency in the Dd2-yDHODH line, such as non-cytochrome bc1 complex inhibitors, like TCMDC-132690 (Fig. 3C). However, we found one compound, TCMDC-124971, that exhibited a >10-fold increase in potency in the Dd2-yDHODH line compared to the parental strain, P. falciparum Dd2-attB (see Table S1 in the supplemental material). A similar case of increased sensitivity in the Dd2-yDHODH line was previously observed with the antimalarial mefloquine (27), whose mode of action is still poorly understood. This observation of increased sensitivity for both mefloquine and TCMDC-124971 suggests they may have multiple targets in the transgenic line. Importantly, we observed that proguanil was able to restore the susceptibility of P. falciparum Dd2-yDHODH to 11 chemical clusters and singletons, including TCMDC-135546, so that their observed EC50s were similar to those of the parental strain, P. falciparum Dd2-attB (Table 1; see Fig. S1 in the supplemental material). Taken together, these results indicate that 30% of the screening actives function by targeting the cytochrome bc1 complex (Fig. 4).
FIG 4.
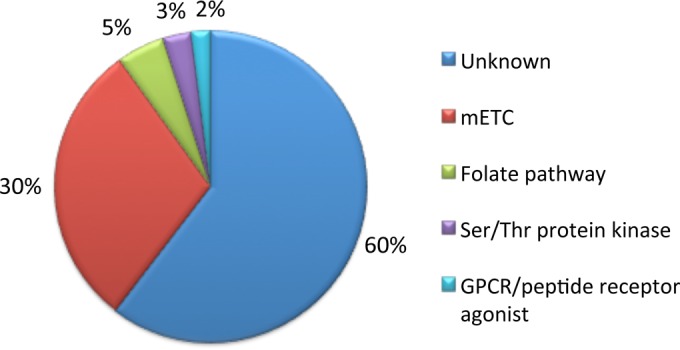
Distribution of mechanisms of action. The relative distribution of target classes for the characterization of active compounds is indicated as percentages. The majority of dual-stage malaria inhibitors identified (60%) have unknown modes of action. mETC, mitochondrial electron transport chain; GPCR, G-protein-coupled receptor.
Next, we investigated the abilities of these compounds to modulate the folate biosynthesis pathway. This pathway is critical to parasite survival, as it contributes to important intermediates that are necessary for nucleotide synthesis (reviewed in reference 28). Common antimalarial agents, such as pyrimethamine and proguanil, are known to inhibit the folate biosynthesis pathway. To identify compounds inhibiting the folate biosynthesis pathway, we utilized the P. falciparum clone 3D7A. This strain is adapted to grow under depleted pABA and FA conditions and thus relies entirely on de novo synthesis of folate metabolites. Therefore, compounds that inhibit the folate biosynthesis pathway exhibit a rightward shift in potency of parasite inhibition (i.e., are less potent) when the culture medium is supplemented with FA (resistance value, >1 fold). As shown in Table 2, we identified two chemical clusters and one singleton (5% of screening actives [Fig. 4]) with decreased potency against parasites in FA-supplemented medium compared to reduced medium (pABA and FA deficient), suggesting a mode of action via inhibition of the folate biosynthesis pathway. The remaining compounds had no significant effects on this pathway (see Fig. S1 and Table S1 in the supplemental material).
TABLE 2.
Growth inhibition of P. falciparum clone 3D7A cultured in complete versus pABA- and FA-depleted medium
| Compound | Chemical cluster no. | IC50 growtha (μM) |
Fold resistance | |
|---|---|---|---|---|
| Complete medium | pABA- and FA-depleted medium | |||
| TCMDC-137381 | 17 | 0.006 | 0.001 | 6 |
| TCMDC-137930 | 17 | 0.14 | 0.017 | 8 |
| TCMDC-124874 | 112 | 0.0055 | 0.0022 | 3 |
| TCMDC-124964 | 112 | 0.003 | 0.0004 | 8 |
| TCMDC-135244 | 1,045 | 1.6 | 0.54 | 3 |
Reported IC50s for clone 3D7A parasites are based on dose-response curves. pABA, para-aminobenzoic acid; FA, folinic acid.
DISCUSSION
Important drug discovery campaigns within the past decade have provided numerous active compounds as starting points for the development of novel antimalarial drugs. In an effort to surmount the increasing emergence of resistance to current drug therapies, the pursuit of new small molecules with novel mechanisms of action in killing Plasmodium parasites is spearheading current efforts for drug discovery. Identifying compounds with new modes of action is also highly desirable for chemical probe development to advance the current understanding of biological processes in Plasmodium parasites. To address this need, a high-throughput liver-stage malaria parasite assay (20) was used to evaluate liver-stage activities of compounds comprising the TCAMS library, a malaria blood-stage parasite inhibitor collection of ∼14,000 compounds. Here, we describe the identification of ∼100 dual-stage inhibitors (0.8% hit rate) with nanomolar to low-micromolar potencies for inhibition of liver-stage malaria parasites. Structural analyses and functional studies were completed to probe the modes of action of these screening actives. While some of the screening actives were found to act through previously identified malarial targets (ETC and the folate biosynthesis pathway), the majority (60%) have unknown mechanisms of action for inhibition of Plasmodium parasite growth.
The observed hit rate of 0.8% is relatively low, considering that all the library members potently killed blood-stage P. falciparum parasites, and this is likely due to many different factors. First, and perhaps most likely, there was a stringent assignment of “active compounds,” which were selected by a >80% reduction of liver-stage parasites at 120 nM. This was also supported by the observation that many compounds had great potency against liver-stage parasites compared to blood-stage parasites (discussed below). Additionally, compounds in liver-stage assays that are susceptible to metabolism by cytochrome P450 are degraded by HepG2 cells, but not by red blood cells. This is likely illustrated by the absence of triazolopyrimidine PfDHODH inhibitors as screening hits. These compounds are present in the TCAMS set, but they are prone to metabolism (29). These facts suggest that the screening hits we identified are not only the most potent, but also metabolically stable. Finally, it is possible that the different Plasmodium species, P. berghei versus P. falciparum, exhibit differential inhibition by the screening compounds. However, since compounds that target essential conserved parasite processes are desired, our screening approach has the added benefit of enriching for compounds with broad activity against multiple Plasmodium parasite species.
The identification of compounds acting through novel modes of action to kill Plasmodium parasites is desirable for chemical probe development. Thus, molecules that target processes distinct from those targeted by current antimalarial agents, such as atovaquone and pyrimethamine, are of interest. Currently, many drugs target metabolic pathways, like ETC, pyrimidine, and folate biosynthesis, which are essential to parasite survival. With the global use of these drugs, many parasite strains have been found that exhibit resistance. Specifically, point mutations in the cytochrome bc1 complex, dihydrofolate reductase, and dihydropteroate synthetase are responsible for parasite drug resistance in clinical isolates (reviewed in reference 6). The cytochrome bc1 complex couples the ETC and the pyrimidine biosynthesis pathway through PfDHODH (Fig. 3A), while dihydrofolate reductase and dihydropteroate synthetase are key enzymes of folate synthesis. Thus, targeting these processes is less attractive for drug development. In this study, we found that 30% of liver-stage malaria parasite active compounds from the TCAMS library exhibit activity against the ETC, whereas only 5% exhibit antifolate activities. Inhibitors of the cytochrome bc1 complex (30–32) and dihydrofolate reductase (30), like atovaquone and pyrimethamine, respectively, have proven activity against liver-stage parasites. Interestingly, we observed that most of these screening actives are more potent in liver- versus blood-stage malaria parasite assays. This selectivity toward liver-stage parasites was not the result of increased cell toxicity, as HepG2 cell viability was unaffected at test concentrations (see Table S1 in the supplemental material). Therefore, this increase in compound potency against liver-stage parasites may be attributed to the greater importance of these metabolic pathways in hepatic-cell infection. During the liver stage, the parasites have a significantly greater proliferation rate (10,000- to 30,000-fold) than in the blood stage (10- to 20-fold) (33). This increased rate likely requires additional energy and nucleotides, making this parasite form particularly susceptible to modulators that affect these pathways.
As noted above, 5% of screening actives were found to target the folate biosynthesis pathway. Interestingly, we observed that compounds from different chemical clusters (17 and 112) that were identified as antifolates exhibited decreased activity against the multidrug-resistant strain P. falciparum Dd2 (IC50 > 2 μM) (see Table S1 in the supplemental material). This cross-resistance against the Dd2 strain suggests an essential role of the folate biosynthesis pathway during the Plasmodium parasite's liver stage. Specifically, this provides evidence that Plasmodium parasites rely on salvaging host pABA and FA to support folate biosynthesis during this metabolically demanding liver stage. While targeting the Plasmodium folate biosynthesis pathway remains an effective antimalarial drug strategy for treatment options (i.e., sulfadoxine-pyrimethamine combination), modulation of the parasite folate salvage mechanisms during the liver stage could offer novel avenues for developing prophylactic therapies. However, the parasite's dependence on salvaged dietary folate in humans needs to be further studied to assess folate salvage during liver-stage infection as a viable drug target.
Determining the molecular target(s) of active compounds, and thus their mechanism(s) of action, remains a challenge for phenotype-based screens. Functional analyses of the screening actives with various parasite strains suggest that the ETC and folate biosynthesis pathway are targeted by 30% and 5% of compounds, respectively. Recent studies suggest that the mechanisms of action of active compounds can also be investigated through the use of large amounts of screening data via in silico activity profiling (17, 34). Here, we have shown through structural analysis that 3% of the screening actives likely target serine/threonine kinases and 2% likely target G protein-coupled receptors. In support of this chemical informatics approach, we observed that similar chemical scaffolds share modes of action in inhibiting Plasmodium growth based on the reported functional assays. However, these assays do not address the possibility of multiple targets.
It is possible that more than one molecular target is involved in the efficiency of screening actives in inhibiting both liver- and blood-stage malaria parasites. Additionally, 60% of the active compounds identified have unknown mechanisms of action, which could help uncover novel pathways essential in Plasmodium parasite development. Finally, our study reveals that out of 103 active compounds, 48 compounds have unique scaffolds, which provides an extensive chemical space for structure-activity optimization to improve potency. These predictions warrant further validation through experimental analyses.
In conclusion, this study shows that investigating the liver-stage malaria parasite activity of known blood-stage malaria inhibitors can identify compounds with potentially novel modes of action across different malaria parasite life stages. While a comprehensive assessment of potential targets through additional experimentation is necessary to determine the precise molecular target(s) involved, our parallel-screening strategy allowed the rapid identification of new small molecules with potentially novel modes of action. We hope the activities of the compounds described in this study will contribute to development of new molecular probes that may advance the current understanding of malaria parasite biology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ralph Mazitschek, Nathanael Gray, and R. Kiplin Guy for useful discussions on screening actives and Maria Mota for expert advice. We also thank Andreia Magalhães and Zachary Margolin for technical assistance and the ICCB-L facility for critical input. The following reagents were obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: MRA-151 for P. falciparum 3D7A, deposited by D. Walliker, and MRA-843 for P. falciparum Dd2-attB, deposited by D. A. Fidock.
We declare that we have no competing interests.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02110-15.
REFERENCES
- 1.WHO. 2014. World malaria report 2014. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Flannery EL, Chatterjee AK, Winzeler EA. 2013. Antimalarial drug discovery: approaches and progress towards new medicines. Nat Rev Microbiol 11:849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ejigiri I, Sinnis P. 2009. Plasmodium sporozoite-host interactions from the dermis to the hepatocyte. Curr Opin Microbiol 12:401–407. doi: 10.1016/j.mib.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ploemen IH, Prudencio M, Douradinha BG, Ramesar J, Fonager J, van Gemert GJ, Luty AJ, Hermsen CC, Sauerwein RW, Baptista FG, Mota MM, Waters AP, Que I, Lowik CW, Khan SM, Janse CJ, Franke-Fayard BM. 2009. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS One 4:e7881. doi: 10.1371/journal.pone.0007881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows JN, Burlot E, Campo B, Cherbuin S, Jeanneret S, Leroy D, Spangenberg T, Waterson D, Wells TN, Willis P. 2014. Antimalarial drug discovery: the path towards eradication. Parasitology 141:128–139. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gamo FJ. 2014. Antimalarial drug resistance: new treatments options for Plasmodium. Drug Discov Today Technol 11:81–88. doi: 10.1016/j.ddtec.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Lim L, McFadden GI. 2010. The evolution, metabolism and functions of the apicoplast. Philos Trans R Soc Lond B Biol Sci 365:749–763. doi: 10.1098/rstb.2009.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coronado LM, Nadovich CT, Spadafora C. 2014. Malarial hemozoin: from target to tool. Biochim Biophys Acta 1840:2032–2041. doi: 10.1016/j.bbagen.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baum J, Papenfuss AT, Mair GR, Janse CJ, Vlachou D, Waters AP, Cowman AF, Crabb BS, de Koning-Ward TF. 2009. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res 37:3788–3798. doi: 10.1093/nar/gkp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. 2010. Spiroindolones, a potent compound class for the treatment of malaria. Science 329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoepfner D, McNamara CW, Lim CS, Studer C, Riedl R, Aust T, McCormack SL, Plouffe DM, Meister S, Schuierer S, Plikat U, Hartmann N, Staedtler F, Cotesta S, Schmitt EK, Petersen F, Supek F, Glynne RJ, Tallarico JA, Porter JA, Fishman MC, Bodenreider C, Diagana TT, Movva NR, Winzeler EA. 2012. Selective and specific inhibition of the plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 11:654–663. doi: 10.1016/j.chom.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, Kim YJ, Lee HK, Cortese JF, Wirth DF, Dignam JD, Rao A, Yeo CY, Mazitschek R, Whitman M. 2012. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol 8:311–317. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Martinez MS, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, Sinden RE, Winzeler SA, Charman EA, Bebrevska L, Gray DW, Campbell S, Fairlamb AH, Willis PA, Rayner JC, Fidock DA, Read KD, Gilbert IH. 2015. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El Mazouni F, Lao Y, Kokkonda S, Tomchick DR, Deng X, Laird T, Bhatia SN, March S, Ng CL, Fidock DA, Wittlin S, Lafuente-Monasterio M, Benito FJ, Alonso LM, Martinez MS, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Haselden JN, Louttit J, Cui Y, Sridhar A, Zeeman AM, Kocken C, Sauerwein R, Dechering K, Avery VM, Duffy S, Delves M, Sinden R, Ruecker A, Wickham KS, Rochford R, Gahagen J, Iyer L, Riccio E, Mirsalis J, Bathhurst I, Rueckle T, Ding X, Campo B, Leroy D, Rogers MJ, Rathod PK, Burrows JN, Charman SA. 2015. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med 7:296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Nagle A, Simon O, Yeung BK, Chatterjee AK, McCormack SL, Manary MJ, Zeeman AM, Dechering KJ, Kumar TR, Henrich PP, Gagaring K, Ibanez M, Kato N, Kuhen KL, Fischli C, Rottmann M, Plouffe DM, Bursulaya B, Meister S, Rameh L, Trappe J, Haasen D, Timmerman M, Sauerwein RW, Suwanarusk R, Russell B, Renia L, Nosten F, Tully DC, Kocken CH, Glynne RJ, Bodenreider C, Fidock DA, Diagana TT, Winzeler EA. 2013. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 17.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. 2008. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A 105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, El Mazouni F, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, Derisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derbyshire ER, Mazitschek R, Clardy J. 2012. Characterization of Plasmodium liver stage inhibition by halofuginone. Chem Med Chem 7:844–849. doi: 10.1002/cmdc.201200045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derbyshire ER, Prudencio M, Mota MM, Clardy J. 2012. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci U S A 109:8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bemis GW, Murcko MA. 1996. The properties of known drugs. 1. Molecular frameworks. J Med Chem 39:2887–2893. [DOI] [PubMed] [Google Scholar]
- 22.Stahl M, Mauser H. 2005. Database clustering with a combination of fingerprint and maximum common substructure methods. J Chem Inf Model 45:542–548. doi: 10.1021/ci050011h. [DOI] [PubMed] [Google Scholar]
- 23.Dong CK, Urgaonkar S, Cortese JF, Gamo FJ, Garcia-Bustos JF, Lafuente MJ, Patel V, Ross L, Coleman BI, Derbyshire ER, Clish CB, Serrano AE, Cromwell M, Barker RH Jr, Dvorin JD, Duraisingh MT, Wirth DF, Clardy J, Mazitschek R. 2011. Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem Biol 18:1602–1610. doi: 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. 2007. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 25.Phillips MA, Rathod PK. 2010. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets 10:226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Cruz FP, Martin C, Buchholz K, Lafuente-Monasterio MJ, Rodrigues T, Sonnichsen B, Moreira R, Gamo FJ, Marti M, Mota MM, Hannus M, Prudencio M. 2012. Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J Infect Dis 205:1278–1286. doi: 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nam TG, McNamara CW, Bopp S, Dharia NV, Meister S, Bonamy GM, Plouffe DM, Kato N, McCormack S, Bursulaya B, Ke H, Vaidya AB, Schultz PG, Winzeler EA. 2011. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem Biol 6:1214–1222. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller IB, Hyde JE. 2013. Folate metabolism in human malaria parasites—75 years on. Mol Biochem Parasitol 188:63–77. doi: 10.1016/j.molbiopara.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. 2011. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem 54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, Sinden RE, Leroy D. 2012. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med 9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, Morrisey JM, Steuten J, Mutka T, Li Y, Wirjanata G, Ryan E, Duffy S, Kelly JX, Sebayang BF, Zeeman AM, Noviyanti R, Sinden RE, Kocken CH, Price RN, Avery VM, Angulo-Barturen I, Jimenez-Diaz MB, Ferrer S, Herreros E, Sanz LM, Gamo FJ, Bathurst I, Burrows JN, Siegl P, Guy RK, Winter RW, Vaidya AB, Charman SA, Kyle DE, Manetsch R, Riscoe MK. 2013. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 5:177ra37. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stickles AM, Ting LM, Morrisey JM, Li Y, Mather MW, Meermeier E, Pershing AM, Forquer IP, Miley GP, Pou S, Winter RW, Hinrichs DJ, Kelly JX, Kim K, Vaidya AB, Riscoe MK, Nilsen A. 2015. Inhibition of cytochrome bc1 as a strategy for single-dose, multi-stage antimalarial therapy. Am J Trop Med Hyg 92:1195–1201. doi: 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bejon P, Andrews L, Andersen RF, Dunachie S, Webster D, Walther M, Gilbert SC, Peto T, Hill AV. 2005. Calculation of liver-to-blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial quantitative polymerase chain reaction studies of volunteers challenged with malaria sporozoites. J Infect Dis 191:619–626. doi: 10.1086/427243. [DOI] [PubMed] [Google Scholar]
- 34.London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermancic P, Bonnet R, Shoichet BK, Taunton J. 2014. Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Biol 10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.