

Abstract
Cryptococcus gattii isolates from the Pacific Northwest have exhibited higher fluconazole MICs than isolates from other sites. The mechanism of fluconazole resistance in C. gattii is unknown. We sought to determine the role of the efflux pumps Mdr1 and Pdr11 in fluconazole susceptibility. Using biolistic transformation of the parent isolate, we created a strain lacking Mdr1 (mdr1Δ) and another strain lacking Pdr11 (pdr11Δ). Phenotypic virulence factors were assessed by standard methods (capsule size, melanin production, growth at 30 and 37°C). Survival was assessed in an intranasal murine model of cryptococcosis. Antifungal MICs were determined by the M27-A3 methodology. No differences in key virulence phenotypic components were identified. Fluconazole susceptibility was unchanged in the Mdr1 knockout or reconstituted isolates. However, fluconazole MICs decreased from 32 μg/ml for the wild-type isolate to <0.03 μg/ml for the pdr11Δ strain and reverted to 32 μg/ml for the reconstituted strain. In murine models, no difference in virulence was observed between wild-type, knockout, or reconstituted isolates. We conclude that Pdr11 plays an essential role in fluconazole susceptibility in C. gattii. Genomic and expression differences between resistant and susceptible C. gattii clinical isolates should be assessed further in order to identify other potential mechanisms of resistance.
INTRODUCTION
Cryptococcosis is an invasive fungal infection most commonly caused by one of two species of encapsulated yeasts: Cryptococcus neoformans and Cryptococcus gattii. C. neoformans has a worldwide distribution, while C. gattii has traditionally been associated with tropical and subtropical regions (1–3). The recent identification of C. gattii in the temperate climate of the Pacific Northwest (4) has caused investigators to question the true distribution of this emerging pathogen, and additional cases have since been reported throughout the United States, the Mediterranean region, and northern Europe (5–8).
The Infectious Diseases Society of America (IDSA) treatment guidelines outline similar treatment principles for C. gattii infection and C. neoformans infection, although they do confirm the need for additional investigation into the role of drug resistance in infections caused by C. gattii (9). Elevated fluconazole MICs are most commonly observed for C. gattii isolates from the Pacific Northwest, particularly those of the VGII molecular type (261/449 [58%] VGII isolates exhibited fluconazole MICs of ≥8 μg/ml) (10–13), with VGIIc subtype isolates exhibiting the highest fluconazole MICs (for 41/42 VGIIc isolates, fluconazole MICs were ≥8 μg/ml) (10).
Past studies examining the molecular mechanisms behind triazole resistance in C. neoformans have demonstrated that resistance most commonly occurs due to a modification in the quality or quantity of the target enzyme, reduced access of the drug to the target, or some combination of these mechanisms (10). In the first instance, point mutations in the gene (ERG11) encoding the target enzyme, 14-α-demethylase, lead to an altered target with decreased affinity for azoles (14–16). In the second, overexpression of ERG11 or a change in the ERG11 copy number results in the production of high concentrations of the target enzyme, creating the need for higher intracellular azole concentrations to effectively inhibit the target enzyme present in the cell (17). The last major mechanism described for cryptococcal isolates involves active efflux of azole antifungal agents from the cell through the action of multidrug efflux transporters, and there is evidence that these mechanisms may act individually or in concert (18).
The ABC transporters are a large class of drug efflux pumps that use ATP hydrolysis to translocate compounds across cell membranes. Mdr1 is a drug efflux pump that belongs to the multidrug resistance (MDR) subclass of ABC transporters (19). Overexpression of Mdr1 promotes azole resistance in C. neoformans. A second subclass of ABC transporters is the pleiotropic drug resistance (PDR) family, which includes the Afr1 protein. The Afr1 drug efflux transporter has been linked to azole resistance and heteroresistance in C. neoformans. The C. gattii orthologs of C. neoformans MDR1 (CnMDR1) and CnAFR1 have been identified as C. gattii MDR1 (CgMDR1) and CgPDR11. Whether Mdr1 or Pdr11, or both, are the mechanisms responsible for the fluconazole resistance of C. gattii is not known.
To elucidate the mechanisms responsible for decreased fluconazole susceptibility in C. gattii, we performed a series of experiments focused on the mechanisms most likely to be responsible for resistance and constructed mutants lacking drug efflux pumps that are likely to play a role in altered triazole susceptibility.
MATERIALS AND METHODS
Isolates, growth media, and susceptibility testing.
Deletion strains were generated from isolate GRT78, a VGII clinical isolate obtained from a patient seen at the UC Davis Medical Center. This patient was a 76-year-old male with no underlying medical problems who presented with a right upper lobe infiltrate that was found on bronchoscopy to be C. gattii. He was treated with 200 mg voriconazole every 12 h for 6 months. He remains symptom free 3 years after the cessation of antifungal therapy.
Prior work on C. gattii has focused primarily on the outbreak strains R265 (VGIIa) and R272 (VGIIb); however, susceptibility testing using the M27-A3 broth microdilution reference method (20) revealed fluconazole MICs of 8 and 4 μg/ml, respectively, for these isolates (21), while GRT78 yielded a fluconazole MIC of 32 μg/ml on repeated testing and thus was our choice for use in this assessment of molecular mechanisms responsible for the elevated fluconazole MICs for some C. gattii VGII strains.
The media used during this study included yeast extract-peptone-dextrose (YPD, comprising 1% yeast extract, 2% peptone, and 2% glucose) liquid medium and agar (2%) plates. YPD medium containing 200 μg/ml hygromycin or 100 μg/ml nourseothricin was used to select transformants.
In vitro susceptibility to fluconazole was also determined by Etest (bioMérieux) according to the manufacturer's instructions and was used in the screening and evaluation of mutants constructed during the conduct of this study. Isolates were subcultured at least twice on Sabouraud dextrose agar (Becton Dickinson, Sparks, MD) prior to in vitro susceptibility testing. The Etest MICs were read as the points at which a prominent reduction in growth intersected the Etest strip, as described previously (40). All isolates used for analysis then underwent broth microdilution susceptibility testing for MIC confirmation using the standard M27-A3 methodology as mentioned above. For posaconazole, voriconazole, itraconazole, fluconazole, and flucytosine, the MIC was defined as a 50% reduction in turbidity from that for growth controls and was read at 72 h. For amphotericin B, the MIC was defined as 100% inhibition relative to growth controls. Briefly, minimum fungicidal concentrations (MFCs) were determined by subculturing 20-μl aliquots from each well that showed complete inhibition after MIC determination (optically clear well) onto Sabouraud dextrose agar. The plates were incubated at 35°C, and the MFCs, defined as the lowest drug concentrations that resulted in no growth, were read. Candida krusei ATCC 6258 and Candida parapsilosis ATCC 22019 were used as quality controls.
Identification of C. gattii MDR1 and PDR11 orthologs.
The C. gattii orthologs of the genes encoding C. neoformans Mdr1 and Afr1 were identified by reciprocal BLAST searches for similarities between these two cryptococcal species. The BLAST “hit” orthologs in C. gattii were identified as MDR1 (locus tag CGB_A9140C; sequence identification [ID] XP_003191799.1) and PDR11 (locus tag CGB_N1170W; sequence ID XP_003197471.1), respectively. MDR1 in C. gattii shares 93% identity at the amino acid level with its ortholog in C. neoformans, while PDR11 in C. gattii shares 96% identity at the amino acid level with AFR1 in C. neoformans.
Disruption of the MDR1 and PDR11 genes. (i) MDR1 disruption.
The mdr1Δ mutant was generated by overlap PCR as described previously (22). A 5,600-bp fragment of the wild-type MDR1 gene was amplified by PCR in a 50-μl master mix containing 0.25 μl of TaKaRa HS DNA polymerase, 5 μl of 10× HS Ex Taq buffer, 4 μl of a 2.5 mM deoxynucleoside triphosphate (dNTP) mixture, 1 μl of each primer (gMDR1_F57 and gMDR1_R5637), 1 μl of 50-ng/μl genomic DNA, and 38.75 μl of nuclease-free water (see Table S1 in the supplemental material). Immediately following PCR, 5 μl of the reaction mixture was aliquoted and was confirmed by agarose electrophoresis, while 0.2 μl of Qiagen Taq polymerase (nonproofreading) and 0.5 μl of a dNTP mixture were added to the remaining reaction mixture, which was then incubated at 72°C for 10 min. The fragment was cloned into plasmid pCR2.1 by following the manufacturer's instructions for the TOPO TA cloning kit (Life Technologies), generating the pCR2.1::MDR1 construct. More than 50 white colonies were screened via PCR using the same primers as those used to amplify the genes and M13 primers. Plasmids were extracted from the transformants via the QIAprep Spin Miniprep kit and were submitted to the UC Davis core sequencing facility with M13 primers. The sequences were analyzed using Geneious software (version 7.7.1), and the identity of the cloned fragment was confirmed.
The MDR1 disruption construct (pCR2.1::mdr1Δ::HYG) was generated by replacing the middle region of the MDR1 gene in the pCR2.1::MDR1 construct with the selectable marker HYG. To ligate the two fragments, two unique restriction sites (AvrII and PacI) were introduced into the primers. The fragment from the pCR2.1::MDR1 construct was amplified with primers MDR_topo PacI F5098 and MDR_topo AvrII R1323, and the HYG fragment was amplified with primers pJAF15&1 AvrII 001F and pJAF15&1 PacI 001R, as described above (see Table S1 in the supplemental material). The two PCR fragments were purified using a QIAquick purification kit (Qiagen), digested with the DpnI, AvrII, and PacI restriction enzymes, and ligated via T4 DNA ligase according to the manufacturer's instructions (New England BioLabs). This generated the pCR2.1::mdr1Δ::HYG construct, which was then transformed into chemically competent Escherichia coli TOP10 cells (from the TOPO TA cloning kit). The transformants were confirmed by PCR and restriction digestion. The A14 transformant was selected as the MDR1 disruption construct (pCR2.1::mdr1Δ::HYG), propagated, and biolistically transformed into 3- to 4-day-old cells of the C. gattii parental strain GRT78. The mutants were confirmed by PCR and reverse transcriptase PCR (RT-PCR).
(ii) PDR11 disruption.
The PDR11 disruption construct pCR2.1::pdr11Δ::HYG was generated by following the same protocol as that for the mdr1Δ strain discussed above. A 5,622-bp fragment of the wild-type PDR11 gene was amplified from the parental strain GRT78 with primers cgPDR11_F2192 and cgPDR11_R7814 (see Table S1 in the supplemental material) and was cloned into pCR2.1. To replace the middle region of the PDR11 gene in the pCR2.1::PDR11 construct, a 5,116-bp fragment was amplified with primers CgPDR11_PacI F5166 and CgPDR11_AvrII R2584, digested with DpnI, AvrII, and PacI, and ligated with the same HYG fragment that was used for mdr1Δ mutants. This generated the PDR11 disruption construct (pCR2.1::pdr11Δ::HYG), which was then transformed into chemically competent E. coli TOP10 cells. Once the construct was confirmed, 2.5 μg to 5 μg of the purified construct was biolistically transformed into the parental strain GRT78. pdr11Δ mutants were confirmed by PCR and RT-PCR.
Reconstitution of MDR1 and PDR11 genes.
The nourseothricin selectable marker (NAT) from plasmid pCH233 (23) was used to reconstitute the mutant strains with the wild-type MDR1 and PDR11 genes. The entire wild-type MDR1 gene (7,207-bp fragment), including the promoter and terminator, was amplified from the parental strain GRT78 with primers gMDR_F912 and gMDR_R8119 by following the same PCR protocol as that described above (see Table S1 in the supplemental material), confirmed by agarose electrophoresis, and purified. A 2.5-μg portion of the purified fragment and 2.5 μg of plasmid pCH233 (NAT selectable marker) were cobiolistically transformed into the knockout strains. Transformants were selected on YPD with 100 μg/ml of NAT.
In order to create the PDR11 reconstituted strain, the PDR11 gene (7,097-bp fragment), including the promoter and terminator, was PCR amplified from the parental strain GRT78 with primers cgPDR11_F1011 and cgPDR11_R8108. Reconstituted strains were confirmed by PCR and Etest.
RNA extraction and RT PCR.
RNA was extracted using the MasterPure yeast RNA purification kit (Epicentre). The quality of the RNA was confirmed via denatured agarose formaldehyde electrophoresis. cDNA was synthesized from RNA using the SuperScript III First-Strand Synthesis system (Invitrogen), followed by PCR using the same primers used to clone the genes into pCR2.1.
Phenotypic testing.
Capsule formation, melanin production, growth at 30°C and 37°C, and resistance to H2O2 were evaluated using previously described protocols (21, 24, 25). The development of heteroresistance was assessed by taking subclones from each strain and using stepwise exposure to YPD plates containing various concentrations of fluconazole (0.5 to 512 μg/ml). Growth was read after 72 h of incubation at 30°C. In order to obtain highly resistant subclones, the clones that grew on different concentrations of fluconazole were isolated and plated on YPD agar with stepwise (2-fold) increases in the fluconazole concentration (1 to 512 μg/ml). The culture plates of each passage were incubated at 30°C for 72 to 96 h as described previously (26).
Inhalational model.
Virulence was assessed using female A/JCr mice (15 to 20 g; Jackson Laboratory). Strains were cultured in YPD broth for 18 to 20 h at 30°C, harvested, washed three times with sterile phosphate-buffered saline (PBS), and counted using a hemocytometer to determine cell concentrations. Inocula for all experiments were confirmed by plating on YPD and counting CFU. Six mice per strain were infected by intranasal inhalation with 5 × 104 CFU in 50 μl PBS as described previously (27). During the conduct of all studies, animals that appeared moribund or in pain were euthanized. All experiments were in compliance with the ethical regulations established by the UC Davis Institutional Animal Care and Use Committee (IACUC).
Statistical analysis.
Mortality and time to mortality were evaluated for statistical significance by Kaplan-Meier survival analysis and the Mann-Whitney U test. P values were obtained from a log rank test. Tissue fungal burdens were assessed using analysis of variance with Tukey's posttest. A P value of <0.05 was considered statistically significant.
RESULTS
Generation of PDR11- and MDR1-deficient and reconstituted strains of C. gattii.
We generated a set of isogenic C. gattii strains from isolate GRT78, a VGII strain with a fluconazole MIC of 32 μg/ml. The PDR11 gene was disrupted as described above, and the resulting disruption allele, pdr11::HYG, was introduced into the wild-type strain GRT78. Of the 60 transformants produced, 4 with presumptive evidence of PDR11 disruption were evaluated by RT-PCR. These analyses confirmed the absence of PDR11 transcripts in one of these transformants, and this pdr11Δ mutant was selected for subsequent experiments. Similarly, the MDR1 gene was disrupted, and the resulting disruption allele, mdr1::HYG, was introduced into the initial GRT78 strain. Three mutants with presumptive evidence of MDR1 disruption were evaluated by RT-PCR, and one was chosen for inclusion in this study.
Deletion of PDR11 but not MDR1 has a significant effect on susceptibility to fluconazole.
Deletion mutations of both PDR11 and MDR1 were created. The susceptibilities of the corresponding knockout mutants to fluconazole, screened by Etest (Fig. 1), revealed a significant reduction in fluconazole MICs for the pdr11Δ mutants (<0.06 μg/ml) but no corresponding reduction in fluconazole MICs for the mdr1Δ isolates (≥32 μg/ml). Fluconazole MICs returned to 32 μg/ml, the same as that for the wild-type isolate, for the pdr11::PDR11 and mdr1::MDR1 reconstituted strains.
FIG 1.

MICs of fluconazole, obtained by Etest, for GRT78 strains. (A and D) Wild-type GRT78 strains with fluconazole MICs of 32 μg/ml. (B) mdr1Δ mutant strain (MIC, 32 μg/ml). (C) MDR1 reconstituted strain (mdr1::MDR1) (MIC, 32 μg/ml). (E) pdr11Δ mutant strain, for which the fluconazole MIC was significantly reduced, to 0.06 μg/ml. (F) PDR11 reconstituted strain (pdr11::PDR11), with reversion to wild-type fluconazole susceptibility (MIC, 32 μg/ml).
Additional susceptibility testing of GRT78 revealed the following MICs: for itraconazole and posaconazole, 2 μg/ml; for voriconazole, 1 μg/ml; and for amphotericin B and flucytosine, 0.125 μg/ml. For the pdr11Δ mutant, the itraconazole, posaconazole, and voriconazole MICs decreased to 0.5, 0.5, and 0.25 μg/ml, respectively, while the amphotericin B and flucytosine MICs were not significantly changed from those for the parental strain. Susceptibility testing of the mdr1Δ mutant and the reconstituted isolates yielded results identical to those for GRT78 (Table 1).
TABLE 1.
MICs and MFCsa for clones used during the conduct of this study
| Isolateb | MIC (MFC) (μg/ml) of: |
|||||
|---|---|---|---|---|---|---|
| Fluconazole | Itraconazole | Posaconazole | Voriconazole | Amphotericin B | Flucytosine | |
| GRT78 | 32 (>128) | 2 | 2 | 1 | 0.125 | 0.125 |
| pdr11Δ strain | 0.06 (4) | 0.5 | 0.5 | 0.25 | 0.125 | 0.125 |
| PDR1 strain | 32 (>128) | 2 | 2 | 1 | 0.125 | 0.125 |
| mdr1Δ strain | 32 (>128) | 2 | 2 | 1 | 0.125 | 0.125 |
| MDR1 strain | 32 (>128) | 2 | 2 | 1 | 0.125 | 0.25 |
MFCs, minimum fungicidal concentrations.
The MDR1 and PDR11 strains are reconstituted strains (mdr1::MDR1 and pdr11::PDR11, respectively).
Minimum fungicidal concentrations (MFCs) were also determined for each isolate in order to ascertain whether gene disruption and/or reconstitution played any role. Except for the pdr11Δ mutant (fluconazole MFC, 4 μg/ml), all mutants and reconstituted isolates exhibited fluconazole MFCs of ≥128. MDR1 thus had no effect on fluconazole MICs or MFCs (Table 1).
In vitro phenotypic analysis of PDR11 and MDR1 deletion and reconstituted strains.
There were no statistically significant differences in capsule formation, melanin production, growth at 30°C or 37°C, or resistance to H2O2 between any of the strains (21). All isolates examined in this study also underwent serial passage in a fluconazole-containing medium for assessment of heteroresistance. Reduced growth was not observed for GRT78 subclones until a fluconazole concentration of 256 μg/ml was reached, and growth was completely inhibited at 512 μg/ml. The pdr11Δ mutants had visibly fewer colonies at a fluconazole concentration of 2 μg/ml and were unable to grow at 4 μg/ml. The mdr1Δ mutant and both reconstituted isolates exhibited a level of fluconazole tolerance unchanged from that of the wild-type isolate (visibly fewer colonies at 256 μg/ml and complete inhibition of growth at 512 μg/ml).
Inhalational model (virulence and in vivo treatment effects).
The possible involvement of PDR11 and MDR1 in the virulence of C. gattii was assessed in an inhalational model of infection. As shown in Fig. 2, there were no significant differences in survival time between any of the strains through 60 days postinfection. Necropsies revealed disseminated infection to the brain, spine, kidneys, spleen, and liver and showed no significant differences between strains (P, >0.05 for all groups).
FIG 2.
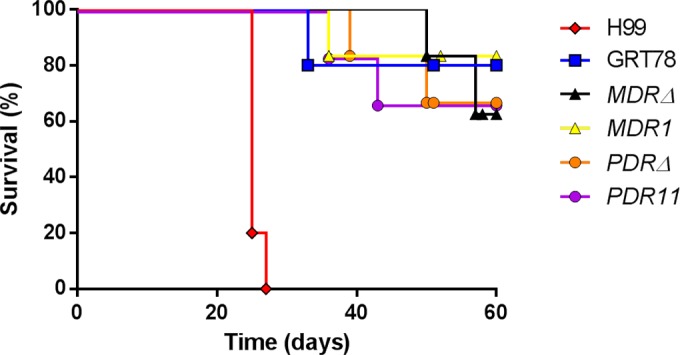
The wild-type strain (GRT78), the mdr1Δ and pdr11Δ mutant strains, and the mdr1::MDR1 and pdr11::PDR11 reconstituted strains do not differ significantly in a murine intranasal inhalational model of virulence. All C. gattii strains evaluated showed lower levels of virulence than C. neoformans strain H99 (P < 0.01).
DISCUSSION
The emergence of C. gattii as an important pathogen has resulted in numerous studies to define its epidemiology and clinical features, as well as the virulence components responsible for the lower treatment response rates than those observed with C. neoformans (21, 28–32). In some regions, C. gattii has been observed to exhibit reduced in vitro susceptibility to fluconazole and other triazole agents (33). Susceptibility differs between C. gattii molecular types, and several studies have shown that VGII isolates frequently have higher fluconazole MICs than other genotypes (11–13, 34, 35). Despite the potential clinical importance of reduced susceptibility, the molecular mechanism behind elevated MICs in selected VGII strains has not been established previously.
We have demonstrated that elevated fluconazole MICs for a clinical C. gattii VGII isolate are dependent on PDR11 expression. Deletion of this ABC transporter resulted in a fluconazole MIC markedly reduced (from 32 to 0.06 μg/ml) from that for the parent isolate, and the elevated fluconazole MIC was completely restored with the reconstitution of the PDR11 gene. In contrast, MDR1 appeared to play no role in in vitro or in vivo resistance to fluconazole in this isolate.
These findings suggest that PDR11 alone may be responsible for the triazole-resistant C. gattii isolates from the Pacific Northwest. Prior work has revealed that neither overexpression nor mutations within ERG11, the triazole drug target, could explain the marked elevation in fluconazole MICs associated with Pacific Northwest C. gattii isolates (36). Subsequent work by the same group used [3H]fluconazole to examine the role of drug efflux pumps in C. gattii and found an association between a decline in intracellular radiolabeled fluconazole and fluconazole MICs (37). Expression of C. gattii and C. neoformans drug efflux pumps in a Saccharomyces cerevisiae strain allowed for additional scrutiny of these pumps and confirmed that in a model system, they may increase triazole MICs. Our study adds to this work by confirming the essential role of PDR11 in a clinical C. gattii isolate with elevated triazole MICs. In contrast to the work by Basso et al. (37), we did not confirm a role for MDR1 in antifungal drug resistance.
The kinetics of C. gattii ABC transporters are also noteworthy. The Km values of these enzymes are similar for C. neoformans and C. gatiii; however, the Vmax values of both species exceed those of Cdr1p and Cdr2p in Candida albicans, and the Vmax values of C. gattii efflux pumps are substantially higher than those observed for C. neoformans (37). However, it remains unclear whether these species-specific differences are related to the quantity of protein expressed and the stability of the enzyme, or whether subtle sequence differences in these regions confer added affinity and thereby heightened efflux of antifungal agents. These efflux pumps also likely exhibit variable affinity and efficiency in the transport of different triazoles—differences that account for the divergent triazole MICs in our mutant and reconstituted strains.
The MICs of other triazoles (itraconazole, posaconazole, and voriconazole) were also elevated for our initial isolate. In results similar to those observed with fluconazole, deletion of PDR11, but not MDR1, played a significant role in altering fungal susceptibility to these mold-active triazoles as well to all agents experiencing at least a 2-dilution reduction in their MICs for the pdr11Δ mutant. The importance of elevated MICs of other extended-spectrum triazoles for C. gattii isolates should not be overlooked. The emergence of elevated fluconazole MICs for VGII strains has become well known, and some patients may be treated empirically with alternative triazoles based on epidemiologic studies demonstrating lower MICs of itraconazole, posaconazole, and voriconazole than of the more commonly used agent fluconazole (12). In view of this, we recommend susceptibility testing for all C. gattii isolates from areas known to harbor VGII isolates prior to, or concurrently with, the initiation of antifungal therapy.
Drug efflux transporters not only are important for resistance to antifungal agents and xenobiotics but also have been implicated in fungal pathogenesis and virulence (38). The PDR family is one of the largest families of ABC transporters in pathogenic fungi, and most ABC transporters have been demonstrated to have multiple substrates, including phospholipids, steroids, host-specific toxins, and host-derived compounds that would inhibit fungal growth in addition to providing resistance to antifungal agents. However, in our strains, no differences were found between the parental isolates, disruption mutants, and reconstituted strains in virulence studies. This is in contrast to the results of a previous study examining the role of AFR1 (PDR11 ortholog) in a murine model of C. neoformans infection (39). However, the study by Sanguinetti et al. used a fluconazole-susceptible C. neoformans isolate and a fluconazole-resistant isolate created by serial passage in a fluconazole-containing medium (39). This study occurred prior to the recognition of chromosomal aneuploidy as a major mechanism behind the stress response in cryptococcal species, and although the authors carefully attempted to assess the quantity of known virulence factors in different isolates by RT-PCR, aneuploidy likely played a large role in the virulence of the strains used in their study, as has been noted by others (26).
In conclusion, we present evidence that the efflux pump gene PDR11 plays a critical role in the antifungal resistance of a VGII clinical strain. These results demonstrate, for the first time, that antifungal resistance in VGII isolates may be entirely dependent on PDR11 alone. Future work should focus on a quantitative assessment of efflux pump expression in C. gattii isolates of different molecular types obtained from different locations and with different triazole susceptibility patterns. With the declining costs of whole-genome and transcriptome-sequencing (RNA-seq) analysis, we are hopeful that potential differences in the gene sequence, expression, and drug affinity of efflux pumps from different C. gattii molecular types will soon be determined.
Supplementary Material
ACKNOWLEDGMENTS
Existing department funds were used for this work.
We are grateful to Joe Heitman (Duke University) for kindly providing the hygromycin plasmid.
Funding Statement
Existing department funds were used for this work.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01777-15.
REFERENCES
- 1.McMullan BJ, Sorrell TC, Chen SC. 2013. Cryptococcus gattii infections: contemporary aspects of epidemiology, clinical manifestations and management of infection. Future Microbiol 8:1613–1631. doi: 10.2217/fmb.13.123. [DOI] [PubMed] [Google Scholar]
- 2.Springer DJ, Chaturvedi V. 2010. Projecting global occurrence of Cryptococcus gattii. Emerg Infect Dis 16:14–20. doi: 10.3201/eid1601.090369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson GR III, Wiederhold NP, Najvar LK, Bocanegra R, Kirkpatrick WR, Graybill JR, Patterson TF. 2012. A murine model of Cryptococcus gattii meningoencephalitis. J Antimicrob Chemother 67:1432–1438. doi: 10.1093/jac/dks060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacDougall L, Kidd SE, Galanis E, Mak S, Leslie MJ, Cieslak PR, Kronstad JW, Morshed MG, Bartlett KH. 2007. Spread of Cryptococcus gattii in British Columbia, Canada, and detection in the Pacific Northwest, USA. Emerg Infect Dis 13:42–50. doi: 10.3201/eid1301.060827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrnes EJ III, Li W, Ren P, Lewit Y, Voelz K, Fraser JA, Dietrich FS, May RC, Chaturvedi S, Chaturvedi V, Heitman J. 2011. A diverse population of Cryptococcus gattii molecular type VGIII in southern Californian HIV/AIDS patients. PLoS Pathog 7:e1002205. doi: 10.1371/journal.ppat.1002205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrnes EJ III, Li W, Lewit Y, Perfect JR, Carter DA, Cox GM, Heitman J. 2009. First reported case of Cryptococcus gattii in the Southeastern USA: implications for travel-associated acquisition of an emerging pathogen. PLoS One 4:e5851. doi: 10.1371/journal.pone.0005851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris JR, Lockhart SR, Sondermeyer G, Vugia DJ, Crist MB, D'Angelo MT, Sellers B, Franco-Paredes C, Makvandi M, Smelser C, Greene J, Stanek D, Signs K, Nett RJ, Chiller T, Park BJ. 2013. Cryptococcus gattii infections in multiple states outside the US Pacific Northwest. Emerg Infect Dis 19:1621–1627. doi: 10.3201/eid1910.130441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen F, Colom MF, Swinne D, Tintelnot K, Iatta R, Montagna MT, Torres-Rodriguez JM, Cogliati M, Velegraki A, Burggraaf A, Kamermans A, Sweere JM, Meis JF, Klaassen CH, Boekhout T. 2012. Autochthonous and dormant Cryptococcus gattii infections in Europe. Emerg Infect Dis 18:1618–1624. doi: 10.3201/eid1810.120068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perfect JR, Dismukes WE, Dromer F, Goldman DL, Graybill JR, Hamill RJ, Harrison TS, Larsen RA, Lortholary O, Nguyen MH, Pappas PG, Powderly WG, Singh N, Sobel JD, Sorrell TC. 2010. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 50:291–322. doi: 10.1086/649858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espinel-Ingroff A, Aller AI, Canton E, Castanon-Olivares LR, Chowdhary A, Cordoba S, Cuenca-Estrella M, Fothergill A, Fuller J, Govender N, Hagen F, Illnait-Zaragozi MT, Johnson E, Kidd S, Lass-Florl C, Lockhart SR, Martins MA, Meis JF, Melhem MS, Ostrosky-Zeichner L, Pelaez T, Pfaller MA, Schell WA, St-Germain G, Trilles L, Turnidge J. 2012. Cryptococcus neoformans-Cryptococcus gattii species complex: an international study of wild-type susceptibility endpoint distributions and epidemiological cutoff values for fluconazole, itraconazole, posaconazole, and voriconazole. Antimicrob Agents Chemother 56:5898–5906. doi: 10.1128/AAC.01115-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagen F, Illnait-Zaragozi MT, Bartlett KH, Swinne D, Geertsen E, Klaassen CH, Boekhout T, Meis JF. 2010. In vitro antifungal susceptibilities and amplified fragment length polymorphism genotyping of a worldwide collection of 350 clinical, veterinary, and environmental Cryptococcus gattii isolates. Antimicrob Agents Chemother 54:5139–5145. doi: 10.1128/AAC.00746-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lockhart SR, Iqbal N, Bolden CB, DeBess EE, Marsden-Haug N, Worhle R, Thakur R, Harris JR, Cryptococcus gattii PNW Public Health Working Group. 2012. Epidemiologic cutoff values for triazole drugs in Cryptococcus gattii: correlation of molecular type and in vitro susceptibility. Diagn Microbiol Infect Dis 73:144–148. doi: 10.1016/j.diagmicrobio.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iqbal N, DeBess EE, Wohrle R, Sun B, Nett RJ, Ahlquist AM, Chiller T, Lockhart SR, Cryptococcus gattii Public Health Working Group. 2010. Correlation of genotype and in vitro susceptibilities of Cryptococcus gattii strains from the Pacific Northwest of the United States. J Clin Microbiol 48:539–544. doi: 10.1128/JCM.01505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodero L, Mellado E, Rodriguez AC, Salve A, Guelfand L, Cahn P, Cuenca-Estrella M, Davel G, Rodriguez-Tudela JL. 2003. G484S amino acid substitution in lanosterol 14-α demethylase (ERG11) is related to fluconazole resistance in a recurrent Cryptococcus neoformans clinical isolate. Antimicrob Agents Chemother 47:3653–3656. doi: 10.1128/AAC.47.11.3653-3656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sionov E, Chang YC, Garraffo HM, Dolan MA, Ghannoum MA, Kwon-Chung KJ. 2012. Identification of a Cryptococcus neoformans cytochrome P450 lanosterol 14α-demethylase (Erg11) residue critical for differential susceptibility between fluconazole/voriconazole and itraconazole/posaconazole. Antimicrob Agents Chemother 56:1162–1169. doi: 10.1128/AAC.05502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamb DC, Corran A, Baldwin BC, Kwon-Chung J, Kelly SL. 1995. Resistant P45051A1 activity in azole antifungal tolerant Cryptococcus neoformans from AIDS patients. FEBS Lett 368:326–330. doi: 10.1016/0014-5793(95)00684-2. [DOI] [PubMed] [Google Scholar]
- 17.Ngamskulrungroj P, Chang Y, Hansen B, Bugge C, Fischer E, Kwon-Chung KJ. 2012. Characterization of the chromosome 4 genes that affect fluconazole-induced disomy formation in Cryptococcus neoformans. PLoS One 7:e33022. doi: 10.1371/journal.pone.0033022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sionov E, Lee H, Chang YC, Kwon-Chung KJ. 2010. Cryptococcus neoformans overcomes stress of azole drugs by formation of disomy in specific multiple chromosomes. PLoS Pathog 6:e1000848. doi: 10.1371/journal.ppat.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cannon RD, Lamping E, Holmes AR, Niimi K, Baret PV, Keniya MV, Tanabe K, Niimi M, Goffeau A, Monk BC. 2009. Efflux-mediated antifungal drug resistance. Clin Microbiol Rev 22:291–321. doi: 10.1128/CMR.00051-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 21.Thompson GR III, Albert N, Hodge G, Wilson MD, Sykes JE, Bays DJ, Firacative C, Meyer W, Kontoyiannis DP. 2014. Phenotypic differences of Cryptococcus molecular types and their implications for virulence in a Drosophila model of infection. Infect Immun 82:3058–3065. doi: 10.1128/IAI.01805-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu G, Kronstad JW. 2006. Gene disruption in Cryptococcus neoformans and Cryptococcus gattii by in vitro transposition. Curr Genet 49:341–350. doi: 10.1007/s00294-005-0054-x. [DOI] [PubMed] [Google Scholar]
- 23.Fraser JA, Subaran RL, Nichols CB, Heitman J. 2003. Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2:1036–1045. doi: 10.1128/EC.2.5.1036-1045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pukkila-Worley R, Gerrald QD, Kraus PR, Boily MJ, Davis MJ, Giles SS, Cox GM, Heitman J, Alspaugh JA. 2005. Transcriptional network of multiple capsule and melanin genes governed by the Cryptococcus neoformans cyclic AMP cascade. Eukaryot Cell 4:190–201. doi: 10.1128/EC.4.1.190-201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ngamskulrungroj P, Serena C, Gilgado F, Malik R, Meyer W. 2011. Global VGIIa isolates are of comparable virulence to the major fatal Cryptococcus gattii Vancouver Island outbreak genotype. Clin Microbiol Infect 17:251–258. doi: 10.1111/j.1469-0691.2010.03222.x. [DOI] [PubMed] [Google Scholar]
- 26.Sionov E, Chang YC, Garraffo HM, Kwon-Chung KJ. 2009. Heteroresistance to fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob Agents Chemother 53:2804–2815. doi: 10.1128/AAC.00295-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fraser JA, Giles SS, Wenink EC, Geunes-Boyer SG, Wright JR, Diezmann S, Allen A, Stajich JE, Dietrich FS, Perfect JR, Heitman J. 2005. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437:1360–1364. doi: 10.1038/nature04220. [DOI] [PubMed] [Google Scholar]
- 28.Franco-Paredes C, Womack T, Bohlmeyer T, Sellers B, Hays A, Patel K, Lizarazo J, Lockhart SR, Siddiqui W, Marr KA. 2015. Management of Cryptococcus gattii meningoencephalitis. Lancet Infect Dis 15:348–355. doi: 10.1016/S1473-3099(14)70945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith RM, Mba-Jonas A, Tourdjman M, Schimek T, DeBess E, Marsden-Haug N, Harris JR. 2014. Treatment and outcomes among patients with Cryptococcus gattii infections in the United States Pacific Northwest. PLoS One 9:e88875. doi: 10.1371/journal.pone.0088875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Speed B, Dunt D. 1995. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis 21:28–34. doi: 10.1093/clinids/21.1.28. [DOI] [PubMed] [Google Scholar]
- 31.Chen S, Sorrell T, Nimmo G, Speed B, Currie B, Ellis D, Marriott D, Pfeiffer T, Parr D, Byth K. 2000. Epidemiology and host- and variety-dependent characteristics of infection due to Cryptococcus neoformans in Australia and New Zealand. Australasian Cryptococcal Study Group. Clin Infect Dis 31:499–508. doi: 10.1086/313992. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell DH, Sorrell TC, Allworth AM, Heath CH, McGregor AR, Papanaoum K, Richards MJ, Gottlieb T. 1995. Cryptococcal disease of the CNS in immunocompetent hosts: influence of cryptococcal variety on clinical manifestations and outcome. Clin Infect Dis 20:611–616. doi: 10.1093/clinids/20.3.611. [DOI] [PubMed] [Google Scholar]
- 33.Torres-Rodríguez JM, Alvarado-Ramirez E, Murciano F, Sellart M. 2008. MICs and minimum fungicidal concentrations of posaconazole, voriconazole and fluconazole for Cryptococcus neoformans and Cryptococcus gattii. J Antimicrob Chemother 62:205–206. doi: 10.1093/jac/dkn132. [DOI] [PubMed] [Google Scholar]
- 34.Trilles L, Meyer W, Wanke B, Guarro J, Lazera M. 2012. Correlation of antifungal susceptibility and molecular type within the Cryptococcus neoformans/C. gattii species complex. Med Mycol 50:328–332. doi: 10.3109/13693786.2011.602126. [DOI] [PubMed] [Google Scholar]
- 35.Chong HS, Dagg R, Malik R, Chen S, Carter D. 2010. In vitro susceptibility of the yeast pathogen Cryptococcus to fluconazole and other azoles varies with molecular genotype. J Clin Microbiol 48:4115–4120. doi: 10.1128/JCM.01271-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gast CE, Basso LR Jr, Bruzual I, Wong B. 2013. Azole resistance in Cryptococcus gattii from the Pacific Northwest: investigation of the role of ERG11. Antimicrob Agents Chemother 57:5478–5485. doi: 10.1128/AAC.02287-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basso LR Jr, Gast CE, Bruzual I, Wong B. 2015. Identification and properties of plasma membrane azole efflux pumps from the pathogenic fungi Cryptococcus gattii and Cryptococcus neoformans. J Antimicrob Chemother 70:1396–1407. doi: 10.1093/jac/dku554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coleman JJ, Mylonakis E. 2009. Efflux in fungi: la pièce de résistance. PLoS Pathog 5:e1000486. doi: 10.1371/journal.ppat.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanguinetti M, Posteraro B, La Sorda M, Torelli R, Fiori B, Santangelo R, Delogu G, Fadda G. 2006. Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect Immun 74:1352–1359. doi: 10.1128/IAI.74.2.1352-1359.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson GR III, Fothergill AW, Wiederhold NP, Vallor AC, Wickes BL, Patterson TF. 2008. Evaluation of Etest method for determining isavuconazole MICs against Cryptococcus gattii and Cryptococcus neoformans. Antimicrob Agents Chemother 52:2959–2961. doi: 10.1128/AAC.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.