

Abstract
There is a growing interest in repurposing mycobacterial efflux pump inhibitors, such as verapamil, for tuberculosis (TB) treatment. To aid in the design of better analogs, we studied the effects of verapamil on macrophages and Mycobacterium tuberculosis-specific T cells. Macrophage activation was evaluated by measuring levels of nitric oxide, tumor necrosis factor alpha (TNF-α), interleukin-1 beta (IL-1β), and gamma interferon (IFN-γ). Since verapamil is a known autophagy inducer, the roles of autophagy induction in the antimycobacterial activities of verapamil and norverapamil were studied using bone marrow-derived macrophages from ATG5flox/flox (control) and ATG5flox/flox Lyz-Cre mice. Our results showed that despite the well-recognized effects of verapamil on calcium channels and autophagy, its action on intracellular M. tuberculosis does not involve macrophage activation or autophagy induction. Next, the effects of verapamil and norverapamil on M. tuberculosis-specific T cells were assessed using flow cytometry following the stimulation of peripheral blood mononuclear cells from TB-skin-test-positive donors with M. tuberculosis whole-cell lysate for 7 days in the presence or absence of drugs. We found that verapamil and norverapamil inhibit the expansion of M. tuberculosis-specific T cells. Additionally, three new verapamil analogs were found to inhibit intracellular Mycobacterium bovis BCG, and one of the three analogs (KSV21) inhibited intracellular M. tuberculosis replication at concentrations that did not inhibit M. tuberculosis-specific T cell expansion. KSV21 also inhibited mycobacterial efflux pumps to the same degree as verapamil. More interestingly, the new analog enhances the inhibitory activities of isoniazid and rifampin on intracellular M. tuberculosis. In conclusion, KSV21 is a promising verapamil analog on which to base structure-activity relationship studies aimed at identifying more effective analogs.
INTRODUCTION
Verapamil (VER) is a calcium channel blocker used clinically for hypertension, atrial fibrillation or atrial flutter, cluster headache, and angina (1, 2). Recently, there has been a growing interest in repurposing verapamil and other efflux pump inhibitors for the treatment of tuberculosis (TB). The Mycobacterium tuberculosis genome encodes >100 transporters, and some efflux pumps are transcriptionally induced in macrophages (3). M. tuberculosis is primarily an intracellular pathogen, and interference with its efflux pump function decreases its intracellular survival (4). Efflux pump inhibitors, such as verapamil, do not have marked activity on extracellular M. tuberculosis; in fact, the MIC may be as high as 600 μM (5). Interestingly, 5- to 10-times-lower concentrations inhibit intracellular or intracellular-conditioned M. tuberculosis (4, 6). More interestingly, verapamil is known to enhance the anti-TB activities of other drugs (4, 6), making it a very attractive candidate for shortening the duration of anti-TB treatment and improving the management of drug-resistant TB. However, repurposing verapamil for the treatment of TB requires a better understanding of its effects on macrophage function and M. tuberculosis-specific T cell immunity. In this study, we tested the antimycobacterial activities of verapamil and norverapamil on intracellular M. tuberculosis, identified the effects of verapamil on macrophage activation and expansion of M. tuberculosis-specific T cells, investigated the roles of autophagy in the anti-TB function of verapamil, and conducted comparative studies between verapamil and selected new analogs.
The overall goal of this study was to develop optimized verapamil derivatives for specific efflux pump inhibition in M. tuberculosis through the identification of analogs with improved potency and a desirable profile. As we previously reported (7), the initial exploratory structure-activity relationship (SAR) investigation on verapamil was focused on the replacement of the methyl substituent on the tertiary nitrogen with other substituents, varying of the length of the methylene spacer between the tertiary nitrogen and aromatic ring, replacement of the 2-(3,4-dimethoxyphenyl)-N-methylethanamine group with various heterocycles, and replacement of the isopropyl group at the stereogenic carbon center with a hydrogen atom and other alkyl substituents. Verapamil derivatives with various substituents on the tertiary nitrogen demonstrated various effects on the antimycobacterial activity of rifampin. In the presence of rifampin, an unsubstituted derivative resulted in a 2-fold reduction in the MIC of rifampin but did not exhibit any synergistic interactions, while ethyl- and propyl-substituted derivatives did not show any reduction in the rifampin MIC, and neither did they exhibit synergistic interactions with rifampin. On the other hand, a 4-fold reduction in the MIC of rifampin in the presence of an N-benzyl-substituted analog was observed, with an attendant synergistic interaction with rifampin. Relative to verapamil, the derivatives in which the aminoethyl aromatic group of verapamil was replaced with substituted aniline and aminomethyl aromatic group did not exhibit any effect on the antimycobacterial activity of rifampin when evaluated in combination. The derivatives obtained by replacing the aminoethyl aromatic group of verapamil with various heterocyclic groups demonstrated varied effects on the antimycobacterial activity of rifampin. This ranged from no effect to a 2- and 4-fold reduction in the MIC of rifampin, which is comparable to that of verapamil but with no interaction with rifampin. The replacement of the isopropyl group at the stereogenic carbon center with hydrogen and other alkyl groups also yielded compounds with varied effects on the MIC of rifampin. An unsubstituted derivative, a methyl-substituted derivative, and a cyclopentyl-substituted derivative all resulted in a 2-fold reduction in the MIC of rifampin, while a 4-fold drop in the MIC of rifampin was observed with ethyl-, propyl-, and cyclohexyl-substituted derivatives. Once again, all combination studies suggested the absence of any interaction between rifampin and these derivatives. All compounds that led to a 4-fold reduction in the MIC of rifampin on extracellular M. tuberculosis were selected for further experiments described in this paper.
MATERIALS AND METHODS
Drugs.
Verapamil, norverapamil, isoniazid, and rifampin were purchased from Sigma. New analogs of verapamil were synthesized at the Department of Chemistry and South African Medical Research Council Drug Discovery and Development Research Unit (University of Cape Town, South Africa). Stock solutions of the analogs were prepared in dimethyl sulfoxide (DMSO) or ethanol, and aliquots were stored at −80°C until required for use in the experiments. Working solutions were prepared in the appropriate cell culture medium.
Antibodies, media, and reagents.
Ethidium bromide, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), saponin, human AB (HAB) serum, fetal calf serum (FCS), Tween 80, and apoptosis detection kits were purchased from Sigma (St. Louis, MO, USA). Components of the mycobacterial growth media, such as Middlebrook 7H9, Middlebrook 7H10, and supplements (albumin, dextrose, catalase [ADC] and oleic acid with ADC [OADC]) were purchased from Becton Dickinson. [3H]-uridine was obtained from PerkinElmer, Inc., USA. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Life Technologies. Interleukin-1 beta (IL-1β), tumor necrosis factor alpha (TNF-α), and gamma interferon (IFN-γ) primary and secondary antibodies for enzyme-linked immunosorbent assay (ELISA) were obtained from R&D Systems.
Antibiotic-free RPMI 1640 (catalog no. 11875; Gibco) supplemented with 10% heat-inactivated human AB serum and 200 mM l-glutamine (Lonza BioWhittaker) was used for cell culture. MTT working solutions were prepared in phosphate-buffered saline (PBS). MTT is converted into a blue formazan in the presence of live cells. Ten percent sodium dodecyl sulfate in a 40% aqueous solution of dimethyl formamide was used as a formazan solubilization buffer.
Monocytes and bacterial cultures.
Mycobacterium bovis strain BCG Connaught (ATCC 35745) and M. tuberculosis Erdman (ATCC 35801) were grown in a suspension with constant gentle rotation in roller bottles containing Middlebrook 7H9 broth supplemented with 10% ADC enrichment and 0.05% Tween 80 (Sigma-Aldrich). Stock vials of BCG and M. tuberculosis were prepared from 2-week-old logarithmic-phase cultures, and aliquots were frozen at −80°C until needed for the different experiments. The number of bacteria (CFU per milliliter) in the frozen vials was quantified by plating samples on 7H10 Middlebrook agar supplemented with OADC. On the day of macrophage infection, bacteria were thawed and sonicated for l min in a water bath sonicator (W385; Heat Systems Ultrasonics, Farmingdale, NY) to obtain a single-cell suspension and diluted appropriately in complete RPMI 1640 medium. A leukemic monocyte line (THP-1) obtained from the ATCC (TIB-202) was cultured in RPMI medium supplemented with 200 mM l-glutamine and 10% FCS and used for the cytotoxicity assays. Human peripheral blood mononuclear cells (PBMCs) stored in liquid nitrogen were used to generate monocyte cultures and study the effects of drugs on M. tuberculosis-specific immunity. These PBMCs were obtained from six healthy purified protein derivative (PPD) skin test-positive donors, according to a protocol approved by the Saint Louis University (St. Louis, MO) institutional review board. Monocytes were purified from PBMCs by plastic adherence based on the unique adhesion properties of monocytes/macrophages (8, 9). Briefly, 1.5 × 105 cells were plated in each well of 96-well round-bottom microtiter plates (Corning, Inc., USA). Nonadherent cells were washed off with complete RPMI medium after overnight incubation at 37°C and 5% CO2. In general, approximately 10% of the initial number of PBMCs plated were retained as adherent monocytes (>90% CD14+), which were then cultured for 2 to 4 days before infection with M. bovis BCG or M. tuberculosis (Erdman).
Measuring cytotoxicity of verapamil, norverapamil, and analogs.
THP-1 cells in RPMI with l-glutamine were cultured in round-bottom 96-well plates at a concentration of 1.5 ×104/well. Drugs were added at different concentrations, and cultures were incubated at 37°C. After 24 h, MTT assays were performed, as described previously (10). The amount of formazan formation was quantified by measuring the absorbance at 570 nm using an SLT Rainbow plate reader (Tecan, US, Inc.). Percent cytotoxicity was calculated using relative optical densities (OD), as follows: 100 − (100 × [OD with drug/OD without drug]).
Measuring the effects of verapamil, norverapamil, and analogs on intracellular mycobacteria.
Adherent monocytes were infected with BCG or M. tuberculosis for 4 h at a multiplicity of infection (MOI) of 3. Unphagocytosed bacteria were removed by washing three times with warm serum-free RPMI. Infected monocytes were cultured in complete RPMI medium with and without drugs for 72 h and then lysed with 0.2% saponin (Sigma) in RPMI 1640 medium. After cell lysis, residual bacteria were quantified using a [3H]uridine incorporation assay, as described previously (8, 9). Briefly, the uridine incorporation assay was performed by adding 1 μCi of [3H]uridine in 7H9 Middlebrook broth into each well and incubating the plates for another 72 h at 37°C and 5% CO2. Bacteria were then harvested on glass fiber filter mats (PerkinElmer Wallac, Inc.), using a Tomtec Mach III cell harvester 96 (Tomtec, Inc., Hamden, CT). The filter mats were air dried, scintillation fluid was added, and the mycobacterial incorporation of tritiated uridine was quantified using a MicroBeta scintillation counter (PerkinElmer Wallac, Inc.). Mycobacterial growth inhibition was determined using the following formula: percent inhibition = 100 − (100 × [dpm in the drug-treated cultures/dpm in the untreated cultures]) (dpm, disintegrations per minute). The experiments on each drug were performed 4 to 5 times in triplicate wells. When obtaining new lots of [3H]uridine (PerkinElmer), the results from the uridine incorporation assay were confirmed by culturing pooled aliquots from triplicate wells of at least one of the multiple experiments on each drug on 7H10 medium and counting the CFU every week for 3 weeks.
CFSE-based flow cytometry assay to measure effects of verapamil, norverapamil, and analogs on M. tuberculosis-specific T cells.
A CFSE-based flow cytometry assay was performed, as described previously (11, 12), using M. tuberculosis whole-cell lysate (WL) at a final concentration of 10 μg/ml to stimulate M. tuberculosis-specific T cells during 7 days of culture. Flow-cytometric acquisition was performed using a multicolor BD FACSCanto II instrument, and analyses were done using the CellQuest and FlowJo (Tree Star) softwares. A minimum of 10,000 CD3+ events were acquired. Cells with decreased CFSE fluorescence were considered to be proliferated cells. The absolute numbers of effector CD3+ T cells were calculated by multiplying the total number of viable cells recovered times the percentage of the specific T cell subset detected by flow-cytometric analysis. Mann-Whitney U tests were used to analyze differences in the expansions of CD3+ T cells in PBMCs cultured in the presence and absence of drugs.
Identifying effects of verapamil and norverapamil on activation of BCG-infected human monocytes.
Human monocytes were plated (3 × 104) in 96-well round-bottom microtiter plates (Corning, Inc., USA) and allowed to mature for 5 days at 37°C and 5% CO2. The monocytes were then infected with BCG for 4 h (MOI, 3). Extracellular bacteria were removed by washing three times in RPMI medium, and different concentrations of verapamil or norverapamil were added. Supernatants were collected 24 h later. IL-1β, TNF-α, and IFN-γ levels were quantified by ELISA. The nitric oxide level was quantified with a detection kit (Enzo Life Sciences, Inc., NY).
Identifying the role of autophagy in the anti-TB activities of verapamil or norverapamil.
Bone marrow-derived macrophages (BMDM) from ATG5flox/flox (control) and ATG5flox/flox Lyz-Cre mice were kind gifts from Christina Stallings (Washington University, MO). These mice were bred and genotyped as described previously (13, 14). BMDM resuspended in Dulbecco's modified Eagle medium (DMEM) with 20% FCS, 10% macrophage colony-stimulating factor (M-CSF), and 1% penicillin-streptomycin were added to flat-bottom 96-well plates (2.5 × 104/well) and allowed to mature at 37°C. On day 11, the cells were washed, culture medium was changed to DMEM with 20% FCS, and the cells were infected with BCG or M. tuberculosis for 4 h (MOI, 3). Extracellular bacteria were removed by washing with medium three times. Different concentrations of verapamil and norverapamil were added, and the cells were cultured. After 72 h, the number of intracellular bacteria was quantified as described above.
Ethidium bromide-based flow cytometry to measure efflux pump activity.
Efflux pump activity was measured based on methods described previously (15, 16). The conditions for this assay were first optimized using in vitro BCG cultures. Briefly, the turbidity of logarithmic-phase cultures of BCG in 7H9 Middlebrook broth was adjusted to the standard turbidity of 0.5 McFarland. Aliquots of 100 μl were transferred into three sets of 2-ml Sarstedt tubes. Ethidium bromide and verapamil were added (at 2.5 μM and 250 μM, respectively), and the cultures were incubated at room temperature for 1 h. Bacteria were pelleted by centrifugation, washed once with 1 ml of PBS, and resuspended in PBS with 0.4% glucose with and without various efflux pump inhibitors or analogs. These cultures were incubated at 37°C for 1 h, and the aliquots were transferred to a round-bottom 96-well plate. Flow-cytometric acquisition was performed using a Guava easyCyte instrument (Guava Technology, Hayward, CA), and a minimum of 5,000 events were acquired. Analyses were done using the FlowJo (version 6.2.1; Tree Star) software.
Design, synthesis, and characterization of analogs.
Twenty verapamil analogs were designed and synthesized. Five analogs were selected for further studies based on the ability of compounds to potentiate the in vitro antimycobacterial effect of rifampin against M. tuberculosis. Analogs showing a 4-fold reduction in the MIC of rifampin were used for further evaluation in this study (Table 1). Norverapamil was selected because it has been used as a standard for comparison in various studies on verapamil (VER).
TABLE 1.
In vitro antimycobacterial data, docking score, and physiochemical properties of verapamil analogs

a MIC99, MIC for 99% inhibition;
b Fold reduction in rifampin MIC99 when evaluated in combination with analogs.
c PPB, plasma protein binding.
d Predicted results using StarDrop version 5.0, Optibrium Ltd., Cambridge, United Kingdom.
e QPPCaco, predicted apparent Caco-2 cell permeability in nm/s. Caco-2 cells are a model for the gut-blood barrier. QikProp predictions (QPP) are for nonactive transport (>500 is great); QPloghERG, predicted 50% inhibitory concentration (IC50) for blockage of hERG K+ channels (below −5.0 is a concern). Predicted results using QikProp, version 3.8, Schrödinger, LLC, New York, NY.
f Exp., experimental. ND, not determined.
Solubility, lipophilicity, plasma protein binding, Caco2 and human ether-à-go-go-related gene (hERG) channel liability, and binding to Rv1258C have been determined and/or predicted. The QikProp program (version 3.8; Schrödinger, LLC, New York, NY, 2013) was used to predict QPPCaco and QPloghERG, while the StarDrop program (version 5.0; Optibrium Ltd., Cambridge, United Kingdom) was used to predict 90% plasma protein binding (PPB90), log distribution coefficient at pH 7.4 (D7.4), and log water solubility at pH 7.4 (S7.4). ChemDraw sketches were converted to MOLfiles, fed into the respective program (QikProp and StarDrop), and processed for the desired properties.
Binding to Rv1258c, an efflux pump protein, was predicted by docking studies (7). Briefly, The Preparation Wizard version 5.8 software was used to prepare a model of Rv1258c. Five putative binding sites on the prepared model protein were identified using SiteMap (version 2.6). Docking studies of verapamil and its analogs were performed with each binding site using Extra Precision (XP) Glide, and the overall interaction with Rv1258c was described in terms of gliding scores. Higher numbers indicate better predictive binding. A more detailed description of the docking studies is found in the supplemental Methods and in Fig. S1 to S4 in the supplemental material.
Measuring intracellular drug levels using liquid chromatography-mass spectrometry.
Plate-adherent human macrophages were treated with verapamil or KSV21 for 2 h. After 2 h, the supernatants were removed and macrophages lysed with water and ethanol. The lysates were passed through 0.2-μm-pore-size filters.
Samples (100 μl total volume) were diluted with control matrix (RPMI plus 10% fetal bovine serum [FBS] or macrophage cell lysate), as appropriate, to bring samples into the dynamic of the standard curve (1 to 1,000 ng/ml). Internal standard was added to all samples. The samples were capped, mixed on a multiplate vortex for 5 min, and centrifuged for 5 min at 3,200 rpm. The supernatants were transferred to a 96-well sample plate and capped for liquid chromatography-tandem mass spectrometry (LC/MS/MS) analysis. The multiple-reaction monitoring (MRM) transitions for verapamil and KS21 were m/z 455.3→165.2 and 495.4→165.1, respectively. The mobile phases consisted of 0.1% formic acid in water (aqueous) and 100% acetonitrile (organic) with an Amour C18 reverse-phase column (2.1 by 30 mm; pore size, 5 μm) at a flow rate of 0.35 ml/min. The starting phase was 10% acetonitrile for 0.9 min and was then increased to 95% acetonitrile over 0.4 min, which was maintained for an additional 0.3 min before returning to 10% acetonitrile over 0.4 min. The 10% acetonitrile was held for an additional 1.6 min. Peak areas were integrated using Analyst 1.5.1 (AB Sciex, Foster City, CA). The percent intracellular accumulation of drugs was calculated as follows: [intracellular drug concentration (macrophage lysate)/drug concentration in supernatants + drug concentration in lysate] × 100.
Identifying the interactions of selected verapamil analogs with isoniazid and rifampin.
The MICs of isoniazid and rifampin for intracellular M. tuberculosis were determined by testing ranges of concentrations of each drug separately. Eight different concentrations ranging from 0.0012 to 7.3 μM were added to M. tuberculosis-infected macrophages in triplicate. After 72 h, the cells were lysed with saponin, and the number of intracellular bacteria was quantified by subculturing different dilutions on 7H10 agar. The number of bacteria from cultures containing drugs was compared with the number of bacteria from drug-free cultures. The MIC was defined as the lowest concentration that inhibits at least 90% of intracellular M. tuberculosis cells. The effects of combinations of verapamil analogs with sub-MICs of isoniazid (INH) or rifampin on intracellular M. tuberculosis were studied using infected human macrophages, as described above.
Statistical analysis.
Mann-Whitney U tests (GraphPad Software, Inc., San Diego, CA, USA) were used for nonparametric analysis of differences between drug-free controls and conditions with drugs. A P value of <0.05 was considered statistically significant.
RESULTS
Verapamil and norverapamil inhibit intracellular mycobacterial growth.
It is known that verapamil does not inhibit the growth of extracellular M. tuberculosis (6). However, the exposure of M. tuberculosis to the intracellular environment of macrophages increases the susceptibility of M. tuberculosis to the efflux pump inhibitory activities of verapamil (4, 6). This is important, because M. tuberculosis is an intracellular bacterium residing primarily inside macrophages. In this study, we used a range of verapamil concentrations, including concentrations that were much lower than those previously tested. Figure 1 shows that verapamil and norverapamil inhibit intracellular BCG and intracellular M. tuberculosis in a dose-dependent manner. The highest concentrations selected had no or minimal cytotoxicity on THP-1 cells.
FIG 1.

Activities of verapamil and norverapamil on intracellular BCG and M. tuberculosis. Human monocytes were infected with BCG or M. tuberculosis (Mtb) for at least 4 h before washing and adding different concentrations of verapamil or norverapamil. The infected monocytes were cultured for 72 h. Next, the monocytes were lysed with saponin, and the released bacteria were quantified using a [3H]uridine incorporation assay. Percent inhibition was calculated as follows: 100 − (100 × [dpm in the drug-treated cultures/dpm in the untreated cultures]). (A) Activities of verapamil on intracellular BCG. (B) Activities of norverapamil on intracellular BCG. (C and D) Activities of verapamil and norverapamil on intracellular M. tuberculosis, respectively. The error bars represent the standard error of mean (SEM) values from the results from 5 experiments. *, P < 0.05; **, P < 0.01 (Mann-Whitney U test).
Verapamil and norverapamil do not enhance activation of M. tuberculosis-infected macrophages.
Available evidence indicates that efflux pumps are important for the intracellular survival of M. tuberculosis. Thus, efflux pump inhibition might be the major mechanism by which verapamil and norverapamil inhibit intracellular mycobacteria (4, 6, 17). Verapamil is a calcium channel blocker, which may interfere with the cellular functions of Mycobacterium-infected macrophages. This suggests that other mechanisms that lead to the activation of macrophages may indirectly affect the intracellular survival of mycobacteria.
Activated macrophages release effector molecules, which help them control the growth of intracellular mycobacteria (18–23). In view of the possibility that verapamil activates macrophages, we assessed the activation levels of BCG-infected macrophages cultured in the presence or absence of 100 μM verapamil or norverapamil by measuring the levels of nitric oxide and several cytokines present in the culture supernatants. We found that verapamil and norverapamil did not increase the levels of nitric oxide, IL-1β, TNF-α, or IFN-γ (see Fig. S5 in the supplemental material), suggesting that the inhibition of intracellular mycobacteria by these drugs was not related to a change in macrophage activation.
Autophagy is not involved in the activities of verapamil and norverapamil on intracellular mycobacteria.
Autophagy is a cellular process that aids in the acidification of the mycobacterial phagosome, leading to the inhibition of intracellular mycobacterial growth (24, 25). Verapamil is a known autophagy inducer (26). Verapamil increases microtubule-associated protein 1 light chain 3 expression and enhances the metabolism of abnormal proteins in an atg5-dependent manner (26). On this basis, we next addressed whether or not verapamil uses autophagy as part of its mechanism(s) of intracellular M. tuberculosis inhibition. We used BMDM from ATG5flox/flox (control) and ATG5flox/flox Lyz-Cre (atg5atg5-deficient) mice. Figure 2 shows that verapamil is similarly potent in Mycobacterium-infected BMDM from control and atg5-deficient atg5mice, indicating that the anti-TB effects of verapamil are not due to the induction of autophagy. Similar results were obtained using norverapamil.
FIG 2.

Inhibition of intracellular M. tuberculosis by verapamil does not involve autophagy. BMDM from Atg5flox/flox (control) and Atg5flox/flox Lyz-CreA plated on 96-well plates were infected with BCG (A) or M. tuberculosis (Mtb) (B) for 4 h. Extracellular bacteria were removed by washing. Different concentrations of verapamil were added, and the cells were cultured for 72 h. Next, BMDM were lysed with saponin and the released bacteria quantified by a [3H]-uridine incorporation assay. Percent inhibition was calculated as follows: 100 − (100 × [dpm in the drug-treated cultures/dpm in the untreated cultures]). The percent inhibition rates of the different concentrations of verapamil on BCG- or M. tuberculosis-infected BMDM from Atg5flox/flox Lyz-CreA mice were not significantly different from the percent inhibition rates of matching concentrations of verapamil on infected BMDM from Atg5flox/flox (control) mice. Results are shown as means ± standard errors.
Verapamil and norverapamil inhibit expansion of M. tuberculosis-specific T cells.
M. tuberculosis-specific immunity is important to limit the progression of TB and improve the cure rate of anti-TB treatment (27–31). Because previous reports have shown that verapamil inhibits tumor-specific T cells (32), we next studied the effects of verapamil and norverapamil on M. tuberculosis-specific T cells. T cells from exposed individuals proliferate and produce IFN-γ following stimulation with M. tuberculosis or M. tuberculosis antigens (12). We stimulated CFSE-labeled PBMCs from PPD-positive donors with M. tuberculosis whole-cell lysate (WL), and after 7 days, we measured the number of proliferated and IFN-γ-producing T cells using flow cytometry. Figure 3 demonstrates that low concentrations of both verapamil and norverapamil significantly inhibit the expansion of M. tuberculosis-specific CD3+ T cells.
FIG 3.
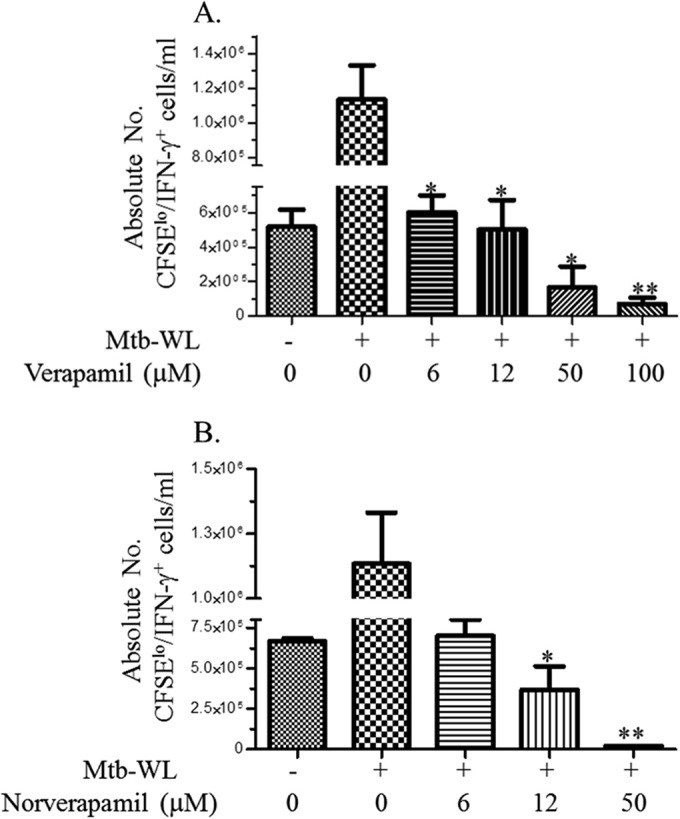
Verapamil and norverapamil limit the expansion of M. tuberculosis-specific T cells. PBMCs from PPD-positive subjects (n = 5) were labeled with CFSE and stimulated with M. tuberculosis WL for 7 days in the presence or absence of different concentrations of efflux pump inhibitors. On day 7, viable cells were estimated after counting of cells in aliquots mixed with trypan blue dye. In parallel, all cells were restimulated with phorbol myristate acetate (PMA) and ionomycin in the presence of GolgiStop for 2 h, and then stained for surface CD3 and intracellular IFN-γ. Flow-cytometric acquisition was performed by use of a BD LSR II instrument. (A and B) Effects of different concentrations of verapamil (A) and norverapamil (B) on the expansion of M. tuberculosis-specific T cells. *, P < 0.05; **, P < 0.01, by Mann-Whitney U test. Results are shown as means ± standard errors.
New verapamil analogs possess anti-TB activities without affecting M. tuberculosis-specific T cells.
New analogs were designed and synthesized for structure-activity-relationship studies, with the goal of identifying analogs with improved potency for use at lower concentrations, which would address the liabilities associated with high dosages of verapamil or norverapamil (i.e., hypotension and other toxicities). This is in view of the fact that verapamil was optimized for its calcium channel-blocking properties and not efflux pump and/or antibacterial inhibition. Specific modifications were made in the first iteration, which included altering one of the side chains of the verapamil molecule. The activities of new analogs on intracellular mycobacteria were tested using concentrations that have no or minimal cytotoxicities on THP-1 cells (see Fig. S6 in the supplemental material). The antimycobacterial effects of new analogs on intracellular BCG are shown in Fig. 4. The inhibitory effects at the highest concentrations of three analogs, KSV21, MKV4, and MKV9, on intracellular BCG were not significantly different from those seen with equivalent concentrations of verapamil or norverapamil. Therefore, these three analogs were next tested for the inhibition of intracellular M. tuberculosis. Figure 5A shows that while all three molecules inhibited the growth of intracellular M. tuberculosis at concentrations of 2.5 μM, KSV21 was significantly more potent than MKV4 (P < 0.05). The activities of KSV21 and MKV9 were similar.
FIG 4.

Activities of verapamil analogs on intracellular BCG. (A) KSV10. (B) KSV21. (C) MKV4. (D) MKV8. (E) MKV9. Infected monocytes were cultured in the presence or absence of drugs for 72 h. Next, monocytes were lysed with saponin, and the released bacteria were quantified using a [3H]uridine incorporation assay. Percent inhibition was calculated as follows: 100 − (100 × [dpm in the drug-treated cultures/dpm in the untreated cultures]). (F) The activity of the highest concentration of each drug was compared with the activity of a similar concentration of verapamil. KSV10 and MKV8 had significantly lower activity than verapamil (P < 0.05, Mann-Whitney U test). The activities of KSV21, MKV4, and MKV9 were comparable with the activity of verapamil. Results are shown as means ± standard errors.
FIG 5.

Effects of new verapamil analogs on intracellular M. tuberculosis and M. tuberculosis-specific immunity. (A) Activities of selected analogs on intracellular M. tuberculosis. Each drug was used at a final concentration of 2.5 μM. KSV21 had significantly better activity than MKV4 (*, P < 0.05, Mann-Whitney U test). (B) Effects of new analogs on the expansion of M. tuberculosis-specific T cells. The experimental setup was similar to that described in the Fig. 3 legend. MKV9 at a concentration of 2.5 μM significantly inhibited M. tuberculosis-specific T cells (*, P < 0.05). Similar concentrations of MKV4 and KSV21 did not inhibit the expansion of M. tuberculosis-specific T cells. MR, medium rested. Results are shown as means ± standard errors.
New verapamil analogs KSV21 and MKV4 do not affect the expansion of M. tuberculosis-specific T cells.
We next studied the effects of selected analogs on M. tuberculosis-specific T cell immunity. We stimulated CFSE-labeled PBMCs from PPD-positive donors with M. tuberculosis WL for 7 days and measured the proliferation of M. tuberculosis-specific effector T cells using flow cytometry. MKV9 inhibited M. tuberculosis-specific T cell proliferation and IFN-γ production (P < 0.05), whereas KSV21 and MKV4 showed no effect on M. tuberculosis-specific immunity at concentrations of 2.5 μM (Fig. 5B). Concentrations of KSV21 as high as 25 μM did not interfere with the expansion of M. tuberculosis-specific T cells and were similar to the results with 2.5 μM.
New verapamil analog KSV21 inhibits mycobacterial efflux pumps.
We next measured the efflux pump inhibitory activities of verapamil, KSV21, and MKV4 using a modified ethidium bromide assay. Ethidium bromide-stained mycobacteria remove the dye through efflux pumps (33). Efflux pump inhibitors, owing to their inhibitory effect on the efflux of ethidium bromide, maintain the fluorescence of stained bacteria, which can easily be identified by flow cytometry. Figure 6A demonstrates that KSV21 prevents mycobacteria from removing ethidium bromide to the same degree as verapamil. This may indicate that KSV21, like verapamil, inhibits intracellular M. tuberculosis by inhibiting mycobacterial efflux pumps. MKV4 did not significantly inhibit mycobacterial efflux pumps at the concentration tested. Similar to verapamil, autophagy did not play a role in the mechanism of killing of intracellular mycobacteria by KSV21 (see Fig. S7 in the supplemental material).
FIG 6.

KSV21 has efflux pump inhibitory activity and enhances the activities of two key first-line drugs on intracellular M. tuberculosis. (A) Efflux pump inhibitory activities of drugs were measured using an ethidium bromide-based flow-cytometric assay. The results show that BCG cultured in the presence of verapamil (100 μM) or KSV21 (40 μM) had significantly higher fluorescence than the drug-free control (P < 0.01), indicating that KSV21 is a potent efflux pump inhibitor. MKV4 (100 μM) did not show efflux pump inhibitory activity. (B and C) Infected monocytes were cultured in the presence or absence of drugs for 72 h. Next, the monocytes were lysed with saponin and the released bacteria quantified using a [3H]uridine incorporation assay. Percent inhibition was calculated as follows: 100 − (100 × [dpm in the drug-treated cultures/dpm in the untreated cultures]). (B) KSV21 enhances the activity of isoniazid on intracellular M. tuberculosis. KSV21 (2.5 μM) was studied alone or in combination with a sub-MIC of isoniazid (0.006 μM). The anti-TB activities of the combination were significantly better than the activity of KSV21 or isoniazid alone (*, P < 0.05, Mann-Whitney U test). (C) KSV21 enhances the activity of rifampin (0.001 μM) on intracellular M. tuberculosis. KSV21 (2.5 μM) was studied alone or in combination with a sub-MIC of rifampin. The anti-TB activity of the combination was significantly better than the activity of KSV21 or rifampin alone (**, P < 0.01, Mann-Whitney U test). Results are shown as means ± standard errors.
KSV21 penetrates macrophages better than verapamil.
The liquid chromatography-mass spectrometry (LC-MS) results showed that KSV21 accumulates within macrophages better than verapamil, with intracellular accumulation rates of 26% and 11%, respectively (see Fig. S8 in the supplemental material). A higher intracellular accumulation rate may make KSV21 a more attractive efflux pump inhibitor, since M. tuberculosis is primarily an intracellular pathogen.
KSV21 enhances the inhibitory activities of isoniazid and rifampin on intracellular M. tuberculosis.
KSV21 is a unique efflux pump inhibitor with potent activity on intracellular BCG and M. tuberculosis (Fig. 4 and 5). Additionally, unlike verapamil and norverapamil, KSV21 was shown not to interfere with the proliferation of M. tuberculosis-specific T cells (Fig. 5B). These desirable features make KSV21 a good candidate for studies in combination with key first-line TB drugs. To that end, we cultured M. tuberculosis-infected macrophages in the presence of multiple concentrations of KSV21 alone and in combination with sub-MICs of isoniazid and rifampin. The number of intracellular M. tuberculosis cells was quantified 3 days later. We found that concentrations of KSV21 of ≥2.5 μM are required to enhance the anti-TB activities of isoniazid and rifampin. Figure 6 shows that KSV21 (2.5 μM) enhanced the anti-TB activities of isoniazid (Fig. 6B) and rifampin (Fig. 6C).
DISCUSSION
Several reports have shown that efflux pump inhibitors, including verapamil, inhibit the survival of intracellular M. tuberculosis and enhance the anti-TB activities of other drugs (4, 6). In addition, it was recently shown that norverapamil has anti-TB activities similar to those of verapamil (6). This is important, since norverapamil is one of the major metabolites of verapamil, and its serum levels may be equivalent to those of verapamil after first-pass metabolism (34, 35). The anti-TB activities of concentrations of verapamil close to maximum achievable serum levels have not been tested. At normal doses, the achievable serum levels of verapamil may not exceed 1 μM (35, 36). Higher serum concentrations, particularly concentrations of >9 μM, may cause serious side effects, with high mortality (37). In this study, we used various concentrations of verapamil and norverapamil, including concentrations that are close to the reported maximum achievable serum drug levels. Our results showed that even the lowest concentration tested (12.5 μM) had 36% and 40% inhibition on intracellular BCG and M. tuberculosis, respectively (Fig. 1). The effects of verapamil and norverapamil on intracellular BCG are concentration dependent, but the effects on intracellular M. tuberculosis do not appear to be concentration dependent.
The design of new and more effective analogs of verapamil requires a better understanding of the possible mechanisms of action. Verapamil is a potent efflux pump inhibitor, and this appears to be the likely mechanism responsible for enhancing the anti-TB activities of other drugs (4, 6). However, the fact that verapamil is a calcium channel blocker and a potent autophagy inducer (26) indicates the possibility that other mechanisms may be contributing to the activity of this drug on intracellular M. tuberculosis. Our results showed that macrophage activation and autophagy are unlikely to be the major mechanisms of the anti-TB activity of verapamil.
M. tuberculosis-specific immunity is essential to limit the progression of TB, and it enhances anti-TB treatment and cure (27–31). Available evidence indicates that verapamil inhibits the proliferation of T and B cells, leading some to consider this drug to be an immunosuppressive agent (38–40). It has been demonstrated that verapamil inhibits the proliferation of T and B cells following polyclonal stimulation with concanavalin, phytohemagglutinin, pokeweed mitogen, and alloantigens (39, 40). The effects of verapamil on M. tuberculosis-specific T cells have not yet been investigated. Our results clearly show that both verapamil and norverapamil inhibit the expansion of mycobacterial antigen-specific T cells. To our surprise, even low concentrations of these drugs significantly inhibited the expansion of M. tuberculosis-specific T cells. Mycobacterial efflux pump inhibitors may also inhibit host cell efflux pumps, which may, therefore, interfere with at least some normal cellular functions (41–44), including proliferation. These results were important in our selection criteria for new analogs in which we focus on those that retain efflux pump inhibitory and anti-TB activities at concentrations that do not interfere with T cell expansion.
Of the five analogs tested, three (KSV21, MKV4, and MK9) had activities comparable to those of verapamil and norverapamil on intracellular BCG. These three compounds were also found to potently inhibit intracellular M. tuberculosis. MKV9 inhibited the expansion of M. tuberculosis-specific T cells and was not studied further. The mycobacterial efflux pump inhibitory properties of KSV21 and MKV4 were tested using pure BCG culture. MKV4 did not show efflux pump inhibitory activities, and the mechanisms for how it may act on intracellular M. tuberculosis are not clear. KSV21 was found to be unique in that in addition to inhibiting intracellular M. tuberculosis, it retained mycobacterial efflux pump inhibitory activities at concentrations that did not interfere with the expansion of M. tuberculosis-specific T cells. Mycobacterial efflux pumps are important for survival in a stressful environment inside macrophages (4), and the expression of many efflux pumps is transcriptionally upregulated inside macrophages (3). Therefore, interference with the functions of mycobacterial efflux pumps is expected to limit survival inside macrophages and enhance the activities of other anti-TB drugs. In fact, efflux pump inhibitors, such as verapamil and reserpine, have been shown to enhance the activities of rifampin, bedaquiline, and moxifloxacin (4, 45). Synergistic interaction with key first-line or new drugs can be useful to shorten TB treatment and effectively manage drug-resistant TB. Because of its activities on mycobacterial efflux pumps, we tested the effects of interactions of KSV21 with key first-line anti-TB drugs, isoniazid and rifampin, on intracellular M. tuberculosis. KSV21 enhanced the effects of isoniazid and rifampin on intracellular M. tuberculosis, likely due to mycobacterial efflux pump inhibition.
KSV21 is a promising analog that can be used as a basis to design more analogs, which will help improve mycobacterial efflux pump inhibitory activities and enhance the effects on intracellular M. tuberculosis without untoward effects on TB-specific immunity. Our results suggest that the in vivo evaluation of KSV21 in relevant animal models is warranted.
Supplementary Material
ACKNOWLEDGMENTS
The project was supported in part by a startup fund to G.A. from Saint Louis University. We gratefully acknowledge the University of Cape Town, South African Medical Research Council, and South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation, for support (to K.C).
BMDM from ATG5flox/flox (control) and ATG5flox/flox Lyz-Cre mice were kindly provided by Christina Stallings (Washington University, St. Louis, MO).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01567-15.
REFERENCES
- 1.Yedinak KC. 1993. Use of calcium channel antagonists for cardiovascular disease. Am Pharm 33:49–64; quiz 64–66. [DOI] [PubMed] [Google Scholar]
- 2.Weaver-Agostoni J. 2013. Cluster headache. Am Fam Physician 88:122–128. [PubMed] [Google Scholar]
- 3.Szumowski JD, Adams KN, Edelstein PH, Ramakrishnan L. 2013. Antimicrobial efflux pumps and Mycobacterium tuberculosis drug tolerance: evolutionary considerations. Curr Top Microbiol Immunol 374:81–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues L, Machado D, Couto I, Amaral L, Viveiros M. 2012. Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect Genet Evol 12:695–700. doi: 10.1016/j.meegid.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Adams KN, Szumowski JD, Ramakrishnan L. 2014. Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J Infect Dis 210:456–466. doi: 10.1093/infdis/jiu095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh K, Kumar M, Pavadai E, Naran K, Warner DF, Ruminski PG, Chibale K. 2014. Synthesis of new verapamil analogues and their evaluation in combination with rifampicin against Mycobacterium tuberculosis and molecular docking studies in the binding site of efflux protein Rv1258c. Bioorg Med Chem Lett 24:2985–2990. doi: 10.1016/j.bmcl.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 8.Spencer CT, Abate G, Sakala IG, Xia M, Truscott SM, Eickhoff CS, Linn R, Blazevic A, Metkar SS, Peng G, Froelich CJ, Hoft DF. 2013. Granzyme A produced by γ(9)δ(2) T cells induces human macrophages to inhibit growth of an intracellular pathogen. PLoS Pathog 9:e1003119. doi: 10.1371/journal.ppat.1003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Worku S, Hoft DF. 2003. Differential effects of control and antigen-specific T cells on intracellular mycobacterial growth. Infect Immun 71:1763–1773. doi: 10.1128/IAI.71.4.1763-1773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mshana RN, Tadesse G, Abate G, Miörner H. 1998. Use of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide for rapid detection of rifampin-resistant Mycobacterium tuberculosis. J Clin Microbiol 36:1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abate G, Eslick J, Newman FK, Frey SE, Belshe RB, Monath TP, Hoft DF. 2005. Flow-cytometric detection of vaccinia-induced memory effector CD4(+), CD8(+), and gamma delta TCR(+) T cells capable of antigen-specific expansion and effector functions. J Infect Dis 192:1362–1371. doi: 10.1086/444423. [DOI] [PubMed] [Google Scholar]
- 12.Hoft DF, Blazevic A, Abate G, Hanekom WA, Kaplan G, Soler JH, Weichold F, Geiter L, Sadoff JC, Horwitz MA. 2008. A new recombinant bacille Calmette-Guérin vaccine safely induces significantly enhanced tuberculosis-specific immunity in human volunteers. J Infect Dis 198:1491–1501. doi: 10.1086/592450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, Mizushima NN, Denison MR, Virgin HW IV. 2007. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- 15.Paixão L, Rodrigues L, Couto I, Martins M, Fernandes P, de Carvalho CC, Monteiro GA, Sansonetty F, Amaral L, Viveiros M. 2009. Fluorometric determination of ethidium bromide efflux kinetics in Escherichia coli. J Biol Eng 3:18. doi: 10.1186/1754-1611-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colangeli R, Helb D, Sridharan S, Sun J, Varma-Basil M, Hazbon MH, Harbacheuski R, Megjugorac NJ, Jacobs WR Jr, Holzenburg A, Sacchettini JC, Alland D. 2005. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol Microbiol 55:1829–1840. doi: 10.1111/j.1365-2958.2005.04510.x. [DOI] [PubMed] [Google Scholar]
- 17.Amaral L, Martins M, Viveiros M. 2007. Enhanced killing of intracellular multidrug-resistant Mycobacterium tuberculosis by compounds that affect the activity of efflux pumps. J Antimicrob Chemother 59:1237–1246. doi: 10.1093/jac/dkl500. [DOI] [PubMed] [Google Scholar]
- 18.Stenger S. 2005. Immunological control of tuberculosis: role of tumour necrosis factor and more. Ann Rheum Dis 64(Suppl 4):iv24–iv28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan J, Xing Y, Magliozzo RS, Bloom BR. 1992. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 175:1111–1122. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. 1997. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun 65:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law K, Weiden M, Harkin T, Tchou-Wong K, Chi C, Rom WN. 1996. Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. Am J Respir Crit Care Med 153:799–804. doi: 10.1164/ajrccm.153.2.8564135. [DOI] [PubMed] [Google Scholar]
- 22.Kleinnijenhuis J, Oosting M, Joosten LA, Netea MG, Van Crevel R. 2011. Innate immune recognition of Mycobacterium tuberculosis. Clin Dev Immunol 2011:405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fenton MJ, Vermeulen MW, Kim S, Burdick M, Strieter RM, Kornfeld H. 1997. Induction of gamma interferon production in human alveolar macrophages by Mycobacterium tuberculosis. Infect Immun 65:5149–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradfute SB, Castillo EF, Arko-Mensah J, Chauhan S, Jiang S, Mandell M, Deretic V. 2013. Autophagy as an immune effector against tuberculosis. Curr Opin Microbiol 16:355–365. doi: 10.1016/j.mib.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu X, Li C, Hong W, Pan W, Xie J. 2013. Autophagy during Mycobacterium tuberculosis infection and implications for future tuberculosis medications. Cell Signal 25:1272–1278. doi: 10.1016/j.cellsig.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. 2008. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arentz M, Pavlinac P, Kimerling ME, Horne DJ, Falzon D, Schunemann HJ, Royce S, Dheda K, Walson JL, ART-DR-TB Study Group . 2012. Use of anti-retroviral therapy in tuberculosis patients on second-line anti-TB regimens: a systematic review. PLoS One 7:e47370. doi: 10.1371/journal.pone.0047370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axelsson-Robertson R, Rao M, Loxton AG, Walzl G, Bates M, Zumla A, Maeurer M. 2015. Frequency of Mycobacterium tuberculosis-specific CD8+ T-cells in the course of anti-tuberculosis treatment. Int J Infect Dis 32:23–29. doi: 10.1016/j.ijid.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 29.Di Perri G, Danzi MC, De Checchi G, Pizzighella S, Solbiati M, Cruciani M, Luzzati R, Malena M, Concia E, Bassetti D. 1989. Nosocomial epidemic of active tuberculosis among HIV-infected patients. Lancet ii:1502–1504. [PubMed] [Google Scholar]
- 30.Daley CL, Small PM, Schecter GF, Schoolnik GK, McAdam RA, Jacobs WR Jr, Hopewell PC. 1992. An outbreak of tuberculosis with accelerated progression among persons infected with the human immunodeficiency virus. An analysis using restriction-fragment-length polymorphisms. N Engl J Med 326:231–235. [DOI] [PubMed] [Google Scholar]
- 31.Parida SK, Madansein R, Singh N, Padayatchi N, Master I, Naidu K, Zumla A, Maeurer M. 2015. Cellular therapy in tuberculosis. Int J Infect Dis 32:32–38. doi: 10.1016/j.ijid.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Degasperi GR, Zecchin KG, Borecky J, Cruz-Hofling MA, Castilho RF, Velloso LA, Guimarães F, Vercesi AE. 2006. Verapamil-sensitive Ca2+ channel regulation of Th1-type proliferation of splenic lymphocytes induced by Walker 256 tumor development in rats. Eur J Pharmacol 549:179–184. doi: 10.1016/j.ejphar.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 33.Rodrigues L, Ramos J, Couto I, Amaral L, Viveiros M. 2011. Ethidium bromide transport across Mycobacterium smegmatis cell-wall: correlation with antibiotic resistance. BMC Microbiol 11:35. doi: 10.1186/1471-2180-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freedman SB, Richmond DR, Ashley JJ, Kelly DT. 1981. Verapamil kinetics in normal subjects and patients with coronary artery spasm. Clin Pharmacol Ther 30:644–652. doi: 10.1038/clpt.1981.216. [DOI] [PubMed] [Google Scholar]
- 35.Follath F, Ha HR, Schutz E, Bühler F. 1986. Pharmacokinetics of conventional and slow-release verapamil. Br J Clin Pharmacol 21(Suppl 2):149S–153S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frishman W, Kirsten E, Klein M, Pine M, Johnson SM, Hillis LD, Packer M, Kates R. 1982. Clinical relevance of verapamil plasma levels in stable angina pectoris. Am J Cardiol 50:1180–1184. doi: 10.1016/0002-9149(82)90440-4. [DOI] [PubMed] [Google Scholar]
- 37.Megarbane B, Karyo S, Abidi K, Delhotal-Landes B, Aout M, Sauder P, Baud FJ. 2011. Predictors of mortality in verapamil overdose: usefulness of serum verapamil concentrations. Basic Clin Pharmacol Toxicol 108:385–389. [DOI] [PubMed] [Google Scholar]
- 38.Bruserud O. 1996. In vitro effects of R-verapamil on the cytokine environment and T-lymphocyte proliferation when human T-lymphocyte activation takes place in the presence of acute myelogenous leukemia blasts. Cancer Chemother Pharmacol 39:71–78. doi: 10.1007/s002800050540. [DOI] [PubMed] [Google Scholar]
- 39.Berrebi A, Shtalrid M, Klepfish A, Bassous L, Kushnir M, Shulman L, Vorst E, Hahn T. 1994. Verapamil inhibits B-cell proliferation and tumor necrosis factor release and induces a clinical response in B-cell chronic lymphocytic leukemia. Leukemia 8:2214–2216. [PubMed] [Google Scholar]
- 40.McMillen MA, Lewis T, Jaffe BM, Wait RB. 1985. Verapamil inhibition of lymphocyte proliferation and function in vitro. J Surg Res 39:76–80. doi: 10.1016/0022-4804(85)90164-7. [DOI] [PubMed] [Google Scholar]
- 41.Ludescher C, Pall G, Irschick EU, Gastl G. 1998. Differential activity of P-glycoprotein in normal blood lymphocyte subsets. Br J Haematol 101:722–727. doi: 10.1046/j.1365-2141.1998.00751.x. [DOI] [PubMed] [Google Scholar]
- 42.Park SW, Lomri N, Simeoni LA, Fruehauf JP, Mechetner E. 2003. Analysis of P-glycoprotein-mediated membrane transport in human peripheral blood lymphocytes using the UIC2 shift assay. Cytometry A 53:67–78. [DOI] [PubMed] [Google Scholar]
- 43.Randolph GJ, Beaulieu S, Pope M, Sugawara I, Hoffman L, Steinman RM, Muller WA. 1998. A physiologic function for p-glycoprotein (MDR-1) during the migration of dendritic cells from skin via afferent lymphatic vessels. Proc Natl Acad Sci U S A 95:6924–6929. doi: 10.1073/pnas.95.12.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tainton KM, Smyth MJ, Jackson JT, Tanner JE, Cerruti L, Jane SM, Darcy PK, Johnstone RW. 2004. Mutational analysis of P-glycoprotein: suppression of caspase activation in the absence of ATP-dependent drug efflux. Cell Death Differ 11:1028–1037. doi: 10.1038/sj.cdd.4401440. [DOI] [PubMed] [Google Scholar]
- 45.Andries K, Villellas C, Coeck N, Thys K, Gevers T, Vranckx L, Lounis N, de Jong BC, Koul A. 2014. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9:e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.