

Abstract
The asymmetric outer membrane of Gram-negative bacteria is formed of the inner leaflet with phospholipids and the outer leaflet with lipopolysaccharides (LPS). Outer membrane protein F (OmpF) is a trimeric porin responsible for the passive transport of small molecules across the outer membrane of Escherichia coli. Here, we report the impact of different levels of heterogeneity in LPS environments on the structure and dynamics of OmpF using all-atom molecular dynamics simulations. The simulations provide insight into the flexibility and dynamics of LPS components that are highly dependent on local environments, with lipid A being the most rigid and O-antigen being the most flexible. Increased flexibility of O-antigen polysaccharides is observed in heterogeneous LPS systems, where the adjacent O-antigen repeating units are weakly interacting and thus more dynamic, compared to homogeneous LPS systems in which LPS interacts strongly with each other with limited overall flexibility due to dense packing. The model systems were validated by comparing molecular-level details of interactions between OmpF surface residues and LPS core sugars with experimental data, establishing the importance of LPS core oligosaccharides in shielding OmpF surface epitopes recognized by monoclonal antibodies. There are LPS environmental influences on the movement of bulk ions (K+ and Cl−), but the ion selectivity of OmpF is mainly affected by bulk ion concentration.
Introduction
The outer membrane (OM) of Gram-negative bacteria is an asymmetric bilayer with phospholipids forming the inner leaflet and lipopolysaccharides (LPS) forming the outer leaflet, and various OM proteins (Omps) populate this membrane (1, 2). In Escherichia coli and its relatives, ∼106 LPS molecules per cell cover nearly three-quarters of the OM surface (3). An LPS molecule consists of a lipid A moiety embedded in the OM, linked to the oligosaccharide core sugars and the repeating units of the O-antigen polysaccharide (4, 5). Lipid A and the core oligosaccharide are phylogenetically conserved regions of LPS, whereas the O-antigen is a hypervariable region that determines bacterial antigenic diversity (6, 7). LPS makes the cell surface hydrophilic and acts as a defensive barrier to hydrophobic antibiotics, dyes, and humoral immune responses mediated by antibodies (8, 9).
OmpF porin is one of the major Omps and has cation-selective aqueous pores in the E. coli OM (Figs. 1 and 2 A) (10). Each monomer of its homotrimeric structure contains a 16-stranded β-barrel with eight reverse turns (T1–T8) on the periplasmic side and eight relatively large loops (L1–L8) on the cell surface. OmpF, which is present at >100,000 copies/cell, is a passive channel for translocation of hydrophilic solutes of <600 Da across the OM (11). In the trimeric form, OmpF contains a large inlet vestibule that gives rise to a narrow constriction zone in each monomer, creating the shape of a large funnel with its entrance on the cell surface and three outlets into the periplasm. The constriction zone is lined by two acidic residues (Asp113 and Glu117) and an opposing cluster of three basic residues (Arg42, Arg82, and Arg132) (12, 13). Sites in the surface-exposed loops (epitopes) of OmpF are recognized by antibodies (Fig. 2 A) (14, 15) and also play important roles in the reception and translocation of colicins (bacterial toxins) (16, 17).
Figure 1.

Representative snapshots of an OmpF trimer (barrel, yellow; helix, red; loop and turn, green) embedded in OMs of E. coli rough LPS (EC-lipa, left), K12 core LPS (K12-lps0, center), and R1 core LPS with five repeating units of O6-antigen (R1-lps5, right). Lipid A is represented as pink spheres, core sugars as gray stick model, and O-antigen polysaccharides as orange stick model. The inner leaflet contains PPPE (blue spheres), PVPG (orange spheres), and PVCL2 (magenta spheres); see the main text for the full names of inner-leaflet phospholipids. Ca2+ ions are represented as cyan small spheres, potassium ions as green small spheres, and chloride ions as magenta small spheres. For clarity, some portions of the system have been removed. Snapshots were taken at the ends of simulations (∼300 ns in K12-lps0 and R1-lps5 systems and ∼350 ns in EC-lipa system). To see this figure in color, go online.
Figure 2.

(A) Eight loops of OmpF depicted with the estimated OmpF epitopes for mAbs on L1, L4, and L5. Red and blue spheres are acidic and basic residues in the constriction zone. (B) Sequences of K12 (depicted with different mutations) and R1 LPS cores considered in this study. The inner core consists of rare sugars, 2-keto-3-deoxyoctulosonate (Kdo) and l-glycero-d-manno-heptose (Hep), and the outer core consists mainly of d-glucose (Glc), d-galactose (Gal), and N-acetyl-d-glucosamine (GlcNAc). To see this figure in color, go online.
The bacterial growth environment influences the phenotypic expression of surface characters that may create different protein-LPS and LPS-LPS interactions (9). These different interactions may affect antibody accessibility. For example, based on flow-cytometry immunochemical assays, rfa (deep rough) mutants of E. coli K12 (Fig. 2 B; Fig. S1 in the Supporting Material) that conferred stepwise truncations in the LPS K12 core showed increasing accessibility to different monoclonal antibodies (mAbs) targeting OmpF surface epitopes (Fig. 2 A) (14, 15). Similarly, the permeability of the OM to antibiotics (18) and the binding and translocation of colicins (17, 19) are influenced by bacterial surface determinants. The heterogeneity of O-antigen polysaccharide unit length and the dynamics of their constituent sugars may complicate the accessibility of mAbs to epitopes located either on the cell surface or deep within the LPS leaflet (14, 20). Understanding these protein-LPS interactions at the molecular level is a first step toward explaining how bacteria restrict antibody access or increase antibiotic resistance.
Results of previous experimental and theoretical studies provide insight into the mechanisms of ion and antibiotic permeation through OmpF pores (21, 22, 23). Theoretical models also suggest that the ion selectivity of OmpF is not governed completely by the charged residues at the channel constriction, but by the collective action of a large number of residues along the ion permeation pathway (21, 24). Although previous results from atomic simulation studies of OmpF (mainly in symmetric phospholipid bilayers) are insightful, a critical need exists to explain the role of LPS in shielding OmpF epitopes from antibody recognition, as well as ion permeation and selectivity in the asymmetric OM environment. In this study, molecular dynamics (MD) simulations of OmpF in different asymmetric OMs (Fig. 1; Table 1; Table S1) 1) elucidate the impact of different LPS molecules on the structure and dynamics of OmpF, and 2) determine the importance of protein-LPS interactions in mAb accessibility and ion permeability in different LPS environments.
Table 1.
E. coli OM Systems and Their Outer-Leaflet Composition
| System | Composition of Outer Leaflet |
|---|---|
| EC-lipa | lipid A |
| K12-lps0 | EC-lipa + K12 core oligosaccharides |
| R1-lps0 | EC-lipa + R1 core oligosaccharides |
| R1-lps5 | R1-lps0 + five repeating unit of O6-antigen |
| R1-lipa-lps5 | mixed LPS with EC-lipa and R1-lps5 |
| R1-lps0-lps5 | mixed LPS with R1-lps0 and R1-lps5 |
| R1-lipa-lps0-lps5 | mixed LPS with EC-lipa, R1-lps0, and R1-lps5 |
Materials and Methods
System setup
The OmpF trimer structure (PDB: 2OMF) was embedded in various asymmetric bilayers mimicking E. coli OMs (Fig. 1). Building and assembly of OmpF/OM systems were achieved by the step-by-step protocol developed by Im and co-workers (25, 26, 27) based on the procedure in CHARMM-GUI Membrane Builder (28, 29, 30, 31). The phospholipid composition of the inner leaflet of the E. coli OM is reported to be similar to that of the inner (cytoplasmic) membrane, comprised of 75–85% phosphatidylethanolamine, 10–20% phosphatidylglycerol, and 5–15% cardiolipin (32, 33). Based on these reports, we chose a ratio of 15:4:1 of 1-palmitoyl(16:0)-2-palmitoleoyl(16:1 cis-9)-phosphatidylethanolamine (PPPE, 75%)/1-palmitoyl(16:0)-2-vacenoyl(18:1 cis-11)-phosphatidylglycerol (PVPG, 20%)/1,10-palmitoyl-2,20-vacenoyl cardiolipin with a net charge of –2e (PVCL2, 5%). The outer leaflets vary in composition, and different OM systems are named based on the outer-leaflet composition (Table 1). The system size and composition are summarized in Table S1. As shown in Fig. 1, the complexity increases with involvement of oligosaccharide cores and O-antigen polysaccharides (E. coli O6) compared to a rough LPS system (i.e., EC-lipa). As a reference, we also built and simulated an OmpF system in a symmetric bilayer with 1,2-dimyristoyl(14:0)-phosphatidylcholine (DMPC).
MD simulations
All membrane systems were equilibrated for 450 ps using CHARMM (34) with the C36 force field for lipids (35) and carbohydrates (36, 37, 38) and the TIP3P water model (39). We used a 2-fs time step together with the SHAKE algorithm (40). NVT (constant particle number, volume, and temperature) and NPT (constant particle number, pressure, and temperature) CHARMM equilibrations were followed by a 300- to 350-ns NPT production run for all systems using NAMD (41) with the NAMD input scripts generated by CHARMM-GUI (42). The van der Waals interactions were smoothly switched off at 10–12 Å by a force-switching function (43), whereas the long-range electrostatic interactions were calculated using the particle mesh Ewald method (44). The temperature and pressure were held at 310.15 K and 1 bar, respectively. In CHARMM equilibration simulations, Langevin temperature control was used for NVT dynamics, and a Hoover thermostat (45) and Langevin piston were used to control temperature and pressure for NPT dynamics (46, 47). In the NAMD production run, Langevin dynamics was used to maintain constant temperature with a Langevin coupling coefficient of 1 ps−1, and a Nosé-Hoover Langevin piston (48, 49) was used to maintain constant pressure with a piston period of 50 fs and a piston decay time of 25 fs. The assembled systems were equilibrated by the well-established protocol in Membrane Builder, in which various planar and dihedral restraints were applied to the LPS molecules, phospholipids, and water molecules, and the restraint forces were gradually reduced during this process (see (25, 26, 27) for details). Additional dihedral angle restraints were applied to restrain all sugar rings to the pertinent chair conformation, and these restraints were maintained during the production simulations. The last-200-ns trajectory of each system was analyzed to obtain average structural properties. The profiles of the diffusion constants of K+ and Cl– along the channel axis were calculated using , where , and was set to 10 ps (23).
Results and Discussion
OmpF structure and dynamics in the E. coli OM
The structural stability of OmpF in different LPS environments was measured in terms of the root mean-square deviations (RMSDs) of main structural elements—backbone, side chain, barrel, loop, and turn atoms—with respect to the crystal structure (Table S2; Fig. S2). The entire-backbone RMSDs are ∼2 Å, indicating that OmpF has no significant difference in structural stability in all systems. The transmembrane β-barrel shows smaller RMSDs compared to those in the extracellular loops (L1–L8) and periplasmic turns (T1–T8). Consistent with the RMSD results, there are smaller RMS fluctuations (RMSFs) in the β-barrel residues (Fig. S3), but residues in the extracellular loops show more fluctuations and a slight dependency on the surrounding environment. Compared to the DMPC system, where the OmpF extracellular loops show large fluctuations, some extracellular loops (e.g., L7 and L8) are less flexible in the homogeneous LPS systems, where protein-LPS interactions are tight (mainly due to well maintained salt-bridge interactions of negatively charged and COO– groups from Hep and Kdo inner-core sugars with the Lys and Arg residues that are prominently located on the outer surface of OmpF (Fig. S4)). In addition to OmpF-LPS interactions that are dynamic in nature, intraprotein (within a monomer) and interprotein (monomer-monomer) interactions also contribute to the overall structure and dynamics of the loops in all the systems, including the DMPC system. For example, as is evident from the crystal trimeric structure (11), some residues in loop L2 form strong polar interactions with several residues in loops L3 and L4 of the neighboring monomer. Relative stabilization of both L7 and L8 loops was observed with inclusion of LPS environments in the homogeneous systems. For example, the RMSF peak in loop L7 residue Gly285 in DMPC is 2.13 Å, whereas the values are ∼1.5 Å for the same residue in K12-lps0 and R1-lps0 systems. This is also evident from the interaction pattern analysis showing LPS core sugars interacting with L7 loop residues (vide infra). Similarly, for loop L8, the average RMSF for residues 320–325 is 1.15 Å in DMPC but decreases to ∼0.8 Å in the K12-lps0 and R1-lps0 systems. Even for loop L6, interactions with LPS core sugars partially stabilize residues 243–245, whose average RMSF in DMPC is 2.5 Å (with the peak at 2.81 Å), whereas those in K12-lps0 and R1-lps0 systems are ∼1.5 Å (with the peak at ∼1.6 Å). For similar reasons, the extracellular loops in the homogeneous LPS systems show lesser fluctuation than those in the EC-lipa and heterogeneous mixed LPS systems.
Although the overall RMSFs from the MD simulations and those estimated from the crystallographic B-factors are in good agreement (Fig. S3), some discrepancies are observed, such as larger fluctuations in loop L3 near the constriction zone. The short polypeptide segment 117–121 (EFGGD) in L3 is more flexible in simulations and thus shows deviations relative to the crystallographic structure (Fig. S5). These discrepancies may arise from a different choice of protonation states of Glu296, Asp312, and Asp127, which can lead to restricted motion of L3. In our study, we used protonated Glu296 and Asp312 and charged Asp127 (23), but Varma et al. reported a low RMSF of the L3 loop when they used protonated Glu296 and charged Asp127 and Asp312 for their 10-ns MD simulation (50). Nonetheless, fluctuation in this part of loop L3 was reported to be responsible for changes in the pore size (11).
Dynamics and flexibility of LPS in the E. coli OM
To examine dynamic behaviors of LPS that may complicate accessibility to epitopes (14, 20), we calculated the 2D-density profiles of the centers of mass of LPS molecules (lipid A and core sugars) in the outer leaflet and those of phospholipids in the inner leaflet in the R1-lps5 system (Fig. 3). Within the simulation timescale (∼300 ns), LPS molecules are highly immobile and show little translocation, whereas fast diffusions of phospholipids make the phospholipid density uniform in the inner leaflet, which is similar in other systems and consistent with previous computational studies (26, 51). The rigidity and low mobility of lipid A and core sugars are mainly attributed to the divalent (Ca2+) ion-mediated cross-linking electrostatic interaction networks with negatively charged and COO– groups in the lipid A and core regions. Such immobility may be characteristic for LPS in the OM, as many experiments suggest rigidity in LPS mobility (25, 52). In fact, it has been suggested that divalent cations bridging with LPS are responsible for the strong barrier for hydrophobic molecules in most Gram-negative bacteria (9, 53).
Figure 3.

2D number density plots of the centers of mass of the outer-leaflet molecules (lipid A and core sugars) and the inner-leaflet phospholipids (PPPE, PVPE, and PVCL2) in the R1-lps5 system. The 2D plots were constructed with a grid spacing of 0.5 Å. To see this figure in color, go online.
In contrast to lipid A and core oligosaccharides, O-antigen polysaccharide units are very flexible, as shown in Figs. 4 and S6. In general, the average flexibility of O-antigen repeating units sequentially increases from antigen1 (attached to the outer core) to antigen5, where the width of conformational space increases along the z axis. The flexibility of O-antigen repeating units is dependent on its local environment and its concentration. In R1-lps5, the O-antigen repeating units, which are farther from the gap created by OmpF, are densely packed along the z axis and strongly interact with each other. However, the O-antigen repeating units closer to this gap (i.e., above OmpF) have weaker interactions and thus tilt or bend. This is evident in the broader conformational distributions of some representative O-antigens, as shown in Fig. S7. In mixed LPS systems, the weakly interacting O-antigen repeating units lead to broader conformational distributions. Our previous simulations suggest that the glycosidic torsion angles at the disaccharide level in each O-antigen repeating unit are quite well defined and maintain similar values in homogeneous and heterogeneous LPS bilayer systems (25, 26). A collective change in a large number of glycosidic torsion angles of the O-antigen region, resulting in bending, twisting, and/or tilting, are thus responsible for the increased O-antigen flexibility in going from antigen1 to antigen5, as well as in densely packed versus mixed LPS environments. Such O-antigen flexibility may hinder direct access of mAbs to OmpF surface epitopes.
Figure 4.

Flexibility of the O-antigens. 2D scatter plots of the centers of mass of core and O-antigen repeating units along the z axis and the distance from the center of mass of the OmpF trimer in the R1-lps5 system. The bilayer is recentered to z = 0. To see this figure in color, go online.
Protein-LPS interactions and accessibility of OmpF surface epitopes
Experimental data (Fig. S1; Table 2) indicate that stepwise truncations of the K12 core increase the number of different mAbs that bind to OmpF; i.e., these epitope sites become more accessible to mAbs by removal of core sugars that interact with the sites and thus block mAb binding. To explore the molecular details of OmpF’s surface availability to mAbs, we characterized the interaction patterns of each OmpF residue with each component of lipid molecules, water, or residues of adjacent OmpF monomers. For this analysis, we used a distance cutoff of 5 Å to define an interaction between any heavy atom of each OmpF residue and that of each environmental moiety. The average result of the OmpF trimer in K12-lps0 system is shown in Fig. 5 and those in other systems are given in Fig. S8, A–G. For all LPS systems, the interaction analysis shows three main characteristics: 1) the transmembrane β-strands mainly interact with lipid A, the phospholipid tail, or adjacent protein monomers; 2) the extracellular loops prefer to interact with LPS core oligosaccharides and water, except for loop L2, whose residues also participate in interactions with neighboring monomers; and 3) the turns mainly interact with phospholipid headgroups and water. Table 2 summarizes the comparison of the interaction patterns in the K12-lps0 system with experimental binding activities of three mAb groups (S1, S2, and S3c). Both the experiment and MD simulations show the importance of LPS core sugars in shielding OmpF surface epitopes recognized by various mAbs (14, 15).
Table 2.
Experimental Binding Activities of mAbs Compared to Overall Interaction Patterns of LPS to Epitope Sites in the K12-lps0 System
| Group | mAb | Epitope-Site Residues (Loop) | Experimental Analysis | MD Analysis |
|---|---|---|---|---|
| S1 | 2, 10, 12, 14 | 160–172 (L4) | high binding activities in both wild-type and mutant E. coli K12 strains | epitope-site residues mainly interact with water and not with LPS sugars; some residues also interact with loop L2 of neighboring monomers |
| S2 | 15, 8, 4, 5, 9, 69 | 195–212 (L5) | sequentially increasing activities in wild-type to Re LPS | epitope-site residues interact with both inner and outer core sugars, and some residues are fully exposed to water |
| S3c | 60 | 25–31 (L1) | moderate activities for Rb3 to Rd1 LPS but high activity for truncated Re LPS | epitope-site residues interact with inner- and outer-core sugars, and some are fully exposed to water |
Figure 5.

Interaction patterns of OmpF protein residues with their surrounding environments in K12-lps0. The graph shows, for each residue, the frequency of interactions with another monomer (gray), water molecules (blue), a phospholipid headgroup (yellow), a phospholipid carbon tail (green), a lipid A tail (dark green), a lipid A headgroup (orange), the LPS inner core (cyan), or the LPS outer core (red). An interaction is first counted when the distance between any heavy atom of a residue and that of its interacting partner is <5 Å and is normalized for each interacting partner. The bar below each set of patterns indicates the protein secondary structure. To see this figure in color, go online.
Most residues in loop L4 interact with water molecules instead of LPS sugars (Fig. 5), indicating that this epitope site is available for easy access to group S1 mAbs, which agrees with the experimental data. Certain residues in L4 also interact with loop L2 in adjacent monomers. These interprotein interactions may maintain the functional conformation of the OmpF trimer to which group S1 mAbs bind, which is also evident from the immunological assays, showing weak interactions of these antibodies with monomeric OmpF (15). Different interaction patterns of L5 residues suggest accessibility variations in these residues as targets for group S2 mAbs. These data support the argument that the reported mAb activity differences are attributed to binding of S2 mAbs to different epitope sites on L5 (Fig. 2; Fig. S1). For example, mAb 15 showing full activity against wild-type K12 may bind to L5 residues with full exposure to water, whereas mAb 69 may bind to residues that interact with the deep inner-core sugar residues, as mAb 69 only showed full activity when the LPS core was truncated to the Re level. Similarly, variable interactions of L1 residues with inner- and outer-core sugars (Fig. 5) may explain the distinct level of mAb 60 activities (S3c group): no binding to Rb2 core, low activity in Rb3 to Rd1 core, and full activity with K12 truncated to the Re level (Fig. 2; Fig. S1). In general, the predicted interaction patterns between epitope residues and E. coli K12 core sugars confirm the experimental findings, indicating a sequential increase in the number of mAbs that can access the epitope sites with truncation of the LPS core length.
The interaction patterns in EC-lipa and DMPC systems (Fig. S8, A and B) show complete exposure of epitope-site residues to water, and these sites are always available for mAbs binding. This clearly indicates a role of LPS core sugars in restricting mAb binding; it also suggests a key limitation of these model systems, which shows a lack of influence of protein-LPS interactions on dynamics, permeability, and binding of antibiotics or colicin on the OmpF surface.
The interaction pattern analysis also suggests that mAb binding to surface epitopes in E. coli R1 core environments (Fig. S8 C) is similar to that in E. coli K12 environments (Fig. 5; Fig. S9). Loop L1 Asn30 shows similar H-bonding interactions with the outer core sugars, i.e., α-Glc in the K12-lps0 system (Fig. S9 A) and terminal α-Gal and α/β-Glc in the R1-lps0 system (Fig. S9 C). Similarly, loop L5 Lys209 interacts with the inner-core α-Hep via H-bonding in both systems (Fig. S9, B and D). The two systems also show minor differences (mainly due to outer-core sugars); notably, H-bonding interactions of the K12 outer-core α-Gal with L1 Gly28 and L5 Asn207 (Fig. S9, A and B) are missing in the R1-lps0 system (Fig. S9, C and D). The experimental data show that truncation of α-Gal and α-Hep in the K12 core (Rc mutation) facilitates access of mAbs 5 and 9 to epitope sites on loop L5 (Fig. 2; Fig. S1). Therefore, both mAbs may show different activities in the R1 core, although there is no experimental evidence for specific binding sites, nor are there data for cross reactivity with other cores, for any of the mAbs discussed in this study.
The core sugar residues in the R1-lps5 system (Fig. S8 D) are also found to maintain interaction patterns similar to those in the R1-lps0 system. As expected, an increase in the hydrophilic layer of O-antigen polysaccharides creates more steric hindrance to mAb accessibility to the epitope sites. Previous experimental studies also show that the presence of O-antigen polysaccharides restricts adsorption of mAbs to protein surface epitopes (14, 20). Importantly, in the R1-lps5 system, O-antigen units closer to OmpF are highly flexible and occupy the gap created by OmpF in the LPS layer. In other words, the O-antigen units interact directly with the surface epitopes and provide a denser hydrophilic environment above the OmpF vestibule (Fig. 6; Fig. S8 D and Movie S1). This further ensures strong steric and electrostatic hindrance to mAbs.
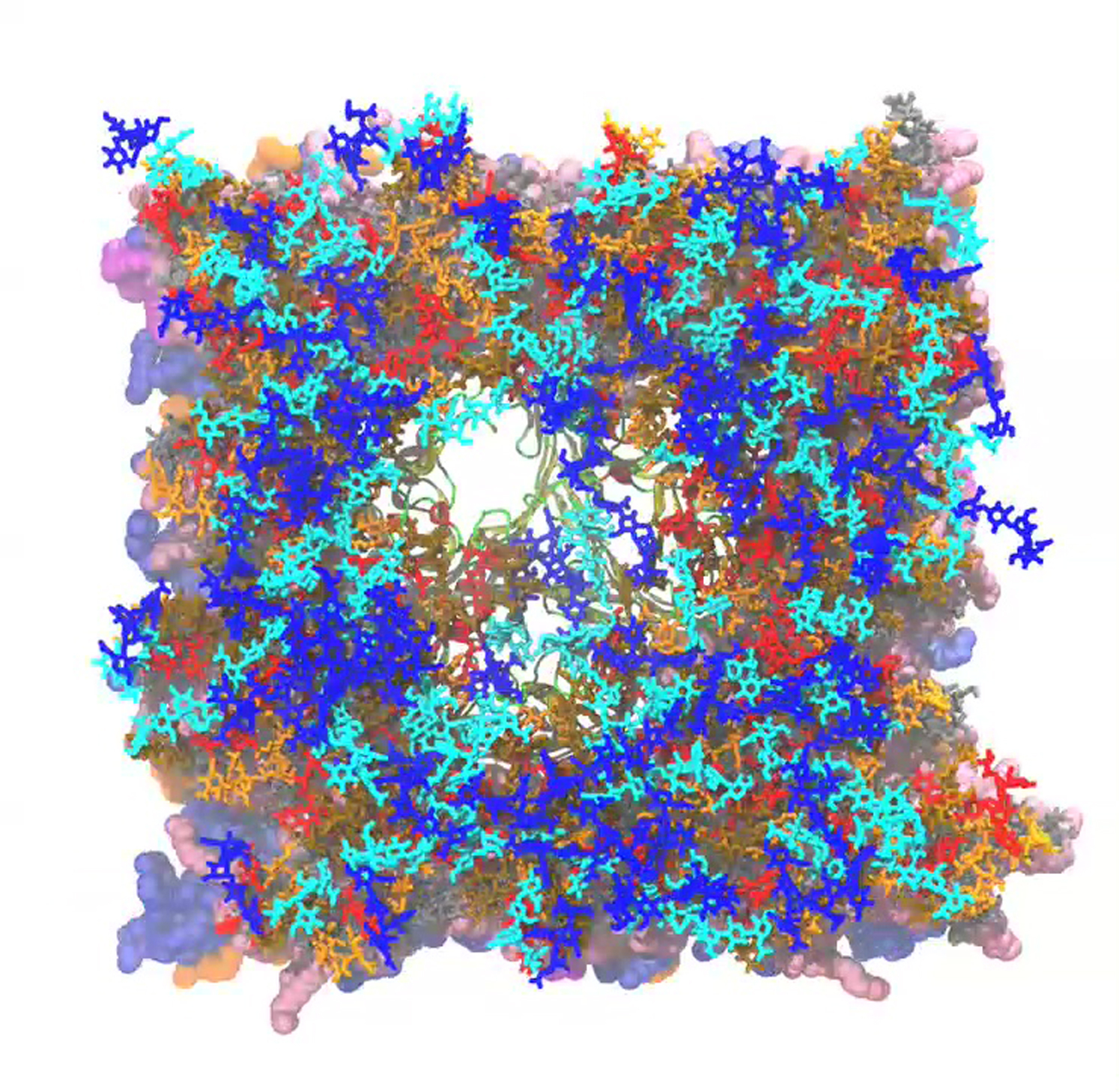
Figure 6.

Snapshots (top views) of O-antigen polysaccharides in R1-lps5, R1-lipa-lps5, R1-lps0-lps5, and R1-lipa-lps0-lps5 systems with inner core (gray), antigen1 (orange), antigen2 (red), antigen3 (ochre), antigen4 (cyan), and antigen5 (blue) stick-model representation. For clarity, ions and water molecules are not shown. Snapshots were taken at the ends of simulations (∼300 ns). To see this figure in color, go online.
For mixed-LPS systems, interactions between epitope sites and LPS (Fig. S8, E–G) are dependent on surrounding LPS molecules and their conformational flexibility to cover the protein surface. Because of the significantly slow translation and rotation of the LPS molecules, as well as limited simulation time, these interaction patterns could depend on the initial placement and the type of LPS composition surrounding the OmpF. In addition, the mixed LPS systems described here certainly do not represent all possible LPS heterogeneities on the OM, in which there are often more than five O-antigen units. In our models, the OmpF vestibule is less covered by O-antigen units in mixed-LPS model systems compared to the R1-lps5 system (Fig. 6). This less dense environment of O-antigen polysaccharides above OmpF pores may be exploited by the antibacterial toxin colicin-N. Such situations provide support for the hypothetical model of colicin-N binding to core sugars envisaged by Johnson et al. (19), because the gap in the mixed-LPS layer may expose the required LPS binding site (inner-core α-Hep and first α-Glc of the outer core) of colicin-N at the protein-LPS interface.
Ion permeation and selectivity of OmpF in the OM
The average numbers of K+ and Cl– ions in each pore and their occupancy ratio (NK/NCl) are given in Table S3 for all systems. The ion distribution inside each pore is similar to those found in earlier OmpF simulation studies, such as slight cation selectivity and a screw-like separation of K+ and Cl– ion permeation pathways (Fig. S10 A) that extend over the height of the β-barrel (21, 22, 23, 54, 55, 56). A larger number of ions is found on the periplasmic side than on the extracellular side because of the asymmetric pore shape.
We note that NK and NCl are slightly different in all systems (Table S3) because of the redistribution of ions initially placed above the LPS layer, leading to different effective bulk KCl concentrations ([KCl]eff). Notably, as shown in Fig. S10, B and C, NK and NCl are linearly correlated with [KCl]eff, suggesting that if all systems were at the identical [KCl]eff, NK and NCl would not be varied, and that LPS contributes very little to NK and NCl. Assuming that NK/NCl in Table S3 is representative of the channel selectivity at a given concentration, this ratio also confirms OmpF’s preferred selectivity for K+ over Cl– in all systems. In addition, the dependence of NK/NCl on [KCl]eff is clearly reflected. For example, in R1-lps5 system (where [KCl]eff is estimated to be 1 M), NK/NCl is 1.36, whereas in R1- lipa-lps0-lps5 (where [KCl]eff is estimated to be 0.35 M), NK/NCl is 2.18. Such concentration dependency of OmpF’s ion selectivity was also observed in previous experimental and computational studies, indicating that OmpF cation selectivity increases as salt concentration decreases, due to less ionic screening of the electrostatic field of OmpF (22).
Consistent with a previously observed trend, the diffusion constants of K+ and Cl– (Fig. S11, A and B) are decreased by >50% in the pore relative to their values in bulk solvent (23). The restricted ion movement in the pores is attributed to the limited hydrodynamic ion permeation pathway, as documented by previous studies (21, 23, 57). It was noteworthy that systems with O-antigen polysaccharides, such as R1-lps5, show further-restricted ion movement in the outer leaflet compared to the inner leaflet. This likely arises from increased hindrance to the ion permeation due to molecular crowding of LPS, as observed in pure-LPS-bilayer simulations (25). On the other hand, less pronounced molecular crowding of O-antigen units may allow less restricted movement of ions in heterogeneous mixed-LPS systems. Therefore, LPS does affect ion (potentially other solutes') diffusion into the OmpF pore, but not the fundamental selectivity (or solute permeation, although LPS O-antigen sugars can shield the OmpF pore (Fig. 6)).
Conclusions
We investigated the influence of different LPS environments on the structure and dynamics of OmpF porin, accessibility of mAbs, and ion permeability and selectivity using all-atom MD simulations. In consideration of the flexibility and dynamics of LPS components, lipid A (headgroup) is the most rigid and O-antigen is the most flexible. In the heterogeneous systems, where the adjacent O-antigen molecules have weak interactions, they show more flexibility and dynamics, whereas the flexibility is greatly reduced for the closely packed O-antigen molecules in homogeneous systems. Consistent with the experimental data, calculated interaction patterns between OmpF residues and LPS K12 core sugars show the importance of LPS core sugars in shielding OmpF surface epitopes recognized by mAbs. With inclusion of O-antigen polysaccharides, access to epitope sites by mAbs is further occluded. Heterogeneous mixed-LPS compositions, which are considered to be more biologically relevant, show that interactions between LPS and epitope sites are dependent on surrounding lipid molecules and their conformational flexibility to cover the protein surface. In addition, interaction patterns in EC-lipa and DMPC systems indicate a key limitation to incorporating genuine protein-LPS (core and O-antigen sugars) interactions, which in turn cannot capture the influence of such interactions on dynamics, permeability, and binding of mAbs, antibiotics, and colicin on the OmpF surface. Ion (K+ and Cl–) movements in the extracellular vestibule are restricted (slower diffusion) due to LPS crowding, but the cation selectivity (NK/NCl) is mainly affected by the bulk concentration of ions.
In summary, bacteria produce OM porins to facilitate passive nutrient uptake. Porin surface epitopes are antigenic and hence potentially disadvantageous in the host as a result of immune recognition. However, LPS sugars sterically hinder antibody access to OM protein epitopes, allowing bacterial invasion into eukaryotic hosts. This point was already well known 25 years ago from experiments on the impact of LPS O-antigen and core structure on the binding of anti-OmpF mAbs (14). Our results computationally explain the ability of LPS sugars to interact with specific regions of the OmpF polypeptide in a way that camouflages and shields its epitopes from immune recognition without compromising OmpF channel activity. Therefore, the findings reported herein provide computational synergy with existing experimental data, explaining in molecular terms how and why LPS shields porin epitopes.
Author Contributions
D.S.P. and W.I. conceived the study and wrote the manuscript; D.S.P., S.R., E.L.W., and Y.Q. performed the research; D.S.P., S.R., E.L.W., Y.Q., Y.S., P.E.K., G.W., M.S.Y., and W.I. analyzed the data; All authors contributed to editing the manuscript.
Acknowledgments
This work was supported in part by grants from the National Science Foundation (MCB-1516154, DBI-1145987, NSF IIA-1359530, and XSEDE MCB070009 to W.I. and MCB0952299 to P.E.K.), the Human Frontier Science Program (HFSP RGP0064/2011 to W.I.), the National Institute of Supercomputing and Networking/Korea Institute of Science and Technology Information, with supercomputing resources including technical support (KSC-2015-C3-004 to M.S.Y.), the Swedish Research Council and Stockholm Center for Biomembrane Research/Swedish Foundation for Strategic Research (to G.W.), and the National Institutes of Health (GM53836 and AI115187 to P.E.K.).
Editor: Scott Feller.
Footnotes
Eleven figures, three tables, and one movie are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)00008-4.
Supporting Material
Movie (top view) of O-antigen polysaccharides in R1-lps5 system with inner core (gray), antigen1 (orange), antigen2 (red), antigen3 (ochre), antigen4 (cyan), and antigen5 (blue) stick model representation. For clarity, ions and water molecules are not shown. The movie represents flexibility of O-anitgens above the OmpF vestibule from the beginning to the end of simulations (∼300 ns).
{kind=link}
References
- 1.Ruiz N., Kahne D., Silhavy T.J. Advances in understanding bacterial outer-membrane biogenesis. Nat. Rev. Microbiol. 2006;4:57–66. doi: 10.1038/nrmicro1322. [DOI] [PubMed] [Google Scholar]
- 2.Bos M.P., Robert V., Tommassen J. Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol. 2007;61:191–214. doi: 10.1146/annurev.micro.61.080706.093245. [DOI] [PubMed] [Google Scholar]
- 3.Rietschel E.T., Kirikae T., Brade H. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 4.Raetz C.R.H., Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erridge C., Bennett-Guerrero E., Poxton I.R. Structure and function of lipopolysaccharides. Microbes Infect. 2002;4:837–851. doi: 10.1016/s1286-4579(02)01604-0. [DOI] [PubMed] [Google Scholar]
- 6.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 7.Wang X., Quinn P.J. Lipopolysaccharide: biosynthetic pathway and structure modification. Prog. Lipid Res. 2010;49:97–107. doi: 10.1016/j.plipres.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Hancock R.E.W. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 1984;38:237–264. doi: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- 9.Nikaido H., Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhakshnamoorthy B., Raychaudhury S., Roux B. Cation-selective pathway of OmpF porin revealed by anomalous x-ray diffraction. J. Mol. Biol. 2010;396:293–300. doi: 10.1016/j.jmb.2009.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowan S.W., Garavito R.M., Schirmer T. The structure of OmpF porin in a tetragonal crystal form. Structure. 1995;3:1041–1050. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- 12.Phale P.S., Schirmer T., Rosenbusch J.P. Voltage gating of Escherichia coli porin channels: role of the constriction loop. Proc. Natl. Acad. Sci. USA. 1997;94:6741–6745. doi: 10.1073/pnas.94.13.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phale P.S., Philippsen A., Schirmer T. Role of charged residues at the OmpF porin channel constriction probed by mutagenesis and simulation. Biochemistry. 2001;40:6319–6325. doi: 10.1021/bi010046k. [DOI] [PubMed] [Google Scholar]
- 14.Bentley A.T., Klebba P.E. Effect of lipopolysaccharide structure on reactivity of antiporin monoclonal antibodies with the bacterial cell surface. J. Bacteriol. 1988;170:1063–1068. doi: 10.1128/jb.170.3.1063-1068.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klebba P.E., Benson S.A., Nikaido H. Determinants of OmpF porin antigenicity and structure. J. Biol. Chem. 1990;265:6800–6810. [PubMed] [Google Scholar]
- 16.Fourel D., Mizushima S., Pagès J.M. Specific regions of Escherichia coli OmpF protein involved in antigenic and colicin receptor sites and in stable trimerization. J. Bacteriol. 1993;175:2754–2757. doi: 10.1128/jb.175.9.2754-2757.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakes K.S. Daring to be different: colicin N finds another way. Mol. Microbiol. 2014;92:435–439. doi: 10.1111/mmi.12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delcour A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson C.L., Ridley H., Lakey J.H. The antibacterial toxin colicin N binds to the inner core of lipopolysaccharide and close to its translocator protein. Mol. Microbiol. 2014;92:440–452. doi: 10.1111/mmi.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van der Ley P., Kuipers O., Lugtenberg B. O-antigenic chains of lipopolysaccharide prevent binding of antibody molecules to an outer membrane pore protein in Enterobacteriaceae. Microb. Pathog. 1986;1:43–49. doi: 10.1016/0882-4010(86)90030-6. [DOI] [PubMed] [Google Scholar]
- 21.Dhakshnamoorthy B., Ziervogel B.K., Roux B. A structural study of ion permeation in OmpF porin from anomalous x-ray diffraction and molecular dynamics simulations. J. Am. Chem. Soc. 2013;135:16561–16568. doi: 10.1021/ja407783a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Im W., Roux B. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 2002;322:851–869. doi: 10.1016/s0022-2836(02)00778-7. [DOI] [PubMed] [Google Scholar]
- 23.Im W., Roux B. Ions and counterions in a biological channel: a molecular dynamics simulation of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution. J. Mol. Biol. 2002;319:1177–1197. doi: 10.1016/S0022-2836(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 24.Aguilella V.M., Queralt-Martín M., Alcaraz A. Insights on the permeability of wide protein channels: measurement and interpretation of ion selectivity. Integr. Biol. (Camb). 2011;3:159–172. doi: 10.1039/c0ib00048e. [DOI] [PubMed] [Google Scholar]
- 25.Wu E.L., Engström O., Im W. Molecular dynamics and NMR spectroscopy studies of E. coli lipopolysaccharide structure and dynamics. Biophys. J. 2013;105:1444–1455. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu E.L., Fleming P.J., Im W. E. coli outer membrane and interactions with OmpLA. Biophys. J. 2014;106:2493–2502. doi: 10.1016/j.bpj.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo S., Wu E.L., Im W. Lipopolysaccharide membrane building and simulation. Methods Mol. Biol. 2015;1273:391–406. doi: 10.1007/978-1-4939-2343-4_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jo S., Kim T., Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jo S., Kim T., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 30.Jo S., Lim J.B., Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu E.L., Cheng X., Im W. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014;35:1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raetz C.R.H. Enzymology, genetics, and regulation of membrane phospholipid synthesis in Escherichia coli. Microbiol. Rev. 1978;42:614–659. doi: 10.1128/mr.42.3.614-659.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vance D.E., Vance J.E. Elsevier; Amsterdam: 2002. Biochemistry of Lipids, Lipoproteins, and Membranes. [Google Scholar]
- 34.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guvench O., Hatcher E.R., Mackerell A.D. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guvench O., Mallajosyula S.S., Mackerell A.D., Jr. CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput. 2011;7:3162–3180. doi: 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guvench O., Greene S.N., Mackerell A.D., Jr. Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 2008;29:2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 40.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of cartesian equations of motion of a system with constraints - molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 41.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J., Cheng X., Im W. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016;12:405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinbach P.J., Brooks B.R. New spherical cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994;15:667–683. [Google Scholar]
- 44.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 45.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 46.Andersen H.C. Molecular dynamics simulations at constant pressure and-or temperature. J. Chem. Phys. 1980;72:2384–2393. [Google Scholar]
- 47.Nosé S., Klein M.L. A study of solid and liquid carbon tetrafluoride using the constant pressure molecular dynamics technique. J. Chem. Phys. 1983;78:6928–6939. [Google Scholar]
- 48.Feller S.E., Zhang Y.H., Brooks B.R. Constant pressure molecular dynamics simulation - The langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 49.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 50.Varma S., Chiu S.W., Jakobsson E. The influence of amino acid protonation states on molecular dynamics simulations of the bacterial porin OmpF. Biophys. J. 2006;90:112–123. doi: 10.1529/biophysj.105.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piggot T.J., Holdbrook D.A., Khalid S. Electroporation of the E. coli and S. Aureus membranes: molecular dynamics simulations of complex bacterial membranes. J. Phys. Chem. B. 2011;115:13381–13388. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- 52.Kastowsky M., Gutberlet T., Bradaczek H. Molecular modelling of the three-dimensional structure and conformational flexibility of bacterial lipopolysaccharide. J. Bacteriol. 1992;174:4798–4806. doi: 10.1128/jb.174.14.4798-4806.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikaido H. Role of permeability barriers in resistance to β-lactam antibiotics. Pharmacol. Ther. 1985;27:197–231. doi: 10.1016/0163-7258(85)90069-5. [DOI] [PubMed] [Google Scholar]
- 54.Pezeshki S., Chimerel C., Kleinekathöfer U. Understanding ion conductance on a molecular level: an all-atom modeling of the bacterial porin OmpF. Biophys. J. 2009;97:1898–1906. doi: 10.1016/j.bpj.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biró I., Pezeshki S., Kleinekathöfer U. Comparing the temperature-dependent conductance of the two structurally similar E. coli porins OmpC and OmpF. Biophys. J. 2010;98:1830–1839. doi: 10.1016/j.bpj.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chimerel C., Movileanu L., Kleinekathöfer U. Transport at the nanoscale: temperature dependence of ion conductance. Eur. Biophys. J. 2008;38:121–125. doi: 10.1007/s00249-008-0366-0. [DOI] [PubMed] [Google Scholar]
- 57.Lee K.I., Jo S., Im W. Web interface for Brownian dynamics simulation of ion transport and its applications to β-barrel pores. J. Comput. Chem. 2012;33:331–339. doi: 10.1002/jcc.21952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie (top view) of O-antigen polysaccharides in R1-lps5 system with inner core (gray), antigen1 (orange), antigen2 (red), antigen3 (ochre), antigen4 (cyan), and antigen5 (blue) stick model representation. For clarity, ions and water molecules are not shown. The movie represents flexibility of O-anitgens above the OmpF vestibule from the beginning to the end of simulations (∼300 ns).