

Abstract
Ionotropic glutamate receptors are postsynaptic tetrameric ligand-gated channels whose activity mediates fast excitatory transmission. Glutamate binding to clamshell-shaped ligand binding domains (LBDs) triggers opening of the integral ion channel, but how the four LBDs orchestrate receptor activation is unknown. Here, we present a high-resolution x-ray crystal structure displaying two tetrameric LBD arrangements fully bound to glutamate. Using a series of engineered metal ion trapping mutants, we showed that the more compact of the two assemblies corresponds to an arrangement populated during activation of full-length receptors. State-dependent cross-linking of the mutants identified zinc bridges between the canonical active LBD dimers that formed when the tetramer was either fully or partially bound by glutamate. These bridges also stabilized the resting state, consistent with the recently published full-length apo structure. Our results provide insight into the activation mechanism of glutamate receptors and the complex conformational space that the LBD layer can sample.
Introduction
Ionotropic glutamate receptors (iGluRs) are essential for neurotransmission at excitatory synapses in brain. Upon binding of glutamate released from presynaptic terminals, they open their integral ion channels, thus coupling a chemical signal to a brief transmembrane ionic current. This mechanism allows neurons to excite their postsynaptic targets with the high temporal precision pivotal to many cognitive processes in the central nervous system.
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor was the first iGluR for which an almost full-length crystal structure was determined: a homotetramer in complex with an antagonist and thus in a resting (closed channel) state (1). More recent structures of both the AMPA and related N-methyl-D-aspartate receptors with full and partial agonists bound displayed again a closed channel (2, 3, 4, 5, 6). Each GluA2 subunit consists of an amino terminal domain (ATD), a ligand binding domain (LBD), a transmembrane region harboring the ion channel, and an intracellular C-terminal domain (Fig. 1 A). Although the transmembrane region of GluA2 maintains fourfold symmetry akin to K+ channels (7), the ATDs and LBDs are arranged as pairs of dimers related to each other by twofold pseudosymmetry, resulting in the overall Y shape of the receptor (Fig. 1 A).
Figure 1.

Crystal structures of tetrameric GluA2 LBDs in complex with agonists or antagonists. (A) GluA2 receptor structure with unliganded (apo) LBDs (PDB: 4U2P). Squared brackets indicate domain layers: ATD, amino terminal domains; LBD, ligand binding domains; TM, transmembrane region. Subunit coloring is as follows: A, green; B, red; C, blue; D, yellow. (B) The crystal packing with two molecules in the asymmetric unit (chains A and B) produces two different tetramers containing identical LBD active dimers. One layer of LBD molecules is shown with the tight and the loose tetramer being boxed with the same coloring. The tight and loose tetramers are built by four molecules of chain B and A, respectively. Subunits are colored according to the full-length receptor (those in the loose tetramer are shown in darker colors). (C) The crystal packing of both tetramers leads to a physiologically plausible tetramer arrangement (shown here for the tight tetramer) with all four ATD linkers (black spheres, Lys393) facing to one side and the four TM linkers (Pro632, orange spheres) facing to the other side. (D–G) Top views of LBD layer. (D) LBD tetramer fully bound by glutamate in the loose arrangement. (E) LBD tetramer fully bound by glutamate in the tight arrangement. The A-C interface is shown, with 2Fo-Fc density contoured at 1 sigma in gray mesh. (F) LBDs from the apo full-length structure (PDB: 4U2P). (G) LBDs in the CA arrangement in complex with DNQX (PDB: 4L17). Right panel shows the interface between A and C subunits from the side. The overall interdimer twofold axes are shown as black ovals; individual subunits are color-coded as indicated in (A); antagonist (DNQX) is shown as orange and agonist (glutamate) as purple spheres. For each structure, the distance (in Ångstroms) between the Cα atoms of A665 in molecules A and C is shown in the inset, magnification of the dotted boxes. To see this figure in color, go online.
High-resolution crystal structures of isolated LBDs showed how the first step in coupling neurotransmitter signal/channel opening proceeds: glutamate binds within a cleft between two lobes of a clamshell, inducing it to close (8). Because the upper lobes of adjacent LBDs are braced back to back in twofold symmetric dimers (9, 10, 11), the two lower lobes separate upon ligand binding. This mechanical action is thought to pry open the channel, permitting ion flow (1, 8, 12). However, iGluR tetramers bound by full agonists have so far been captured only in lower resolution electron microscopy (EM) maps (13, 14), or various inactive forms (2). Thus, further work is required to understand the geometry of receptors during activation.
Our previous study on cross-linked LBDs in complex with antagonist revealed a distinct conformation of the tetramer of LBDs reached by a ∼30° rotation of the LBD dimers (15). This “closed angle” (CA) arrangement was trapped by an interdimer disulfide cross-link between subunits A and C engineered through the A665C mutation (see Fig. 1 G). The functional properties of the A665C mutant suggest that the CA structure represents a partially active conformation. However, EM maps of a putative activated AMPA receptor do not indicate collapse of the interdimer angle (14), consistent with multiple conformations of the LBD tetramer during activation.
Here, we present the crystal structure of an LBD tetramer with glutamate bound to all four subunits. By trapping this arrangement during functional experiments on full-length receptors and through molecular modeling, we provide evidence for this arrangement of LBDs being populated during activation of the AMPA receptor.
Materials and Methods
Protein expression and purification
Rat GluA2 LBD was expressed from pET22b vector (kindly provided by E. Gouaux). Mutagenesis was performed by overlap polymerase chain reaction. Protein was expressed in Origami B (DE3). Following lysis and ultracentrifugation, the soluble LBD was purified on a Ni-NTA column. The His-Tag was cleaved and protein was further purified by cation exchange and size exclusion chromatography.
Crystallization and structure determination
Crystals were grown at 4°C using the sitting drop method in 20% w/v PEG3350 and 200 mM (NH4)2HPO4 (TR mutant) and 20% PEG 3350 and 200 mM KNO3 (wild-type (WT)). Diffraction data were collected on BL14.1 at the BESSY II electron storage ring (Berlin-Adlershof, Germany). Diffraction images were recorded at a wavelength of 0.91814 Å using a Rayonics MX-225 3 × 3 charge-coupled device detector. Both data sets were processed and scaled using the XDS program suite (16). The structure was solved by molecular replacement using one chain of the glutamate-bound LBD (Protein Data Bank (PDB): 1FTJ (8)) as a search probe in Phaser (17). The model was built iteratively in COOT (18) and refined using Phenix (19). Figures were prepared with the Pymol Molecular Graphics System, Version 1.7, Schrödinger (Munich, Germany).
Electrophysiology
All mutants were generated on the GluA2flip background using overlap polymerase chain reaction and confirmed by double-stranded DNA sequencing. WT and mutant AMPA receptors were expressed transiently in HEK-293 cells for outside-out patch recording. The external solution in all experiments contained: 150 mM NaCl, 0.1 mM MgCl2, 0.1 mM CaCl2, and 5 mM HEPES, titrated to pH 7.3 with NaOH, to which we added different drugs. The (pipette) internal solution contained: 115 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 10 mM NaF, 5 mM Na4BAPTA, 10 mM Na2ATP, and 5 mM HEPES, also titrated to pH 7.3 with NaOH. For metal bridging experiments, zinc was buffered with 10 mM tricine as described (20) or simply added (10 μM) to the external solution. To achieve zinc-free conditions, we added EDTA to the external solution (10 μM or 2 mM). Cyclothiazide (CTZ) stock solution was prepared in dimethyl sulfoxide and always added at 100 μM to the external solution. Drugs were obtained from Tocris Bioscience (Bristol, UK), Ascent Scientific (Weston-Super-Mare, UK), or Sigma Aldrich (Munich, Germany). We applied drugs to outside patches via perfusion tools made from custom-manufactured four-barrel glass (Vitrocom, Mountain Lakes, NJ) (15, 21, 22). Patches were clamped at –30 to –60 mV for macroscopic records and at –60 to –80 mV for single-channel currents. Currents were filtered at 1–10 kHz (–3 dB cutoff, 8-pole Bessel) and recorded using Axograph X (Axograph Scientific, Sydney, Australia) via an Instrutech ITC-18 interface (HEKA Electronic Dr. Schultze, Lambrecht, Germany) at 20 kHz sampling rate.
All p-values were determined by a nonparametric randomization test (using ≥105 iterations; DCPyPs suite, https://code.google.com/p/dc-pyps/). A paired randomization test was used where the same patch was compared between different conditions. The spread of the data is indicated as standard deviation of the mean unless stated otherwise.
Molecular modeling
Amino acid substitutions were generated using SCWRL4 (23). Zinc ions were initially modeled by roughly centering them between coordinating histidine side chains. The zinc and coordinating residues were then subjected to energy minimization using CHARMM (24), in which a steepest descent algorithm was followed by an adopted basis Newton-Raphson method. Residues other than those that coordinate zinc were either held fixed or restrained by a root mean-square deviation (RMSD)-restraining potential applied separately to all nonhydrogen atoms of each LBD dimer. For each model, we alternately protonated either the Nδ or Nε atom of the imidazole ring of each substituted histidine.
Results
Structure of the LBD tetramer in complex with glutamate
While performing crystallography experiments on isolated LBDs mutated according to an AMPA receptor variant displaying slow recovery from desensitization (E713T and Y768R) (25), we obtained crystals with space group P2 that diffracted to 1.26 Å resolution (Table S1 in the Supporting Material). The LBDs were arranged in two distinct tetrameric forms, one with a compact packing strikingly similar to the CA form (15). The mutations did not participate in crystal contacts, and we subsequently obtained crystals of the corresponding WT construct in the same space group, which diffracted to 1.45 Å resolution (Table S1), suggesting that this particular LBD arrangement was not a consequence of the initial mutations. The phase problem was solved by molecular replacement and the two models refined to Rwork/Rfree of 12.9%/16.1% and 17.6%/20.8%, respectively. The two structures were almost identical, and we refer in the following to the higher resolution, mutant LBD (E713T/Y768R).
The asymmetric unit of each crystal contained two LBD molecules, each forming a dimer with a crystallographic symmetry mate. The resulting two dimers are almost identical (RMSD 0.35 Å) to the canonical active state of the LBD dimer (represented by PDB: 1FTJ) (8). These dimers further assemble via crystallographic symmetry to form two different tetrameric arrangements (Fig. 1 B). Both tetrameric forms have their subunits in a physiologically plausible arrangement, with the ATD linker on one face and the membrane proximal linker on the opposite face (Fig. 1 C). The main difference between the two tetrameric forms is a lateral displacement of the LBD dimers relative to each other. The loose tetramer has a distance of 23 Å between the Cα atoms of A665 of adjacent dimers, and the tight tetramer a distance of 8 Å (Fig. 1, D and E). The tight tetramer had a similar architecture to the previously described antagonist-bound CA tetramer (Fig. 1, E and G), but it also exhibited several differences in the interdimer interfaces. Thus, the Cα atoms of the upper lobes of these two tetrameric LBD arrangements can be superimposed with RMSD of only 1.3 Å. In the CA structure, A665 was mutated to cysteine to link subunits A and C by an engineered disulfide bond (Fig. 1 G) (15). In the absence of the cross-link, a less compact interface is found in the tight arrangement. This interface is exclusively characterized by water-mediated hydrogen bonds between the backbone atoms of K663 and A665 in the loop between helices F and G (FG loop) (Fig. 1 E). The distance between Cα atoms at position 665 expands from 5 Å in the CA tetramer to 8 Å in the tight tetramer (Fig. 1 E). The FG loops are generally closer in the tight arrangement than in full-length crystal structures. For example, the Cα atoms of I664 (in molecules A and C) are separated by 8 Å in the tight arrangement and by 12.5 Å in the apo full-length structure (PDB: 4U2P; Fig. 1 F). In the loose LBD arrangement, the A-C interface is absent, as are any contacts between the FG loop and neighboring subunits. For example, the Cα of I664 in subunit A sits 11 Å from Cα of K765 in helix K of the laterally opposed subunit B. Overall, these observations are consistent with a dynamic FG loop that can be cross-linked by cysteines at positions 663 to 665 (1, 15, 22, 26, 27).
The lateral interdimer interface (between subunits C and D, and subunits A and B, Fig. 2 A) is more compact in the tight arrangement than in the CA structure (Fig. 2, B and C). For example, the Cα atoms of K765 in subunit D and T672 in subunit C are separated by 8 Å and 12 Å in the tight and the CA structure, respectively. Only a single water-mediated hydrogen bond connecting S676 in subunit C and K765 in subunit D was resolved in the CA crystal structure, likely due to the larger distance between the subunits and, possibly, the lower resolution of the crystallographic data (2.8 Å) (Fig. 2 B). In the tight arrangement, salt bridges were absent but we resolved a complex water network including a water-mediated hydrogen bond between one conformation of R675 and D427 (Fig. 2 C). Consistent with a role for this interface during channel activation, the R675S mutation speeds deactivation of GluA2 (25). For another residue that has a similar speeding effect when mutated, K761, no side-chain density was found after the gamma carbon in the tight tetramer. Furthermore, the double mutant GluA2 R675S K761M exhibited deactivation in response to a 1 ms pulse of glutamate that was at the limit of detection (4000 ± 500 s–1, n = 6; approximately threefold faster than WT GluA2; Fig. 2 D). These observations support the idea that a sparse contact resembling the lateral interface is formed during receptor activation. Accordingly, the same interdimer interface (between subunits C and D, and subunits A and B) is essentially absent in the apo structure due to larger interdimer separation in this LBD tetramer compared to both tight and CA tetramers.
Figure 2.

The tight arrangement and the intersubunit interfaces. (A) Ribbon representation of the tight tetramer with all four subunits bound to glutamate (purple spheres). Interfaces are indicated with an outlined box and named according to the subunits involved (A-C and C-D interface, respectively). (B) Zoom into the interface between subunits C and D in the CA arrangement (PDB: 4L17). Water molecules are depicted as red spheres. Dashed lines represent hydrogen bonds. All distances are in Ångstroms. (C) The same lateral interface in the tight arrangement. Alternative conformations for side chains are indicated in parentheses (A) and (B). One alternate conformation of R675 is stabilized by a phosphate ion (orange sticks) from the crystallization buffer. (D) Patch clamp recording of the rapid deactivation of the GluA2 R675S K761M double mutant (deactivation time constant from the exponential fit (open circles) is 3300 s–1 for this trace). Dashed line shows WT deactivation. The upper trace shows the solution exchange for the nominal 1 ms glutamate pulse. To see this figure in color, go online.
Evaluation of the tight and loose tetrameric LBD arrangements
To test if either of the glutamate-bound LBD tetramers produced by crystal packing represented a functional state in full-length AMPA receptors during activation, we designed bridges that were predicted to coordinate in the tight, but not in the loose arrangement, and vice versa (Table 1). Because cysteine mutants behaved poorly in functional experiments, we turned to zinc bridging sites, which are generally better tolerated. Amino acid side chains involved in a zinc binding site must be within 4–5 Å of each other (28, 29, 30). The preferred tetrahedral geometry of zinc coordination is achieved by at least three ligands, including histidine side chains. Thus, we designed each putative cross-link with three histidine residues at opposing sites across the interdimer interface. We then attempted to bridge these histidines with zinc ions and measured the effect on receptor activation by glutamate (Fig. 3).
Table 1.
His mutants designed to distinguish between tight and loose tetrameric packing arrangement
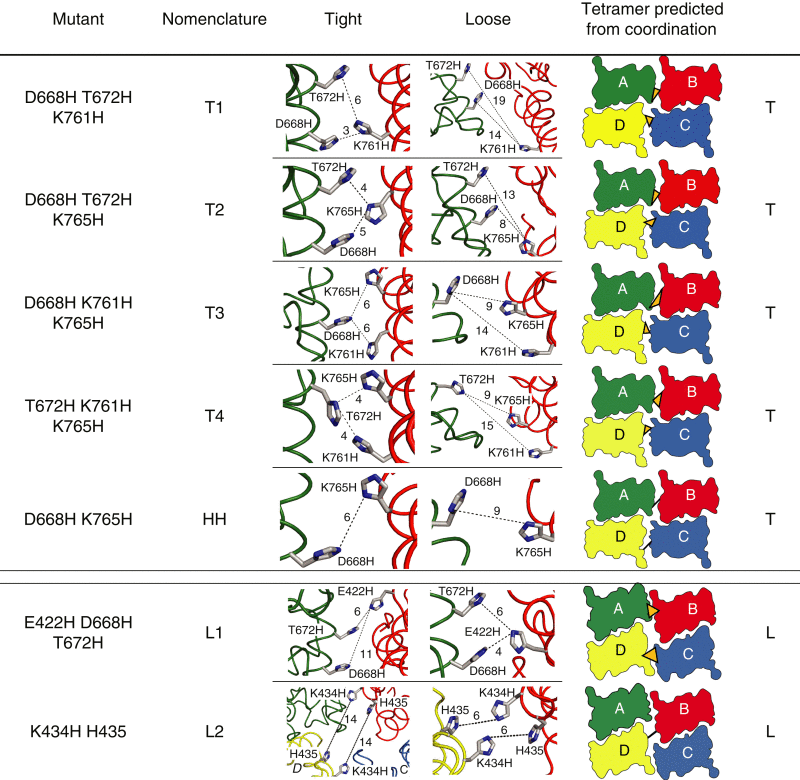 |
Mutated residues are listed in the first column from the left, followed by nomenclature introduced for each mutant. Cartoon models in the third and fourth columns show mutated residues in the tight and loose crystal arrangements, respectively, for the respective mutant (mutant side chains were oriented by eye, without energy minimization). Approximate distances between nitrogen atoms are denoted by dotted lines and expressed in Ångstroms. Individual subunits are color coded as in Fig. 1: A, green; B, red; C, blue; D, yellow. The fifth column contains a top view of the LBDs in the arrangement that predicts cross-linking of the respective mutant (T-tight or L-loose). Black lines and orange triangles indicate expected zinc coordination sites. The HH control mutant, for which no cross-link was predicted, is included for comparison. To see this table in color, go online.
Figure 3.

Functional trapping of glutamate-bound tight and loose tetrameric arrangements. (A) Representative current traces of WT and mutant receptors designed to distinguish between the tight and loose tetrameric arrangements of LBDs observed in the crystal. Patches were exposed to zinc-free (EDTA) solution (triangles), then to 1 μM zinc (open circles) and then washed again with EDTA (gray circles). WT GluA2 was not sensitive to zinc. (B) Summary of peak current responses in zinc, relative to the EDTA control responses. From the four mutants predicted to trap by the tight arrangement (T1-4), three were robustly inhibited by zinc (T1, T2, and T3). Two mutants predicted to cross-link by the loose arrangement (L1-2) showed minimal modification in the tested conditions. The control double mutant HH was unexpectedly inhibited, whereas T4 mutant was not modified despite being predicted by the tight arrangement.
Four mutants were predicted to cross-link in the tight tetrameric arrangement: D668H T672H K761H (designated T1, see Table 1), D668H T672H K765H (T2), D668H K761H K765H (T3), and T672H K761H K765H (T4). Zinc bridges specific to the loose arrangement were more challenging to design because of the greater displacement of the subunits, but we did find one triple mutant E422H T672H K761H (L1) and the single mutant K434H that was predicted to form a quadruplet across the twofold interdimer axis with the native residue H435 (L2, Table 1). In addition, all possible single and double combinations were created, as controls of zinc coordination (Fig. 3 B). Strikingly, the T1, T2, and T3 mutants all showed readily reversible ∼50% peak current reduction in 1 μM zinc, consistent with the formation of a zinc trap between subunits. For the T1 mutant, peak current in zinc was 56 ± 5% of that without zinc (n = 9 patches), for T2 it was 56 ± 6% (n = 3), and for T3, 32 ± 9% (n = 6; for all three constructs p < 0.0001 compared to the WT). In contrast, the two mutants designed to cross-link in the loose arrangement either showed very little inhibition in zinc, similar to that observed in some controls (the peak current of the L2 mutant in zinc was 88 ± 3.5% of that without zinc, n = 6, p = 0.0003 compared to the WT) or no inhibition at all (L1 peak current in zinc was 96 ± 3%, n = 4, p = 0.2 compared to the WT) (Fig. 3 B).
Two further mutants gave unexpected results. Although predicted to trap in the tight arrangement, the mutant T4 was insensitive to zinc (94 ± 5% peak current in the presence of zinc, n = 7, p = 0.1 compared to the WT). Furthermore, the control mutant HH (D668H K765H) showed trapping by zinc, despite only harboring two histidine residues (the presence of zinc reduced the peak current to 57 ± 3%, n = 17, p < 0.0001 compared to the WT). Consequently, current inhibition of triple mutants in this background (T2 and T3) was not immediately informative. To obtain further structural insights, we addressed the mechanistic basis for inhibition in HH-based mutants, and the missing inhibition in the T4 mutant, in subsequent experiments.
Zinc coordination by the HH mutant
Inhibition of the HH control mutant by zinc was surprising because double mutants are rarely able to provide strong enough zinc coordination required to bridge subunits within a protein complex (29). Although zinc coordination is possible with two histidine residues, the apparent zinc affinity increases from 10−5 M for two, 10−7 for three, and 10−9 M for four coordinating ligands (30). In the case of the HH mutant, apparent zinc affinity was high enough to indicate three rather than two ligands (IC50 = 370 nM, Fig. 4 A). Assuming a diffusion-limited association rate (∼108 M–1s–1), the bridge should have a lifetime of at least 10 ms, exceeding that of the open state of GluA2 (1–2 ms (25)). To explain the trapping by this mutant, we looked at potential neighboring residues that could participate in zinc coordination, and identified D769 as the best available candidate, despite its 8 Å separation from D668H in the tight crystal arrangement (Fig. 4 B). Consequently, participation of D769 in a cross-link in the HH mutant could indicate rearrangements of the interdimer interface from the tight structure.
Figure 4.

Zinc coordination by the HH mutant. (A) Half-maximal inhibition of currents activated by 10 mM glutamate (in the absence of CTZ) by zinc was similar for both mutants HH and T1, with IC50 of 370 ± 20 and 380 ± 10 nM, respectively (n = 3 patches for both). (B) Cartoon diagram shows a plausible arrangement of side chains in the HH mutant, modeled into the tight LBD arrangement. Distances are in Ångstroms. Native residue D769 was chosen as the most likely third coordinating partner of zinc in the bridge and tested through a series of mutants. (C) Representative traces of jumps into 10 mM glutamate in the presence of 1 μM zinc (red circles) or its absence (black triangles) for mutants HH, HH D769G, and HH D769H. The inhibition was reversible, as shown by the wash in 2 mM EDTA (open circles). (D) Summary of inhibition of D769 mutants by 1 μM zinc. To see this figure in color, go online.
To test the hypothesis that D769 participates in the coordination of zinc in the HH (D668H K765H) double mutant, we created four mutants on the background of the HH expected to either disrupt or alter the strength of the coordination: D668H K765H D769G (HH D769G), D668H K765H D769K (HH D769K), D668H K765H D769H (HH D769H), and D668H K765H D769E (HH D769E) (Fig. 4). Fig. 4, C and D, shows sensitivity of the “secondary” mutants to 1 μM zinc. Consistent with its participation in zinc coordination of the HH mutant, mutating D769 to glycine (the equivalent residue in kainate receptors) or lysine weakened zinc-dependent modification of currents (p = 0.0003 compared to HH, n = 8 and p < 0.0001 compared to HH, n = 12, respectively). Along this line, the D769H mutant expected to strengthen zinc coordination was more strongly inhibited by zinc (p < 0.0001 compared to HH, n = 5). Inhibition of the mutant HH D769E was similar to that in HH (p = 0.06 compared to HH, n = 7). These results strongly indicate that, in the HH mutant, D668H, K765H, and D769 come into close proximity during receptor activation, consistent with LBD architecture resembling, but not identical to, the tight tetrameric arrangement.
We returned to the triple mutants T2 and T3, which contain the HH site and T672H or K761H, respectively, this time including the D769G mutation to disrupt any bridging specific to the HH mutant alone (T2 D769G and T3 D769G). In the resulting quadruple mutants, D668H T672H K765H D769G and D668H K761H K765H D769G, zinc (1 μM) still robustly inhibited full-length receptors (Fig. 3 B), suggesting that coordination in these triple mutants is not dependent of D769. Thus, we identified that the HH mutant produces a unique zinc bridge, probably corresponding to a distinct conformation of the tetramer that is not trapped in other mutants.
The T1 and HH mutants trap receptors in the apo state
Having established that the HH and T1 bridges trap the receptor through distinct sets of side chains, we hypothesized that the corresponding distinct geometries might be reflected by trapping in different functional states.
To assess trapping in the resting state, we incubated patches in 10 μM zinc, without any agonist present, for 1 s and then exposed them to a pulse of 10 mM glutamate and 10 μM zinc-chelator EDTA. Desensitization was blocked by CTZ throughout the recording (31) (Fig. 5 A). If zinc is bound to the receptors in the resting state, the release of zinc is expected to slow the on-relaxation upon jump into zinc-free glutamate solution. Indeed, this is what is observed for both mutants, T1 and HH (Fig. 5 A), supporting trapping of these geometries in the apo state.
Figure 5.

Apo state trapping. (A) WT receptors are unaffected by 1 s exposure to zinc (10 μM) in the absence of any ligand, but both, the T1 and HH mutants exhibit slower on-relaxation upon jump into (zinc-free) glutamate. For both mutants, the on-relaxation contained two components: τ1 = 5 ± 0.5 ms and τ2 = 91 ± 12 ms (n = 4 patches) for T1 and τ1 = 9 ± 4 ms and τ2 = 90 ± 12 ms (n = 4) for HH. The faster component reflects the solution exchange, and the slower component corresponds to relaxation from zinc trapping, i.e., lifetime of the bridges formed in zinc. The bar graph shows the residual active current after exposure in the apo state to 10 μM zinc. (B) Traces showing WT and mutant receptors jumped into 10 mM glutamate (Glu) or 2 mM quisqualate (Quis) in the absence (10 μM EDTA, black traces) or presence of zinc (10 μM, orange traces) with zinc or EDTA present throughout the recording. CTZ (100 μM) was also present throughout the recording to block desensitization. The effects of zinc are reversible as shown by alternating jumps between zinc-containing and zinc-free solutions. (C) Summary of the effects of zinc on the on-relaxation. For each patch, traces in EDTA and zinc were separately averaged and normalized to their peak. Their difference trace was then integrated to obtain the cumulative difference in charge (Q). We determined the average variability of QON within groups (QON(control)) to control for the inherent variability. QON was then normalized by QON(control). QON was measured 100 ms after the start of the agonist pulse. The trapping mutants, T1 and HH both showed slower on-relaxation in zinc, and thus a deficit of charge during the rise to the peak current, in both glutamate and quisqualate (not shown) ∗∗∗p < 0.001, ∗p < 0.05. (D) Plot summarizing the effects of zinc on the steady-state current in glutamate (white bars) and quisqualate (gray bars) for T1, HH, and the controls (WT and CA-HHH). The steady-state current is the average current of the last 30 ms of the agonist pulse. (E) The HH D769H mutant showed a similar functional profile to the T1 mutant when equilibrated in 10 μM zinc and jumped into 10 mM glutamate in the presence of 100 μM CTZ. The effect was reversible as seen from alternating jumps into 10 μM zinc (orange traces) and zinc-free solution (10 μM EDTA, black traces). The effect of zinc on the HH D769E mutant was similar, and jumps were extended to 500 ms to allow the current to plateau. (F) The apo structure (PDB: 4U2P, with ATDs omitted) with the T1 and HH mutations modeled as gray spheres. Top views of the LBD assembly, with boxed sections expanded to show the side chains forming the sites in gray, stick representation. Distances (dotted lines) between the mutated residues are in Ångstroms. Unlike HH, the T1 site is not expected to be bridged by zinc without rearrangement. To see this figure in color, go online.
If the slowdown in the on-relaxation was a consequence of zinc trapping the resting (apo) state, the time constant of the relaxation (∼100 ms) then relates to the lifetime of the bridges. However, in the experiments in Fig. 5 A, zinc was exchanged for glutamate, meaning that the relaxation could be accelerated by zinc unbinding. Furthermore, our multibarrel system, used in the experiments in Fig. 5 A, has a solution exchange of ∼5 ms compared to ∼200 μs for standard ultrafast perfusion. To get better resolution of the on-relaxation for comparison across the different mutants, and to assess it in constant zinc, we used an ultrafast jump into glutamate in the continuous presence of 10 μM zinc or EDTA (CTZ was again present throughout the recording). The on-relaxations had multiple exponential components, and so to compare the effects across mutants, we expressed the normalized differences in transferred charge (for calculation details, see the legend of Fig. 5 C). In this simplified experiment, zinc had a dual effect on T1 receptors. As before, trapping of apo receptors was evident from a slow on-relaxation at the start of the glutamate jump (Fig. 5, B and C); in addition, because zinc was still present once the patch was in glutamate, a decrease in the current amplitude compared to the zinc-free conditions was revealed (Fig. 5 D). The amplitude responses to glutamate from T1 mutant receptors preequilibrated in zinc were 79 ± 2% of those without zinc (n = 23, p < 0.0001 compared to the WT; n = 13). Decrease in amplitude in the presence of zinc observed here is a consequence of trapping of activated T1 receptors as described below and in Fig. 8. The deficit in charge transfer during the T1 on-relaxation due to zinc was only about twice that of the WT background (ratio: –2.4 ± 0.5, n = 14, p = 0.01 compared to WT; n = 11, Fig. 5 C). The decrease in the transferred charge was more pronounced for HH than for T1 receptors and therefore enough to account for all the inhibition we observed in our initial screening experiments (Figs. 3 B and 5, B and C). For the HH mutant, the fold-deficit of charge transfer was 7 ± 1 (n = 13, P = 0.0003 compared to WT n = 11) (Fig. 5 C). The faster release of trapping of the T1 bridge indicates that it is less stable (that is, with a shorter lifetime) than the HH bridge. Given that IC50 for zinc is similar for the two mutants (Fig. 4 A), this observation might mean that the geometry required for the T1 bridge to form is visited more frequently.
Figure 8.

State dependence of the HH zinc bridge. (A) The traces show an experiment similar to that described in Fig. 6A, but for the HH mutant. Lower panel (zoomed from the top panel) shows relaxation at 500 μM glutamate (light blue) and no trapping at 10 mM glutamate (dark blue). (B) Glutamate concentration-response curves in 10 μM zinc for WT GluA2 (blue open circles; EC50 = 170 ± 40 μM), HH before trapping by zinc (green diamonds; EC50 = 220 ± 50 μM), and HH following trapping (red circles; EC50 = 950 ± 50 μM). The difference between the apparent affinities for glutamate for WT and HH in the absence of zinc was not significant (p = 0.06). The active fraction after trapping shows a shallow bell-shaped distribution with maximum trapping at 215 μM glutamate (n = 6 patches) (yellow triangles). (C) The off-relaxations in Zn2+ and EDTA were quantitated by the difference in charge transfer, normalized to the variability within groups as described for QON in the legend to Fig. 5C. QOFF was determined over the interval up to 200 ms after the end of the agonist pulse. (D) Summary plots of the normalized difference between charge transfer in the glutamate decay between control and zinc. Neither T1 nor HH showed any change in the deactivation decay. (E) As for (D), but following a quisqualate jump. (F) Outward currents from WT GluA2 show little effect of 300 μM zinc. (G) Outward currents activated by 10 mM glutamate for the HH mutant were not modified following a jump into 300 μM zinc. There was no change in the active fraction (arrow). (H) Bar graph showing the active fraction after application of 300 μM zinc was not significantly different from that of WT 93 ± 2 and 93 ± 3% for HH and WT, respectively (p = 0.8, n = 4 patches for both). To see this figure in color, go online.
The slow rise of the current in the HH mutant was maintained in the presence of a more potent agonist, quisqualate (Fig. 5 B) (32) (the fold-deficit was 6 ± 1.5, n = 12, p = 0.001, data not shown), consistent with the resting state trapping being agonist independent. In both quisqualate and glutamate, the current due to the HH mutant equilibrated to approximately the same level in zinc as in EDTA. In zinc, the quisqualate current was 104 ± 2% compared to amplitude in EDTA (n = 12, p = 0.03 compared to the WT; n = 9) and in glutamate 100 ± 2% (n = 23, p = 0.25 compared to the WT; n = 13) (Fig. 5, B and D). Two further histidine mutants made on the background of the HH mutant, HH D769H and HH D769E, also slowed the on-relaxation (Fig. 5 E). These data indicate that apo trapping of HH covers a range of geometries and is not restricted to the particular geometry of the HH cross-link.
Apo trapping was also tested for the triple mutant G437H K439H D456H (CA-HHH), originally created to demonstrate adoption of the CA conformation in partially occupied receptors (15). Zinc failed to trap apo receptors at the CA-HHH site (Fig. 5, B–D). Because CA-HHH mutations are at the tips of the upper lobes of the LBDs, this result indicates that, even though the LBDs are mobile when unliganded, they do not necessarily collapse together in the apo state as would be necessary for the formation of the CA-HHH cross-link. In addition, the current due to the L2 mutant (designed to cross-link in the loose tetramer arrangement) was unaltered in the presence of 10 μM zinc and CTZ (current amplitude in zinc was 100 ± 4%, n = 3, p = 0.7 compared to the WT, data not shown).
The apo state of GluA2 was recently crystallized (Dürr, 2014), and so we modeled the histidine residues into the interface between dimers to assess the likelihood of a zinc binding site forming in the resting state (Fig. 5 F). Whereas the apo structure provided excellent context for a zinc site formed by the HH mutant between subunits C and D, the interface between subunits A and B was less ideal. The relevant residues were too far apart (>15 Å) for the T1 bridge to form. Trapping in the apo state must therefore result from the dynamics and mobility of unliganded LBDs (22).
Trapping of T1 in the presence of glutamate
Given the trapping in apo conditions and 10 mM glutamate by the T1 mutant, we then tested responses to saturating zinc (10 μM) in the presence of different concentrations of glutamate (10 μM to 30 mM; Fig. 6, A and B), to see if trapping was dependent on channel gating. In these conditions, zinc had no effect on the WT receptors (Fig. 6 A). Current modification of the T1 mutant was absent at low concentrations of glutamate (10 μM; Fig. 6, A and B), which seemed to protect against the modification that occurs in the apo state. This result suggests that the binding of one or two glutamate molecules is enough to disrupt the intersubunit binding sites for zinc that are formed in the apo state for the T1 mutant. Inhibition developed at glutamate concentrations above 100 μM, and persisted in saturating glutamate concentrations (up to 30 mM) (Fig. 6, A and B), indicating bridging in glutamate-bound receptors. However, the formation of the bridge was inhibitory, consistent with trapping in a less active state than the receptor can attain without bridging. Because glutamate was in these experiments present in high concentrations (up to 30 mM), we tested for a possible chelation of zinc by glutamate. For this purpose, 29.5 mM D-glutamate was added to 0.5 mM L-glutamate. D-glutamate barely activates AMPA receptors, but can still chelate zinc and should therefore reveal if zinc concentration is affected by chelation (Fig. 6, C and D). In the presence of 29.5 mM D-glutamate, T1 trapping in 0.5 mM L-glutamate was reduced by 9 ± 2% (n = 5, p = 0.06). Thus, zinc chelation does not appear to be a major factor in the inhibition observed at 30 mM glutamate. Nevertheless, we accounted for this chelation when determining the T1 active fraction at 30 mM L-glutamate in Fig. 6 B.
Figure 6.

State-dependence of the T1 zinc bridge. (A) Patch-clamp experiment showing five test pulses (2 + 3) of 10 mM glutamate (blue bars) in the presence of 10 μM EDTA (green bars), flanking the application (1 s) of 10 μM zinc (red) with different concentrations of glutamate (indicated by the black bar): 10 μM (orange trace), 500 μM (light blue), 10 mM (dark blue). CTZ (100 μM) was present throughout the experiment. Middle panel (zoom of the top panel) shows relaxation observed at 500 μM and 10 mM glutamate (τ = 35 ± 2 ms and τ = 32 ± 9 ms, n = 6–9 patches, respectively). The active fraction is indicated in the inset with an orange triangle. The recovery of current in 10 mM glutamate and EDTA following trapping at different concentrations showed a common fast component (τ = 3 ± 0.4 ms, n = 6 patches) corresponding to the exchange of solution and a slow component (τ =106 ± 30 ms, n = 6 and τ = 147 ± 14 ms, n = 6 patches, following trapping in 500 μM and 10 mM glutamate, respectively). WT A2 shows no modification by zinc in 500 μM and 10 mM glutamate, bottom panel. (B) Glutamate concentration-response curves in 10 μM zinc for WT GluA2 (blue circles; EC50 = 170 ± 40 μM), T1 before trapping by zinc (green diamonds; EC50 = 348 ± 80 μM), and T1 following trapping (red circles; EC50 = 6.4 ± 2.9 mM). The difference between the apparent affinities for glutamate for WT and T1 in the absence of zinc was not significant (p = 0.2). The relationship between the active (untrapped) fraction and log-concentration of glutamate followed an exponential function (yellow triangles). (C) Correction for chelation of zinc by high glutamate. Blue trace shows response of mutant T1 to 500 μM L-glutamate in the presence of 10 μM zinc (red bar), flanked by 10 mM L-glutamate test pulses in EDTA (green bars). Orange trace shows the application of 29.5 mM D-glutamate in addition to 500 μM L-glutamate and 10 μM zinc. Following the application of zinc, a larger active fraction (as estimated from the larger instantaneous current activated by 10 mM glutamate) was observed in the presence of 29.5 mM D-glutamate than in its absence (inset arrow), presumably due to zinc chelation. (D) The chelation effect at 30 mM glutamate reduced trapping by 9 ± 2%. Thus, the trapped fraction was underestimated due to a lower free zinc concentration. The 30 mM glutamate value for the active fraction in Fig. 6B, was reduced accordingly by a factor of 1.09. To see this figure in color, go online.
Characterization of active states trapped by the HH zinc bridge
We did not observe any inhibition of responses to saturating glutamate in the absence of desensitization (blocked by CTZ) for the HH mutant. This result suggests the HH bridge does not modulate receptors fully bound by glutamate. However, we noted that, with desensitization intact, preincubation in zinc slowed entry to desensitization for the HH mutant (Fig. 7; in zinc, kdes = 103 ± 9 s–1, and in the absence of zinc, 148 ± 14 s–1; n = 9, p = 0.004). WT desensitization was unaffected by zinc (Fig. 7), meaning that glutamate-bound receptors were trapped by the HH bridge, but that CTZ masked or eliminated the effect in saturating glutamate. For T1 receptors, desensitization was slightly faster in zinc (kdes in zinc 220 ± 30 s–1 and without zinc, 160 ± 10 s–1, n = 7, p = 0.01), for reasons that are unclear. Slowing was specific to the HH mutant accompanied by amino acids with a carboxylate group at position D769, because it was absent in the D769G and D769K variants. We previously showed that interdimer cross-links could reduce desensitization of kainate receptors while also inhibiting the peak response (27), consistent with common mechanisms of LBD dimer rearrangement in AMPA and kainate receptors during desensitization (14).
Figure 7.

The HH bridge alters desensitization. (A) Zinc slows the desensitization decay of the HH mutant (open circles), compared to the desensitization in EDTA (control, black triangles; wash, gray solid circles). Zinc accelerated the decay of the T1 mutant. Insets show the normalized currents. (B) The HH mutant and the HH D769E mutant both have markedly slower desensitization decays. The kinetics of control mutants was not different from WT GluA2.
Distinct from the neighboring T1 bridge, the HH mutant showed inhibition by zinc only at intermediate glutamate concentrations (Fig. 8 A), producing a bell-shaped inhibition profile (Fig. 8 B). This concentration dependence is similar to that previously described for the GluA2 G437H K439H D456H mutant (CA-HHH), which was inhibited by zinc, marking the attainment of the CA activation intermediate (15).
Given these data, we considered the possibility that HH bridge formed in the fully bound tetramer without inhibiting activation. In this case, bridges formed by zinc in the HH mutant could slow down unbinding of glutamate by hindering LBD opening. Even though the glutamate response curve was right shifted in zinc, suggesting that glutamate potency, if anything, decreased, we assessed unbinding by measuring the kinetics of deactivation directly.
The off-relaxation was again measured in terms of transferred charge as described previously for on-relaxation (Fig. 8 C). For the HH mutant, we observed no lengthening of the decay for either agonist (Fig. 8, C–E). Any effect of zinc on charge transfer was less than the variability within each condition (0.2 ± 0.15, n = 14, p = 0.95 compared to WT; n = 12) (Fig. 8 D). This remained unchanged when glutamate was replaced by more potent quisqualate (Fig. 8 E), excluding the possibility that fast glutamate unbinding masked any slowing of the off-relaxation in zinc. In case the stabilization occurred in only a fraction of channels, we examined the HH cross-link at the single-channel level (with desensitization blocked, data not shown), but could not detect any changes in open probability or conductance and no zinc-dependent stabilization of channel opening after the end of the agonist pulse. This result further corroborates the notion that HH does not modify receptors fully bound by agonist when CTZ is present.
Finally, we tested whether the lack of effect at high glutamate concentrations in the HH mutant could have been because inhibition developed too slowly for us to resolve at 10 μM zinc. To test for this possibility, we greatly increased the first order association rate of zinc, closer to that of glutamate, by increasing the concentration of zinc. However, at the maximum concentration of zinc that we were able to test (300 μM; giving no block of WT GluA2 at +60 mV), we still failed to see any modification of the current in 10 mM glutamate (Fig. 8, F–H) following a jump into zinc. Thus, the absence of the zinc effect on HH receptors in saturating agonist concentrations appears to be genuine and is highly unlikely to have been missed due to the experimental design.
Structural bases of T1 and HH functional profiles
To understand how distinct cross-linking geometries relate to the tetrameric LBD conformation, we generated molecular models of the engineered zinc binding sites, modeling a zinc ion between the introduced histidines (details of how the models were built are provided in the Materials and Methods section). For each mutant, the coordination geometry of the histidine substitutions with the modeled zinc ion was optimized either by allowing reconfiguration of only the substituted residues and the zinc ion, or alternatively by also allowing rigid-body movement of the LBD dimers (i.e., A-D and B-C). To determine the “goodness” of zinc coordination across modeled structures, we used the calcium bond-valence sum (CBVS) metric (33). The CBVS metric estimates the goodness of zinc coordination based on the valence of zinc and the coordination distances between zinc and coordinating ligands. For example, CBVS for zinc coordinated by six oxygen atoms at the ideal Zn-O distance is 4.07 (33). The CBVS score was suboptimal for all the sites we modeled, probably because of the lack of water molecules or other ligands to achieve tetrahedral coordination of zinc, and the conservative approach to modeling that aimed for minimal perturbation of the crystal structure template (Fig. 9 A). However, the modeling results show that the T1 mutant provides the best coordination site for zinc in the tight tetrameric arrangement, which cannot be improved by rigid body translation. In contrast, coordination of zinc in the HH mutant was almost nonexistent (CBVS = 0.001), but, following a rigid body displacement of the dimers by 1.2 Å, this coordination improved to match that of the T1 mutant (CBVS = 0.54, Fig. 9, A and B, left panel). We also investigated the triple mutant T4 (Table 1), because this putative bridge unexpectedly failed to show any zinc-dependence effect in full-length receptors. While it is conceivable to that zinc bridging occurred without a functional signature, modeling results indicated that the cross-link fails in the T4 mutant due to steric clashes between T672H and K765H (Fig. 9 B, right panel). These results further support the idea that HH traps a subtly different conformation from that captured by the tight tetramer, and illustrate a mechanism by which zinc bridges can distinguish subtly different geometries of dimer-dimer packing.
Figure 9.

The active conformations and receptor activation. (A) CBVS scores for zinc coordination by mutants modeled into the tight tetramer (gray bars), and the same analysis for tetramers in which the active dimers were allowed to relax as rigid bodies (orange bars). (B) Left panel: alignment of the tight tetramer (gray) and the translated-dimer structure optimized for coordination of zinc (orange sphere) by the HH mutant by rigid body translation of the dimers (orange). Right panel: coordination of zinc by the T4 mutant before and after rigid body translation. Modeling reveals steric clashes between T672H and K765H. (C) Sites of Arg660 (marker for subunits A/C, green spheres), Gln756 (marker for subunits B/D, blue spheres) and Pro632 (LBD-TM3 marker, red spheres) in the full-length receptor (gray, PDB: 4U2P). (D) Lateral positions of Pro632, aligned in plain view, are shown for apo conformation (red), CA in which the subunits B and D are modeled bound by glutamate (orange), and the tight tetramer (cyan). Arrows indicate the direction of Pro632 displacement from apo LBDs toward LBDs partially (CA) and fully (tight) populated by glutamate. (E) Displacements of Pro632, measured diagonally between the subunits (A-C) and (B-D) in the full-length receptor for apo (red), the glutamate bound active state model from EM (Glu_EM, blue), the partly bound conformation (CA, orange), the tight tetramer conformation before (tight, cyan) and after (HH-modeled, green) rigid body movement to accommodate zinc in the HH mutant. (F) Diagonally opposed residues at the core of the LBD tetramer Arg660 and Gln756, with the same color code as in (D). (G) Cartoon of proposed LBD movements during receptor activation, with the amino terminal domain omitted for clarity. The open angle, fully bound form represents the glutamate bound active state model from EM (Glu_EM). The open angle, partly bound form has not been captured in crystal structures, but is included here for completeness (indicated with a question mark). Cartoons are accompanied by ribbon representations of the respective LBDs (PDB IDs in brackets). Subunits are color coded as in Fig. 1A. Black arrows indicate vectors defined for determination of the interdimer angle between dimer pairs A-D and B-C with the vertex at the center of mass of 665 Cα atoms in subunits A and C (15). To see this figure in color, go online.
Discussion
We have used high-resolution structural information from a glutamate-bound LBD tetramer in combination with zinc bridging experiments to map functional conformations of a glutamate receptor with a time resolution of ∼10 ms. Assuming that the glutamate-bound LBD dimers move as rigid bodies, we could verify existing and new states of the LBD tetramer along the activation pathway. Together with existing structural data, we could build upon existing studies of LBD clamshell and dimer dynamics (9, 22, 34, 35) to provide a more complete view of LBD dynamics at the tetramer level and its role in AMPA receptor activation (Fig. 9, C–G).
The crystal lattice contained two distinct tetrameric packing arrangements, which we termed tight and loose, based on the Cα A665 distances. In both packing arrangements, the active dimers were canonical (8). Similar tetrameric forms were captured in a crystal of the LBD in complex with the allosteric potentiator NS5217 bound in the dimer interface (3H6W; Cα RMSD 0.46 Å compared to our structure), consistent with an active state, but the functional significance of the tetramer arrangement was not recognized in this study (36). An arrangement akin to the “loose” packing has previously been discussed based on the molecular packing of LBD dimers bound with the full agonist quisqualate, but was never tested (37). Our data cannot exclude the presence of the loose LBD arrangement during receptor activation, but we were unable to obtain much evidence for it, except for a very minor inhibition of the L2 mutant in the absence of CTZ. The only other structure of an AMPA receptor in complex with glutamate is the recent full-length EM structure resolved at ∼12 Å (14). The structure predicts relaxation of the LBD tetramer upon agonist binding, however, the design of cross-links based on this structure is hampered by its comparatively low resolution. In contrast, the high-resolution, “tight” LBD arrangement proved amenable to cross-linking across a series of bridging mutants in multiple conditions. The tight structure predicted 4 out of 5 bridges (bridges T1-3 and HH were successfully predicted, whereas T4 bridge failed). These bridges enabled us to probe the interdimer interface and, assuming that the canonical active dimers were preserved by CTZ, the overall arrangement of the dimers.
Computational modeling indicated that the T1 mutant traps the tight LBD tetramer conformation, whereas the HH mutant traps a second functional LBD arrangement (HH-modeled) that is related to the tight tetramer by a rigid body movement of two adjacent LBD dimers by ∼1 Å to achieve optimal zinc coordination. Functional experiments showed that HH-modeled arrangement was accessible only to partially bound LBDs, whereas tight arrangement was attained by partially and fully bound LBDs. Both bridges (T1 and HH) caused partial inhibition in the presence of zinc. Their inhibitory nature indicates trapping of functional states that are either completely inactive or not fully active. Single channel recording of trapped receptors may help to resolve the nature of the inhibition. If the bridges fully block activity, partial inhibition could still be explained by the short lifetime of the bridges (Fig. 5 A) and infrequent visits of the receptors to conformations that support trapping. The biexponential character of the recovery from the apo state (for example, in Fig. 5, B and E), suggests multiple cross-linking events. On the other hand, it is also possible that following trapping by one bridge, an asymmetric arrangement results that excludes the second bridge from forming. There were no steric barriers to the formation of both bridges in the structure we solved, but the formation of the HH bridge in the apo structure was only unequivocally possible between subunits C and D.
To visualize and quantitate how these LBD arrangements relate to each other and the membrane ion channel, we determined distances between opposed subunits. We took the Cα of Pro632 as a proxy for the M3-S2 linker, because the entwined M3 helix bundle must be released for the channel to open (Fig. 9, D and E). In line with previous work (4), we also measured the distance between Cα of Q756 in subunits B and D, and R660 in subunits A and C, to assess condensation or expansion of the tetramer (Fig. 9 F). In this analysis, we included the apo state with all LBDs unbound, the CA intermediate state (PDB: 4L17) with subunits B and D modeled as glutamate bound, the two fully bound LBD models presented in this work (tight and HH-modeled), and the model derived from the 12 Å EM map of full-length active GluA2 (Glu_EM; PDB: 4UQ6 (14) (Fig. 9, E and F). In the scheme shown in Fig. 9 D, the linkers would undergo the largest movement as the first agonist molecules bind, i.e., during the transition from the apo into the partially bound state. The first movement (from apo/resting state to the CA/partially bound intermediate) separates the lower lobes of the distal (B and D) subunits by 34 Å (Fig. 9 E). The lower lobes of the proximal subunits also separate (by 9 Å). These movements are similar in extent to within-dimer displacements driven purely by clamshell closure (8). The positions of Pro632 are very similar in HH-modeled compared to CA (in HH-modeled, proximal subunits move 1 Å apart and distal subunits come 4 Å closer compared to CA). This supports the notion that HH mutant traps another intermediate, partially bound state, distinct from the CA conformation, as our electrophysiological measurements suggest. Further glutamate binding leads to two different structures: tight arrangement (captured by fully bound T1 mutant) and Glu_EM structure. The tight arrangement again resembles the CA intermediate with no more than 2 Å displacement for Pro632. This similarity to partially active forms is perhaps the best explanation for why the T1 bridge, even though it can form with glutamate in all four subunits, cannot support maximally conducting channels. If we assume that the Glu_EM LBD structure is indeed linked to an open channel (the pore was not resolved in the study), then the main difference from the CA, HH-modeled, and tight arrangements is the approach of Pro632 (up to 20 Å) of the distal subunits (proximal subunits barely move). In the full-length structures in complex with partial agonists (4, 5, 6), the B-D displacement is 47–53 Å, thus closer to the Glu_EM structure. Diagonal distances at the core of the tetramer, measured at Q756 (for subunits B and D) and R660 (for subunits A and C) are much shorter in the tight tetramer and the CA-intermediate than in the apo structure (Fig. 9 F). These observations suggest that crystal structures of isolated LBDs tend to generate more tightly packed conformations than their full-length counterparts (4). This tighter LBD packing is still attainable in full-length receptors, as indicated by functional experiments. Indeed, the compact arrangement was essential for our cross-linking approach to succeed. However, none of these compact arrangements support maximally active channels.
Even so, the compact arrangement of the LBD tetramer we identified is notable because its condensed interdimer angle is distinct from the open interdimer angle obtained in full-length structures with the ion channel either closed or unresolved (the antagonist-bound, apo, and partial-agonist bound GluA2 structures, the Glu_EM model, and the agonist bound (but inhibited) GluN1/N2 heterodimer structures). Simple geometry suggests that an open angle between dimers allows the channel to be closed even when agonists are bound, because outward displacement of the linkers due to agonist binding is balanced by the lateral approach of the base of each LBD dimer toward the axis of the pore. When the tight arrangement is attained, the outward displacement of the linkers should be substantially more than in any of the full-length structures published to date (Fig. 9 E). Naively, the tight arrangement seems a more likely way to open the channel, if the linkers pull open the channel by a purely mechanical action (38). Such a simple relation between linker tension and channel activation is likely to be too simplistic however, and the tight arrangement might represent a configuration too compact to be stable, because the linkers to the channel are under excessive strain. In this interpretation, the tight arrangement would be occupied for a lesser fraction of the time than other fully bound states. In N-methyl-D-aspartate receptors, the interdimer angle may be controlled more strongly by the ATDs than in AMPA and kainate receptors (2, 3). Despite the use of zinc bridges in detecting proximity of subunits during activation, we cannot escape the limitation of the geometric and steric requirements for bridging, which fail to detect arrangements where sites in the dimers do not make contact, and which may disrupt a more stable arrangement of the four LBDs, in the fully glutamate-bound state. The bridges we report here cannot form in the Glu_EM LBD arrangement (14), or in partial agonist-bound structures (4), because the half-sites are not in close apposition.
HH and T1 bridges unequivocally form in the apo state (Fig. 5 A) (4). Strikingly, even a low concentration of glutamate, which should leave most LBDs unbound, was able to protect against trapping by both these bridges (Figs. 6 and 8). This result indicates that apo state is characterized by mobile LBDs, and this mobility is reduced by binding of even one glutamate molecule to one of the four LBDs. Individual subunits move enough to cross-link a range of geometries across the interdimer interface (T1, HH, HH D769E, and HH D769H), but there is a limit: the inter dimer angle does not collapse enough for the CA-HHH bridge to form in resting receptors.
Taken together, these results show that it is likely that multiple arrangements of the LBD tetramer are accessed during receptor activation, even when the LBDs are all glutamate bound and the active dimers are intact. Distinct LBD quaternary arrangements would be consistent with substate gating, which depends both on concentration of agonist (39, 40), and desensitization (25). The scarcity of interdimer contacts even in the compact arrangement presented here supports the possibility that individual AMPA receptor subunits might gate independently (39, 41).
Conclusion
In summary, analogies to existing structures suggest that condensation of the LBD layer into a compact arrangement is related to activation, possibly mainly being accessed by the LBD layers that are not fully bound by glutamate. Of further interest is how auxiliary subunits such as Stargazin interact (if at all) with these structural transitions through their extracellular loops (42). The burgeoning set of tetrameric iGluR structures will allow the design of experiments and simulations to determine the extent to which each of the conformations is occupied during normal gating.
The GluA2-TR and WT atomic coordinates and structure factors reported in this work have been deposited in the Protein Data Bank under codes PDB: 4YU0 and 4Z0I, respectively.
Author Contributions
J.B. and H.S. performed electrophysiological recordings and analyzed data; M.C. crystallized the ligand binding domains, collected synchrotron data, and solved the structures; A.L.C. recorded the R675S K7761M mutant; K.F. assisted with data collection and structure solution; A.Y.L. performed molecular modeling; A.P. conceived and supervised the project, analyzed data, and wrote the article with J.B., H.S., O.D., and A.Y.L. All authors discussed the interpretation of results and contents of the manuscript.
Acknowledgments
We thank Mark Mayer and Peter Seeburg for the gift of the GluA2 plasmid and Eric Gouaux for the gift of the GluA2 LBD expression vector.We thank Marcus Wietstruk for technical support and Yvette Roske (MDC-Berlin, Germany), Clarissa Eibl, and the BESSY staff at BL14.1 for help with data collection and analysis.
This work was supported by Deutsche Forschungsgemeinschaft (DFG) grants PL619.1, and the Cluster of Excellence NeuroCure (EXC-257) to A.P. H.S. was the recipient of a Long-Term Fellowship from Human Frontier Science Program (HFSP). X-ray data collection was performed at the Joint MX lab, Helmholtz-Zentrum Berlin für Materialien und Energie, Elektronenspeicherring BESSY II, Berlin, Germany.
Editor: Chris Lingle.
Footnotes
Jelena Baranovic, Miriam Chebli, and Hector Salazar contributed equally to this work.
One table is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(16)00006-0.
Supporting Material
References
- 1.Sobolevsky A.I., Rosconi M.P., Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karakas E., Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee C.-H., Lü W., Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dürr K.L., Chen L., Gouaux E. Structure and dynamics of AMPA receptor GluA2 in resting, pre-open, and desensitized states. Cell. 2014;158:778–792. doi: 10.1016/j.cell.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L., Dürr K.L., Gouaux E. X-ray structures of AMPA receptor-cone snail toxin complexes illuminate activation mechanism. Science. 2014;345:1021–1026. doi: 10.1126/science.1258409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yelshanskaya M.V., Li M., Sobolevsky A.I. Structure of an agonist-bound ionotropic glutamate receptor. Science. 2014;345:1070–1074. doi: 10.1126/science.1256508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong N., Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y., Olson R., Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa H., Singh S.K., Gouaux E. Subunit arrangement and function in NMDA receptors. Nature. 2005;438:185–192. doi: 10.1038/nature04089. [DOI] [PubMed] [Google Scholar]
- 11.Plested A.J.R., Mayer M.L. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 12.Lau A.Y., Roux B. The hidden energetics of ligand binding and activation in a glutamate receptor. Nat. Struct. Mol. Biol. 2011;18:283–287. doi: 10.1038/nsmb.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa T., Cheng Y., Walz T. Structure and different conformational states of native AMPA receptor complexes. Nature. 2005;433:545–549. doi: 10.1038/nature03328. [DOI] [PubMed] [Google Scholar]
- 14.Meyerson J.R., Kumar J., Subramaniam S. Structural mechanism of glutamate receptor activation and desensitization. Nature. 2014;514:328–334. doi: 10.1038/nature13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau A.Y., Salazar H., Roux B. A conformational intermediate in glutamate receptor activation. Neuron. 2013;79:492–503. doi: 10.1016/j.neuron.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy A.J., Grosse-Kunstleve R.W., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emsley P., Lohkamp B., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams P.D., Afonine P.V., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paoletti P., Ascher P., Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klippenstein V., Ghisi V., Plested A.J. Photoinactivation of glutamate receptors by genetically encoded unnatural amino acids. J. Neurosci. 2014;34:980–991. doi: 10.1523/JNEUROSCI.3725-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plested A.J., Mayer M.L. AMPA receptor ligand binding domain mobility revealed by functional cross linking. J. Neurosci. 2009;29:11912–11923. doi: 10.1523/JNEUROSCI.2971-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krivov G.G., Shapovalov M.V., Dunbrack R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbone A.L., Plested A.J. Coupled control of desensitization and gating by the ligand binding domain of glutamate receptors. Neuron. 2012;74:845–857. doi: 10.1016/j.neuron.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong N., Jasti J., Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 27.Das U., Kumar J., Plested A.J. Domain organization and function in GluK2 subtype kainate receptors. Proc. Natl. Acad. Sci. USA. 2010;107:8463–8468. doi: 10.1073/pnas.1000838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laitaoja M., Valjakka J., Jänis J. Zinc coordination spheres in protein structures. Inorg. Chem. 2013;52:10983–10991. doi: 10.1021/ic401072d. [DOI] [PubMed] [Google Scholar]
- 29.Harding M.M., Nowicki M.W., Walkinshaw M.D. Metals in protein structures: a review of their principal features. Crystallogr. Rev. 2010;16:247–302. [Google Scholar]
- 30.Elling C.E., Schwartz T.W. Connectivity and orientation of the seven helical bundle in the tachykinin NK-1 receptor probed by zinc site engineering. EMBO J. 1996;15:6213–6219. [PMC free article] [PubMed] [Google Scholar]
- 31.Partin K.M., Patneau D.K., Mayer M.L. Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol. Pharmacol. 1994;46:129–138. [PubMed] [Google Scholar]
- 32.Robert A., Armstrong N., Howe J.R. AMPA receptor binding cleft mutations that alter affinity, efficacy, and recovery from desensitization. J. Neurosci. 2005;25:3752–3762. doi: 10.1523/JNEUROSCI.0188-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Müller P., Köpke S., Sheldrick G.M. Is the bond-valence method able to identify metal atoms in protein structures? Acta Crystallogr. D Biol. Crystallogr. 2003;59:32–37. doi: 10.1107/s0907444902018000. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez J., Du M., Jayaraman V. Role of dimer interface in activation and desensitization in AMPA receptors. Proc. Natl. Acad. Sci. USA. 2010;107:9891–9896. doi: 10.1073/pnas.0911854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahmed A.H., Ptak C.P., Oswald R.E. Dynamics of cleft closure of the GluA2 ligand-binding domain in the presence of full and partial agonists revealed by hydrogen-deuterium exchange. J. Biol. Chem. 2013;288:27658–27666. doi: 10.1074/jbc.M113.495564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hald H., Ahring P.K., Kastrup J.S. Distinct structural features of cyclothiazide are responsible for effects on peak current amplitude and desensitization kinetics at iGluR2. J. Mol. Biol. 2009;391:906–917. doi: 10.1016/j.jmb.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Jin R., Gouaux E. Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry. 2003;42:5201–5213. doi: 10.1021/bi020632t. [DOI] [PubMed] [Google Scholar]
- 38.Kazi R., Dai J., Wollmuth L.P. Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat. Neurosci. 2014;17:914–922. doi: 10.1038/nn.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenmund C., Stern-Bach Y., Stevens C.F. The tetrameric structure of a glutamate receptor channel. Science. 1998;280:1596–1599. doi: 10.1126/science.280.5369.1596. [DOI] [PubMed] [Google Scholar]
- 40.Smith T.C., Howe J.R. Concentration-dependent substate behavior of native AMPA receptors. Nat. Neurosci. 2000;3:992–997. doi: 10.1038/79931. [DOI] [PubMed] [Google Scholar]
- 41.Prieto M.L., Wollmuth L.P. Gating modes in AMPA receptors. J. Neurosci. 2010;30:4449–4459. doi: 10.1523/JNEUROSCI.5613-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomita S., Adesnik H., Bredt D.S. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.