

SUMMARY
The prevalence of autism spectrum disorders (ASDs) is rapidly growing, yet its molecular basis is poorly understood. We used a systems approach in which ASD candidate genes were mapped onto the ubiquitous human protein complexes and the resulting complexes were characterized. The studies revealed the role of histone deacetylases (HDAC1/2) in regulating the expression of ASD orthologs in the embryonic mouse brain. Proteome-wide screens for the co-complexed subunits with HDAC1 and six other key ASD proteins in neuronal cells revealed a protein interaction network, which displayed preferential expression in fetal brain development, exhibited increased deleterious mutations in ASD cases, and were strongly regulated by FMRP and MECP2 causal for Fragile X and Rett syndromes, respectively. Overall, our study reveals molecular components in ASD, suggests a shared mechanism between the syndromic and idiopathic forms of ASDs, and provides a systems framework for analyzing complex human diseases.
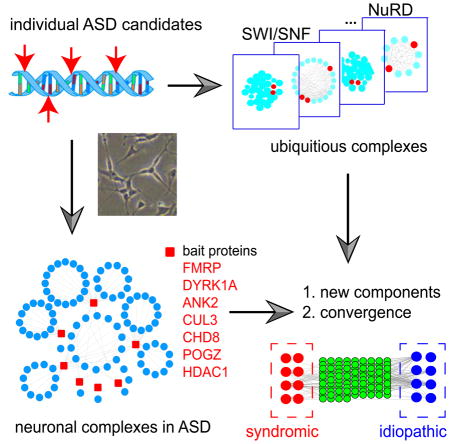
Graphical Abstract
INTRODUCTION
Autism spectrum disorders (ASDs) have a strong genetic component; however identifying the associated genetic elements has been challenging due to extreme locus heterogeneity: combining all of the information obtained thus far reveals a genetic cause for only at most, 25% of ASD cases(Huguet et al., 2013). To date, most ASD-associated genes have been identified from mutation analysis. However, since heritable mutations in the extant human populations have been shaped by mutational stochasticity and natural selection, given the substantially reduced fertility for males with ASD(Power et al., 2013), the heritable mutations associated with ASD might not be able to reach high frequencies, and thus might not be readily captured by typical mutational screens, especially those targeting common variants (such as genome-wide association studies). More importantly, since many fundamentally important bioprocesses are implicated in ASD and ASD-associated genes tend to be essential (Georgi et al., 2013), deleterious mutations in these genes might not be captured by any mutational screen unless the mutations are hypomorphic. Therefore, many molecular components in ASD have remained unidentified, whose effects cannot be readily revealed by regular mutational screens, necessitating the development of new research strategies.
Integrative analyses have been recently performed to uncover the hidden genetic architecture in ASD. These include construction of gene co-expression (or functional co-association) network to identify gene groups relevant to ASD(Gilman et al., 2011; Parikshak et al., 2013; Willsey et al., 2013), and topological deconstruction of the global human protein interactome to reveal molecular pathways in ASD (Hormozdiari et al., 2014; Li et al., 2014). However, these computational approaches were at a high-level description rather than grounded on the detailed mechanisms of action in a specific cellular context. Additional experimental strategies, such as yeast two-hybrid (Y2H) screens, have mapped the binary physical interactions for a selected set of ASD candidates(Corominas et al., 2014; Sakai et al., 2011). Since Y2H assesses the intrinsic binding capacity between interacting proteins in a non-native state, it remains unclear whether the in vitro protein-protein interactions (PPIs) identified from Y2H will also be observed in a cellular context.
Since protein complexes are functional building blocks in a cell, we devised a systems framework to identify human cellular protein complexes associated with ASD (Fig. 1). Unlike previous approaches in which disease-related pathways are directly inferred from a collection of individually identified susceptible loci, our strategy directly investigates protein complexes and is able to reveal the sets of naturally interacting proteins and pathways in ASD. This approach is generalizable and can be easily extended to identifying disease relevant pathways in other complex human diseases.
Fig. 1. An Overview of This Study.

Major procedures, observations and conclusions are summarized in each box from Step 1 to Step 5. We first examined a comprehensive set of ubiquitously expressed human protein complexes and identify the protein subunits co-complexed with ASD candidate proteins (Step 1). We then functionally characterized these co-complexed subunits and characterized their phenotypes in mouse mutants (Step 2a). We studied HDAC1/2 in the NuRD chromatin remodeling complex for their roles in regulating ASD candidate genes in mouse embryonic brain (Step 2b). In neuronal cells, we performed immunoprecipitation followed by mass-spectrometry analysis (IP-MS) to derive the co-complexed subunits with seven key ASD-associated proteins (Step3). These neuronal complexes were then assembled into an high-quality protein complex network (Step 4). This neuronal network was then functionally characterized for their temporal expression dynamics during neocortical development. The network allowed identifying novel ASD-associated components, displayed increased rate of deleterious mutations in ASD cases, and were regulated by the ASD-associated syndromic factors, FMRPI304N and MECP2, which are causal for Fragile X and Rett syndromes, respectively (Step 5).
Our approach involved first analyzing ubiquitously expressed protein complexes and then neuronal complexes to obtain a more comprehensive view of ASD-associated pathways (Fig. 1). Both approaches identified key components in ASD that have not yet been reported previously, and our results revealed the convergent regulation of two key syndromic regulators FMRP (mediating translational inhibition, causal for Fragile X syndrome) and MECP2 (a DNA methylation binding protein, causal for Rett syndrome), which each operate on the protein complex targets identified in this study. Our study thereby provides a systems framework to unravel genetic architecture of complex human diseases and complements the mutation screens in standard sequencing analysis.
RESULTS
Identification of Ubiquitous Human Protein Complexes in ASD
Several fundamental bioprocesses have been implicated in ASD, such as translation(Santini et al., 2012) and chromatin remodeling, suggesting that many ASD candidate genes are likely ubiquitously expressed. We thus first explored the ubiquitously expressed human protein complexes to identify the subunits co-complexed with known ASD candidate proteins. We examined a comprehensive list of 622 soluble stable protein complexes derived from a recent study based on high-throughput complex fractionation followed by tandem mass spectrometry (MS) (Havugimana et al., 2012). This dataset represents an extensive systematic search for human protein complexes. Compared with individually curated protein complexes from the literature, this set of 622 complexes is expected to have significantly less ascertainment bias. The original study showed that these complexes have high abundance across diverse human tissues, including the human brain(Havugimana et al., 2012), which were subsequently validated by our own RNA-Seq data from tissues collected from the postmortem dorsolateral prefrontal cortex (Supplemental Experimental Procedures and Fig.S1).
We analyzed 378 ASD-associated genes affected by different types of ASD-associated mutations, including high-confidence ASD-associated de novo CNVs(Noh et al., 2013), syndromic mutations (from SFARI annotations, Category S), de novo loss-of-function mutations from recent exome-sequencing studies (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012; Willsey et al., 2013)(Supplemental Experimental Procedures and Table S1). The vast majority of these ASD candidates were from unbiased genome or exome-wide screens. These 378 genes mapped to 98 distinct protein complexes (Fig. 2A, Table S2), which implicated several complexes to ASD, including the chromatin remodeling SWI/SNF complex involving the well-known BAF subunits in many neuropsychiatric diseases (Ronan et al., 2013) (Fig. 2A). It is known that ASD candidates are enriched for synaptic and chromatin-remodeling proteins (De Rubeis et al., 2014), many of which form protein complexes to achieve their functions; therefore it is less interesting to simply test whether ASD proteins are preferentially involved in protein complexes. Meanwhile, we cannot test whether a given complex is enriched for subunits encoding ASD genes since a single subunit could associate the entire protein complex with ASD, such as AUTS2 in the AUTS2-Polycomb complex (Gao et al., 2014). We postulated that if protein complexes represent functional units on which disease-associated mutations converge, we expect to see that subunits co-complexed with known ASD candidates are also likely involved in this disease. We thus directly tested this hypothesis.
Fig. 2. Identifying Human Protein Complexes in ASD.

(A). An overview of the ASD-associated proteins in the 622 stable protein complexes, where the Mi-2/NuRD and the SWI/SNF complexes are shown as examples. Nodes represent the ASD-associated proteins (red) and their co-complexed subunits (green). (B). Differential GO term enrichment between the genes co-complexed with ASD and those with non-ASD genes. Each node represented one GO term, and the edges represent term-term similarity. The color gradient indicates the gene percentage difference of each term between the two complex groups. The node size indicates the statistical enrichment of a given node. ClueGO was used to perform this differential GO term analysis. (C). Enriched functions for the subunits co-complexed with proteins affected by de novo mutations identified from unaffected siblings. ** indicates adjusted P≤0.01.
Since the vast majority of the ASD candidates (>86%) in this study were from previous screens for de novo mutations (note that many syndromic mutations were also spontaneous), as a matched control set, we considered 592 genes affected by de novo loss-of-function mutations or exon-affecting CNVs (copy number variants) from unaffected siblings or healthy control subjects in previous studies (Supplemental Experimental Procedures); these mapped to 87 protein complexes. We found that that the sizes of the complexes harboring ASD candidates did not significantly differ from those encompassing the control proteins (P=0.31, Wilcoxon rank-sum test).
Excluding the ASD candidates and the control proteins, we compared the functional differences (Gene Ontology, GO) between the two sets of protein complex subunits based on ClueGo(Bindea et al., 2009). Notably, the enriched GO terms for the subunits co-complexed with the ASD candidates were highly divergent from those of the subunits co-complexed with the control proteins. Specifically, two major functional components (Fig. 2B), intracellular transport (adjusted P=3.8e-15, hypergeometric gest corrected by Bonferroni step-down) and ATP-dependent chromatin remodeling (adjusted P=3.8e-6) exhibited significant functional specificities for the subunits co-complexed with ASD candidates, encompassing the individually enriched terms such as mRNA export (adjusted P=7.8e-4), protein transmembrane transport (adjusted P=3.21e-5) and the NuRD (Nucleosome Remodeling Deacetylase) complex (adjusted P=8.3e-5). Several individual terms outside of the two main functional components also showed enrichment, such as membrane docking (adjusted P=0.03) and exocyst (adjusted P=4e-3). In contrast, the functional specificities (Fig. 2B) of the control subunits largely reflect a background enrichment of the entire set of ubiquitous complexes.
We next performed the same analysis on the morphological traits of the orthologous mouse mutants and observed that the subunits co-complexed with ASD candidates showed significant enrichment for abnormal brain and neuron morphology, as well as for abnormal nervous system and neurodegeneration (Fig. 2C, adjusted P≤0.05, Table S2). These terms were insignificant for the subunits co-complexed with the control proteins (adjusted P≥0.12). Taken together these comparisons suggest ASD-association of the subunits co-complexed with known ASD candidates and a role for intracellular transport and chromatin remodeling underpinning this disease.
HDAC1/2 Regulate ASD-associated Genes in the Embryonic Mouse Brain
To investigate how the ubiquitous protein complexes could achieve brain-specific functions, we specifically chose the NuRD complex because it is ubiquitous in different cell types and has been recently shown to regulate synaptic connectivity in the rodent cerebellar cortex (Yamada et al., 2014). NuRD contains two subunits affected by ASD-associated CNVs (Fig. 2A); one of these Hdac1 in mouse targets the NuRD to specific chromosomal locations and is involved in the differentiation of presynaptic sites (Yamada et al., 2014). In humans, eleven HDACs have been identified, but only HDAC1/2 are subunits of the NuRD complex, highlighting their functional divergence from other HDACs. Although HDACs have been associated with fear, memory, stress, depression, schizophrenia and Alzheimer’s disease (Volmar and Wahlestedt, 2015), their direct role in ASD has not been established. Pharmacological studies have shown that the administration of HDAC inhibitors (pan-specific or those only specific for HDAC1-3) could ameliorate deficits in cognition and social interaction in a rodent model of ASD(Foley et al., 2012). Given the recent availability of Hdac1/2 knockout data in the embryonic mouse brain(Hagelkruys et al., 2014), it has become possible now to study Hdac1/2 for their potential involvement in ASD associated pathways.
Hdac1 and Hdac2 have partially redundant functions in the embryonic mouse brain (Hagelkruys et al., 2014), and we therefore examined the transcriptome response in the embryonic mouse brain when both genes were deleted (Hdac1Δ/ΔnHdac2Δ/Δn) at E14.5, the time point in which severe knockout phenotypes start to emerge (Hagelkruys et al., 2014). We mapped the human ASD candidates onto their unambiguous one-to-one mouse orthologs (341 of 378 ASD candidate genes mapped), and observed that these ASD candidate orthologs displayed significant down-regulation in the mouse brain at E14.5 when both Hdac1 and Hdac2 were knocked out (P=2.4e-7, Wilcoxon rank-sum test, relative to mouse genes with one-to-one human orthologs, Fig. 3A).
Fig. 3. The Regulatory Role of HDAC1 and HDAC2 in ASD.

(A). Log2-fold changes of genes in the embryonic mouse brain (E14.5) upon Hdac1Δ/Δn Hdac2Δ/Δn. Mouse orthologs of human genes implicated in intellectual disability (ID), schizophrenia (SZ) and Alzheimer’s disease (ALZ) and autism (ASD) were compared with all the mouse genes with one-to-one protein-coding human orthologs (transcriptome, X-axis). Statistical significance was derived from Wilcoxon rank-sum test. (B). Representative ASD-implicated genes showing substantial down-regulation of the double knockout (DKO; in triplicate) of HDAC1 and HDAC2 in mouse embryonic brain compared to wild type (WT; in duplicate). Gene expression was converted back from the log2 scale to the original measurements.
Since HDAC2 is implicated in Alzheimer’s disease(Graff et al., 2012), we further examined whether the above down-regulation was specific for ASD. We analyzed genes implicated in intellectual disability (ID, 401 genes), schizophrenia (SZ, 499 genes), and Alzheimer’s disease (ALZ, 613 genes) (Supplemental Experimental Procedures). The down-regulation was absent for the genes implicated in ID, ALZ (Fig. 3A), but milder in SZ (Fig. 3A). Notably, mouse orthologs of the ALZ genes displayed significant up-regulation in the Hdac1Δ/ΔnHdac2Δ/Δn double-knockout mutants (P=1.1e-6, Wilcoxon rank-sum test), suggesting that Hdac1 and Hdac2 negatively regulate ALZ genes, but positively regulate ASD genes. Specially, as exemplified in Fig. 3B, among the down-regulated genes for ASD, mouse orthologs of many well-established ASD genes were significantly involved, including five (Chd8, Cul3, Dyrk1a, Grin2b and Tbr1) of the nine high-confidence recurrent hits from a recent exome-sequencing study with an expanded ASD cohort (Willsey et al., 2013). Genes involved in language disabilities, Foxp2 and Cntnap2, also exhibited remarkable down-regulation. The highly repressed genes also included Mecp2 (causal for Rett syndrome), Ube3a (causal for Angelman syndrome), Nlgn3 and the synaptic scaffolding component Dlg4 (PSD-95, inter-connecting SHANK-neurexin-neuroligin pathway), whose mouse mutant showed severe autistic phenotypes(Feyder et al., 2010). In addition, the BAF complex subunit BAF170 (Smarcc2) also exhibited expression reduction, regulating cerebral cortical size and thickness (Tuoc et al., 2013), and this gene harbored de novo CNV and splicing-site-affecting mutations among individuals with ASD(Neale et al., 2012; Sanders et al., 2011).
We next examined the transcriptome response in mouse brains expressing a single allele of either HDAC1 or HDAC2 at E14.5 (mice with Hdac1Δ/+nHdac2Δ/Δ or with Hdac1Δ/ΔHdac2Δ/+n)(Hagelkruys et al., 2014). Despite the down-regulation in Hdac1Δ/ΔnHdac2Δ/Δn mice, the presence of either single allele restored expression of these ASD candidates to a level similar to that of the transcriptome background (Fig. S2), suggesting that Hdac1 and Hdac2 each individually exert regulation on the ASD candidate genes. Since mice with a single Hdac1 allele (Hdac1Δ/+nHdac2Δ/Δ) die at the first day after birth (P0) accompanied with the dysregulation of genes different from those at E14.5, we examined the response of the ASD candidates at P0 upon Hdac1Δ/+nHdac2Δ/Δ, and found that they did not show significant differential expression (Fig. S2). Taken together, we established that HDAC1 and HDAC2 positively regulate ASD gene candidates during early brain development.
Interactions Mediated by ASD Candidate Proteins in Neuronal cells
To gain more mechanistic insights into ASD, we sought to identify protein complexes in a neuronal context. Large-scale profiling of physical protein interactions in specific tissues or induced pluripotent stem cell (iPSC)-derived neurons is currently challenging due to the large number of cells required. We therefore differentiated SH-SY5Y cells into neuron-like cells (Supplemental Experimental Procedures), which have been frequently used to model neurological and neuropsychiatric diseases including ASD(Chiocchetti et al., 2014). We confirmed the neuronal characteristics of the differentiated cells by visual inspection of neuronal morphology (Fig. 4A) as well as by the overall up-regulation of neuron markers upon differentiation (P=7.7e-5, Wilcoxon rank-sum test) based on our RNA-Seq profiling (Supplemental Experimental Procedures and Table S3).
Fig. 4. Identifying Neuronal Proteins Co-complexed with ASD-associated Proteins.

(A). The flowchart of the affinity purification experiments followed by mass-spectrometry analysis. (B). The prey proteins co-purified with the baits were examined using a Bayesian mixture model. The bait-prey interactions assigned with the highest posterior probabilities (≥80%) as true interactions are highly enriched for known protein interactions in literature. The statistical significance was derived from Fisher’s exact test. (C). The inverse-tangent normalized spectral count of the identified prey proteins co-purified with the bait proteins (X-axis) in two biological replicates. (D). The identified prey proteins exhibited substantially increased expression in neuronal regions including Brodmann areas 9 (BA9), 40 (BA40) and the amygdala (AMY). In each region, a subset of genes with FPKM>0 was used as background control. The statistical significance was derived from Wilcoxon rank-sum test. (E). The identified prey proteins exhibited substantially increased expression correlation with their respective bait proteins during brain development. The temporal transcriptome datasets from Cortecon and BrainSpan were examined. The statistical significance was derived from Wilcoxon rank-sum test.
For the immunoprecipitation experiments, we targeted eleven ASD-associated genes from the following three broad categories: Group 1, comprising nine genes that have arguably high confidence in idiopathic ASD(Willsey et al., 2013): ANK2, CHD8, CUL3, DYRK1A, GRIN2B, KATNAL2, POGZ, SCN2A and TBR1. Group 2, encompassing the gene FMR1 (causal for Fragile X Syndrome, FXS) encoding for an RNA-binding protein (FMRP) that exerts translational repression on its target messengers. More than 30 proteins have been identified as its interacting partners (the BioGrid database); however most of these interactions were identified from Y2H assays in vitro or from non-neuronal cells. Hence, identifying complexes encompassing FMRP in a neuronal context is expected to elucidate its role in neuropsychiatric diseases. Group 3, comprising the aforementioned HDAC1 (co-complexed with HDAC2), which was affected by an ASD-associated de novo CNV (Fig. 2A).
These prioritized eleven proteins as baits were subjected to immunoprecipitation in the differentiated SH-SY5Y neuron-like cells (Table S4), followed by high-resolution mass spectrometry analysis (IP-MS) to identify their endogenous co-complexed neuronal subunits (Fig. 4A, Supplemental Experimental Procedures). A total of 26 independent proteome-wide co-purification experiments were performed, including two biological replicates for each of the eleven bait proteins and four independent negative control experiments (Supplemental Experimental Procedures). Among the eleven bait proteins, we successfully recovered seven baits in both biological replicates in their respective purified immunoisolates (ANK2, CHD8, CUL3, DYRK1A, FMRP, POGZ and HDAC1) and confirmed low abundance of the remaining four unrecovered bait proteins (GRIN2B, TBR1, SCN2A, and KATNAL2) by both RNA-Seq and immunoblot assays (Fig. S3). Therefore, we focused only on the seven ASD-associated bait proteins for their neuronal protein interactions.
After filtering with the peptide identification confidence 90% and removing the most common background contaminants (Supplemental Experimental Procedures), we identified 2812 putative PPI pairs from seven bait proteins. These putative interactions were then scored by SAINTexpress(Mellacheruvu et al., 2013), which assigned each interacting bait-prey pair with a Bayesian posterior probability of true interaction by comparing their MS/MS spectra counts between the seven bait proteins and the negative controls (Supplemental Experimental Procedures). We retained 119 interactions (involving 95 prey proteins, Table S5) above the probability threshold 80%, which exhibited the strongest enrichment (Fig. 4B, P=3.97e-6, Fisher’s exact test) towards known PPIs (Turinsky et al., 2011). The MS/MS spectral counts for the 119 high-confidence interactions are shown in Fig. 4C, and the binding specificities with the seven bait proteins were clearly consistent between the two biological replicates, with an average fold enrichment of 57.46 relative to the negative control experiments.
Coordinated Expression of the Identified Interacting Proteins in the Human Brain
We reasoned that if the identified 119 high-confidence interactions indeed reflect the molecular activities in the human brain, the interacting proteins are expected to be co-expressed during brain development. Co-expression also dictates the quality of the identified PPIs. We first examined the mRNA expression of the 95 identified prey proteins in Brodmann areas 9, 40 (BA9 and BA40) and the amygdala (AMY) using our published dataset(Li et al., 2014). We found that the identified prey proteins were substantially elevated in expression (Fig. 4D, P≤9.9e-9, Wilcoxon rank-sum test), demonstrating their overall activity in these neuronal brain regions.
Next, we examined two sets of temporal transcriptome data for brain development: (1) the CORTECON dataset in which human embryonic stem cells (hESCs) were differentiated into cortical neurons (van de Leemput et al., 2014); and (2) the BrainSpan dataset in which expression profiling was performed in the human neocortex across eight different developmental stages from the post-conceptual week (PCW) 8-10 (early fetal) to the postnatal 12 months(Parikshak et al., 2013). For both datasets, we only considered the protein-coding genes with moderate or high expression in the brain (mean FPKMs ≥1 across developmental stages), and computed pairwise Pearson’s correlation (R) for each of the 119 high-confidence bait-prey interactions across the developmental stages. In both CORTECON and BrainSpan, these interacting proteins showed significantly increased expression correlation relative to the co-expression of the seven bait proteins with all the brain expressed protein-coding genes (P=1.9e-5 and 0.04 for CORTECON and BrainSpan, respectively, Wilcoxon rank-sum test, Fig. 4E). Collectively, our analyses demonstrated the overall quality of the 119 high-confidence bait-prey interactions, which revealed their co-complex membership in a cellular context.
High-Quality Physical Interactions Form an ASD-associated Interactive Network
Although the seven bait proteins were individually identified as ASD-associated candidates, the 119 bait-prey interactions form an interactive network (Fig. 5A, except for POGZ). We observed that 15 prey proteins were shared by at least two bait proteins, and thus we tested the null hypothesis whether the same number of shared proteins would be expected from non-specific protein interactions. We mimicked the IP-MS by randomly sampling (to nullify the binding specificity of interacting proteins) the same number of moderately or highly expressed brain-expressed genes (FPKM>1 in BA9) for each bait protein, and observed that these ASD proteins were more likely to target a common set of genes (P<1e-4, permutation test), suggesting a convergent network underlying the organization of these ASD proteins. One example is ACOT7, which simultaneously interacted with CHD8, ANK2, FMRP, and CUL3. These interactions were specific as our antibody-based co-IPs confirmed the presence of ACOT7 protein in the purified cell lysates of the ANK2, FMRP, CUL3, and CHD8, respectively, but not from the control-IP experiments (Fig. 5B, Supplemental Experimental Procedures). These validations support the high quality of our interactome data and suggest that ACOT7 might be implicated in ASD. Further examination of its expression in the cerebellar vermis, superior gyrus, and prefrontal cortex(Voineagu et al., 2011) revealed a substantial expression reduction only in the prefrontal cortex among the ASD cases relative to the control subjects (P=0.02, Wilcoxon rank-sum test, Fig. 5C), suggesting that the nonsense mutations observed from CHD8, ANK2, FMR1 and CUL3 may have a shared molecular etiology with ACOT7 dysregulation in ASD. Taken together, the identified network represents a set of pathways where ASD-associated factors are likely to converge.
Fig. 5. The Neuronal Protein Interactome of Seven Key ASD Proteins.

A). The identified high-confidence bait-prey interactions, encompassing 7 bait proteins (the squared nodes), 95 distinct prey proteins (the circled nodes), and 119 co-complex interactions. The red and blue colors for the nodes indicate biased brain expression in the early fetal or the postnatal stages, respectively. The white color of the node borders indicate the genes differentially regulated by FMRPI304N causal for Fragile X syndrome. Four interactions mediated by ACOT (highlight in white) with CUL3, CHD8, ANK2 and FMR1 were individually validated using the Western blot assay. (B). Immunoblot validation of ACOT7-mediated interactions with CUL3, CHD8, ANK2 and FMR1. (C). ACOT7 displayed significantly reduced expression in the prefrontal cortex in individuals with ASD relative to the controls, whereas the expression in the superior temporal gyrus and the cerebellar vermis was no longer significant. The statistical significance was derived from the Wilcoxon rank-sum test. (D). The increased mutational burden of deleterious mutations in the network for individuals with ASD. The 95 prey proteins were compared with a set of matched control gene sets with similar CDS length and GC content, and the comparisons were performed separately in individuals with ASD and also in the non-ASD subjects and only the non-synonymous variants specifically observed in each group were considered. The deleterious mutations were predicted by MutationTaster.
In the whole network, these interacting prey proteins displayed a significant enrichment in the proteome of the human postsynaptic density (PSD)(Bayés et al., 2010) relative to genes with moderate to high expression (FPKM>1, Fig. 4D) in the BA9, BA40, AMY regions as well as in the differentiated neuron-like cells (P<0.03, Fisher’s exact test, Table S5). Mammalian phenotypic enrichment test further revealed that mouse mutants for the 95 prey proteins displayed defects in neuron physiology (adjusted P=0.024 by Benjamini-Hochberg’s method), neuron morphology (adjusted P=0.05), and embryogenesis (adjusted P=3.98e-3) relative to wild type mice. Overall, these analyses suggest the significance of the identified interacting proteins in maintaining neuronal functions.
Increased Mutational Burden of the Identified Interaction Network
To assess whether the identified prey proteins harbored a significant number of deleterious mutations in ASD cases, we examined the non-synonymous variants from an exome-sequencing study, which included 505 ASD cases and 491 matched control subjects with unrelated European ancestry (Liu et al., 2013) (Supplemental Experimental Procedures). We reasoned that if the mutations converge onto a molecular network implicated in the disease (rather than individual genes), two predictions immediately follow: (1) compared with other genomic regions (genomic background), genes implicated in the network are more likely to harbor deleterious mutations; and (2) the increased level of deleterious mutations should not be observed in unaffected individuals. We tested these predictions individually.
It is known that subjects with ASD and their matched controls have almost equal frequencies of common variants (Anney et al., 2012). We thus examined the variants observed among individuals with ASD, but not in the control subjects. To determine the statistical significance of the mutational enrichment on the 95 prey proteins, we compiled a list of 9201 genes whose CDS length and GC content are similar with these identified prey proteins (P=0.297 and 0.98 for CDS length and GC content, respectively, Wilcoxon rank-sum test; Supplemental Experimental Procedures). This large set of control genes thus allowed us to more accurately estimate a background distribution of the deleterious non-synonymous mutations in genes presumably not directly associated with ASD.
We studied 40,830 non-synonymous variants with predicted mutational effects by MutationTaster (Schwarz et al., 2010), which were specifically identified in ASD individuals but not in their matched control subjects. We observed a modest but statistically significant increase in the fraction of deleterious (prediction score equal to or greater than 0.99) non-synonymous mutations in the network relative to those detected in the control gene set (Fig. 5D, P=9.3e-3, Fisher’s exact test; the seven bait proteins were excluded from the analysis). Conversely, when considering 31,668 variants specifically observed in the non-ASD control subjects with predicted mutational effects, the fraction of deleterious mutations on this network was almost identical with the negative control gene set (Fig. 5D, P=0.79, Fisher’s exact test). Collectively, this comparative analysis reveals increased mutational burden on these identified prey proteins and further implicates the interaction network in ASD.
Functional Implication of the Network in Early Fetal Brain Development
Given the overall co-expression of the interacting proteins in the neocortex across human brain developmental stages (PCW 8 to postnatal 12-month, analyzed in Fig. 4E), we examined whether the network is active in a specific developmental stage. We used β to denote the ratio of expression in the early fetal development (PCW 8-10) relative to the mean expression in the postnatal stages (4months, 10 months and 12 months), and increased β indicates more biased expression in the early fetal brain development. We observed that the overall network showed a significant increase in β (red nodes in Fig. 5A, and also see the comparison in Fig. 6A, P=1.4e-12, Wilcoxon rank-sum test) relative to all the 12,140 genes with moderate or high expression in the pre-frontal cortex (represented by genes with FPKM>1 in BA9). This enrichment was particularly pronounced for proteins interacting with FMRP, HDAC1 and DYRK1A (P=6.9e-4, 2.2e-6 and 6.5e-5, respectively, Wilcoxon rank-sum test, Fig. 6A). For HDAC1, members of the NuRD complex all showed substantial expression bias towards the early fetal brain development (HDAC1/2, CHD4, MTA1/2 and GATAD2A etc. Fig. 5A). Overall, the strongly biased expression in the early fetal stage suggests an early origin of this disease.
Fig. 6. Analysis of the Identified Neuronal Protein Interaction Network.

(A). Analysis of expression bias β (Y-axis) in early neocortical developmental stage (PCW 8) relative to the postnatal stages (4, 10 and 12 months). β values of all the prey proteins or of the interacting proteins with their respective ASD-associated bait proteins (listed on X-aixs) were compared with that of brain expressed genes, represented by genes with FPKM >1 in BA9. The statistical significance was derived from the Wilcoxon rank-sum test. (B). Expression dynamics in the neocortex for the identified prey proteins interacting with each bait across 8 different brain developmental stages (X-axis). (C). Positive correlation between β and FMRP site density for the proteins interacting with HDAC1. FMRP site density is the number of FMRP PAR-CLIP sites in CDS or UTRs per Kb. (D). The identified prey proteins is significantly enriched for genes harboring FMRP binding sites ablated by FMRPI304N. The comparisons were made between each gene group and their matched control gene set with similar expression level and cDNA length. Literature curated SFARI genes, ASD-associated genes in this study, and genes affected by de novo CNVs in ASD probands were also analyzed together the identified prey proteins in the network. The statistical significance was derived from Fisher’s exact test. (E). MECP2 repression on the 95 prey proteins in the network. The statistical significance of the up-regulation of the orthologous prey proteins in mouse cortical neurons upon anti-MECP2 shRNA knockdown was determined by Wilcoxon rank-sum test. The significance increase relative to the transcriptome background was absent from the mock control. The log2-fold change (y-axis) was determined by gene expression after shRNA treatment relative to that after transfection of an anti-luciferases shRNA control. (F) A proposed model for the shared molecular basis of the idiopathic and syndromic forms of ASD. The green nodes represent the shared interaction partners of the red and blue nodes for proteins implicated in the syndromic and idiopathic forms of ASD, respectively. The interactions (grey edges) indicate functional dependencies (e.g. physical, regulatory or epistatic interactions).
We further examined the expression dynamics of each bait protein together with their interacting partners at individual brain developmental stages (Fig. 6B). CUL3 and its interacting partners exhibited tight expression correlation across brain developmental stages, reflecting high dosage sensitivity of these interacting proteins, and their expression levels were significantly repressed in the postnatal 12-month brain. HDAC1 however showed a different pattern, in which its expression was specifically up-regulated at PCW 8-10, and was then repressed across all the other stages (corresponding to its high β value in Fig. 6A). Its interacting proteins showed a similar pattern, but were more gradually shifted from the highest level in the early fetal stages to lower expression in the postnatal stages, corresponding to their elevated expression bias β in Fig. 6A. FMR1 showed the opposite trend; in the prenatal stages its expression co-fluctuated with its interacting partners, but became discordant in the postnatal stages by up-regulating the FMR1 level (Fig. 6B).
FMRP Preferentially Regulates Components of the Network
Since FMRP post-transcriptionally represses protein translation of its target messengers(Darnell et al., 2011), the observation of the significant up-regulation of FMR1 and the overall down-regulation of many other proteins in the network at the same postnatal stages (Fig. 6B) led us to hypothesize that FMRP likely post-transcriptionally regulates genes in this network. This was not only because of the role of FMRP as a repressor, but also because such a post-transcriptional regulatory mechanism is often employed in eukaryotic cells to reinforce transcriptional logic, serving as a surveillance system to suppress “leaky” transcripts, which otherwise might exhibit dosage fluctuation that is highly deleterious(Tsang et al., 2007).
We therefore examined FMRP-mediated regulation identified from the PAR-CLIP system (Ascano et al., 2012). Compared with FMRP target genes identified by other platforms, only PAR-CLIP was able to identify the exact binding sites at the nucleotide resolution. Notably, although the PAR-CLIP experiments were performed on HEK293 cells, many of these PAR-CLIP results have also been validated using the human brain tissues, and comparisons also showed that 90% of genes expressed in these cells were also expressed in human brain(Ascano et al., 2012). Considering FMRP binding sites are mostly localized in coding regions (CDS) or the un-translated regions (3′ UTRs and 5′ UTRs) and that genes with greater length may have more binding sites, we computed FMRP site density for each gene in the network, where the number of sites in CDS and UTRs were normalized by the cDNA length of each gene. Different from other bait proteins, we observed that the HDAC1-interacting proteins displayed a significant positive correlation (R=0.42, P=0.02) between the FMRP site density (number of sites per Kb) and their β values (expression bias towards the early fetal stage PCW 8-10 relative to the postnatal stages, Fig. 5A and Fig. 6A), where genes highly expressed in the early fetal stage (fold change >2 relative to the postnatal stages) had the highest density of FMRP binding sites (Fig. 6C). This observation thus suggests a post-transcriptional role of FMRP in controlling the HDAC1-mediated interactions.
Perturbation of the Interaction Network by the Syndromic FMRPI304N
Since FMRP is causal for fragile X syndrome (FXS), we asked whether the identified PPI network (seeded by the ASD genes for idiopathic ASD) could bridge the gap between the idiopathic and syndromic forms of ASD. The pathogenic mutation FMRPI304N causing FXS has been analyzed by the PAR-CLIP platform (Ascano et al., 2012), where the mutant protein exhibited attenuated RNA-binding affinity due to the mutation in its KH2 RNA-binding domain. With the same set of data, a recent study has developed a hidden Markov model that identified 9,549 transcriptomic locations strongly bound by FMRPWT, but not by FMRPI304N(Wang et al., 2014). We mapped these sites onto RefSeq genes, and identified 1,925 genes harboring at least one such site in their CDS or UTRs. Among the 95 prey proteins in our network, 35 were affected by FMRPI304N (i.e. with substantially reduced binding affinity by FMRPI304N, Fig. 5A), as were 5 (HDAC1, DYRK1A, CUL3, CHD8, and POGZ) of the 7 bait proteins. Our statistical analyses further determined that the number of genes affected by FMRPI304N was significantly enriched in our network compared with control genes (matched with similar cDNA length and expression levels; see Supplemental Experimental Procedures) (Fig. 6D, P=0.01, Fisher’s exact test), and the enrichment was specific for our network, but absent in other ASD-associated genes from multiple sources (Fig. 6D, Supplemental Experimental Procedures). Overall, these results suggest that the interacting proteins (Fig. 5A) constitute a specific molecular network under the post-transcriptional control of FMRP, and the pathogenic mutation I304N in FMRP significantly ablates the regulatory mechanism on this network, contributing to FXS.
Regulation of MECP2 on the Network in Mouse Cortical Neurons
To further establish the association of the network with syndromic forms of ASD, we tested the regulation of MECP2 (methyl CpG binding protein 2, causal for Rett syndrome) onto the interaction network. We re-analyzed the published data(Lanz et al., 2013), namely the transcriptomic responses in murine cortical neurons upon individually knocking down eight ASD associated genes using shRNAs (short hairpin RNAs) against Mecp2, Mef2a, Mef2d, Fmr1, Nlgn1, Nlgn3, Pten and Shank3. The knockdown efficiency had been reported to achieve at least 75% expression reduction in quadruplicate experiments. These cortical neurons were obtained from the murine embryonic brain at E16 (corresponding to human PCW 8, 56–60 days), which makes it possible to draw parallels to the humans as the network identified in our study exhibited an overall increased transcriptional dynamics at PCW 8-10 (the increased β for all the prey proteins, Fig. 6A).
We mapped the 95 prey proteins in the network onto their one-to-one mouse orthologs, and determined their response to shRNA treatment against each of the eight ASD genes. Fold changes were computed for gene expression after individual shRNA treatment relative to expression after transfection of an anti-luciferase shRNA control. We observed that mouse orthologs of the prey proteins exhibited significant up-regulation upon Mecp2 knock-down (Fig. 6E, FDR=7.6e-3, Wilcoxon rank-sum test relative to the transcriptome background, Benjamini-Hochberg correction for all the eight shRNA experiments), whereas the differential expression was absent for all the other seven ASD-associated genes nor for the mock control (FDRs≥0.2, Wilcoxon rank-sum test). Close examination further revealed that the up-regulation upon Mecp2 knockdown was substantial (Fig. 6E, median fold change of 2.11) with Hdac2 and Gatad2a in the NuRD complex changing greater than 2.5- fold and the neuronal signaling protein Flot2 more than 3-fold. Overall, these observations suggest considerable Mecp2 repression on the network in mouse embryonic cortical neurons in mouse.
DISCUSSION
A hallmark of ASD is its extreme locus heterogeneity, where the recurrence of causal mutations is typically rare within individuals with ASD. Therefore, it is unlikely that it will be possible to infer the complete genetic architecture of ASD merely based on individually identified ASD-associated mutations. Here we provide a systems framework for analyzing complex diseases by identifying naturally occurring protein complexes encompassing disease-associated proteins. This allowed us to uncover novel disease-associated components and revealed many new aspects of complex diseases.
In a previous study, we leveraged a human PPI network to derive the high-level functional modules involving proteins associated with ASD (Li et al., 2014). In the present study we provide a finer-grained framework by directly identifying and analyzing protein complexes that are ubiquitously expressed in human cells. We then used this information to identify complexes specifically expressed in human neuronal culture. This analysis not only allowed us to identify new components and mechanisms involved in ASD, but also uncovered the molecular convergence underlying the highly heterogeneous mutations across ASD spectrum disorder.
We first analyzed the ubiquitously expressed protein complexes and then identified ASD-associated protein complexes in neuronal cells, which to our knowledge has not been reported previously. Importantly, the identified network displayed convergent regulation of FMRP and MECP2 in ASD, suggesting shared molecular basis between the syndromic and idiopathic forms of ASD. Although previous studies have observed an overall enrichment of ASD-associated genes among hundreds to thousands of FMRP (wild-type) targets(De Rubeis et al., 2014; Iossifov et al., 2014; Iossifov et al., 2012; Parikshak et al., 2013), our study provides several additional insights: (1) we identified FMRP interacting proteins in neuronal culture (Fig. 5A), which are largely distinct from those identified from in vitro Y2H screens (detailed comparison shown below); (2) our analysis showed that the neuronal FMPR-interacting proteins are preferentially expressed in the early fetal brain (Fig. 6A), suggesting an early onset of the FMRP-mediated diseases; (3) we showed the effect of syndromic mutation FMRPI304N on the identified network, suggesting its etiological contribution to FXS; (4) our study also revealed a significant correlation between FMRP regulation and fetal brain expression of the HDAC1-mediated interaction network (Fig. 6B). More importantly, our study shows a heretofore unreported convergence between the ASD-associated pathways and the regulatory network governed by MECP2 causal for Rett syndrome. Taken together, as depicted in Fig. 6F, we propose that genes implicated in the idiopathic and the syndromic forms of ASD have a shared molecular basis through physical or regulatory interactions with a common set of genes, which might help explain their overlapping phenotypes on the ASD spectrum.
In comparison to the Y2H screens(Corominas et al., 2014; Sakai et al., 2011), our IP-MS approach allowed us to examine the disease in a more biologically relevant context (i.e. neuronal cell culture). Consequently, our screen uncovered a number of interactions not previously identified from Y2H. For example, the most recent Y2H screen identified five proteins interacting with FMRP(Corominas et al., 2014), whereas an earlier Y2H screen identified 22 with FMRP(Sakai et al., 2011), with only one overlapping (FXR2) between the two Y2H studies. Except for the well-known FMRP-interacting proteins FXR1 and FXR2, none of these Y2H screens captured the remaining 30 prey proteins identified in our IP-MS in the neuronal culture, highlighting the importance of studying autism-associated protein interactions in a relevant cellular context.
Our investigation specifically pinpointed roles of two histone deacetylases (HDAC1 and HDAC2) in the NuRD complex. While their membership in NuRD complex had been identified in earlier studies (Havugimana et al., 2012; Xue et al., 1998; Yamada et al., 2014), by targeting HDAC1, our IP-MS recovered the NuRD complex in differentiated neuronal culture. We also performed a separate IP-MS screen for HDAC2 (with six biological replicates) in the neuronal cell line, and confirmed that the overlapping proteins that interact with both HDAC1 and HDAC2 were mostly the subunits of the NuRD complex (Table S6). However, for the prey proteins that specifically interacted with HDAC2 (and not with HDAC1), none interacted with the interaction network identified in this study (Fig. 5A), nor were they previously associated with ASD (the SFARI gene list). Therefore, for ASD association, HDAC2 likely has a compensatory role for HDAC1 in the NuRD complex, which might explain the compensatory behavior of the two paralogous copies observed in the embryonic mouse brain (Hagelkruys et al., 2014).
In conclusion, our integrative analysis revealed molecular components in ASD and suggests the etiological convergence of the idiopathic and syndromic forms of ASD. The framework presented here complements the classic mutational analysis in sequencing studies, and highlights the importance of using integrative analyses to reveal the molecular components and biological pathways underlying complex diseases. As such, this strategy will be valuable for the analyses of many other complex diseases.
EXPERIMENTAL PROCEDURES
SH-SY5Y cells (ECACC, Sigma-Aldrich) were differentiated into neuron-like cells by the addition of retinoic acid (RA, Sigma-Aldrich; R2625) and brain-derived neurotrophic factor (BDNF, eBioscience). ScriptSeq™ Complete Gold Kit (Epicentre) was used for RNA-seq library preparation, followed by sequencing with Illumina’s HiSeq 2000. The peptide mixtures that co-immunoprecipitated with the bait proteins were analyzed by a high-resolution Orbitrap Elite mass spectrometer. All these experiments were performed using two different cell batches that were separately cultured, grown and differentiated (i.e. two biological replicates). Full experimental procedures can be found in the Supplemental Experimental Procedures.
Supplementary Material
Acknowledgments
Special thanks to the families of the brain tissue donors for their contribution to advancing autism research. The autism exome-sequencing data analyzed in this manuscript were obtained from dbGAP with the accession identifier phs000298.v2.p2. We thank the funding agency (NIMH), contributing investigators and NIH dbGAP data repository for this valuable dataset. M.S. was supported by grants from NIH and Stanford Department of Genetics. This work was also supported by the National Institutes of Health (R01GM106019) and Parkinson Society of Canada (2014-673) grants to M.B. MB holds a CIHR New Investigator award. JL is supported by Banting Postdoctoral Fellowship.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, three figures and six tables.
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
JL and MS conceived the project. JL, MS, MB, JH, AU, ZZ and DPW designed the study. JL performed the analysis. MS (Minyi Shi) and ZM (Zhihai Ma) performed the experiments. RHM, HA, KJ, SP, and ZM performed the proteomic experiments. JL, MB and MS wrote the manuscript. JL, MS, MB, JH, and AU revised the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, et al. Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Human molecular genetics. 2012;21:4781–4792. doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Jr, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayés À, van de Lagemaat LN, Collins MO, Croning MDR, Whittle IR, Choudhary JS, Grant SGN. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nature Neuroscience. 2010;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocchetti AG, Kopp M, Waltes R, Haslinger D, Duketis E, Jarczok TA, Poustka F, Voran A, Graab U, Meyer J, et al. Variants of the CNTNAP2 5′ promoter as risk factors for autism spectrum disorders: a genetic and functional approach. Molecular Psychiatry. 2014 doi: 10.1038/mp.2014.103. [DOI] [PubMed] [Google Scholar]
- Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L, Broly M, Rodriguez M, Tam S, Trigg SA, et al. Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nature Communications. 2014;5 doi: 10.1038/ncomms4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014 doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyder M, Karlsson RM, Mathur P, Lyman M, Bock R, Momenan R, Munasinghe J, Scattoni ML, Ihne J, Camp M, et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am J Psychiatry. 2010;167:1508–1517. doi: 10.1176/appi.ajp.2010.10040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley AG, Gannon S, Rombach-Mullan N, Prendergast A, Barry C, Cassidy AW, Regan CM. Class I histone deacetylase inhibition ameliorates social cognition and cell adhesion molecule plasticity deficits in a rodent model of autism spectrum disorder. Neuropharmacology. 2012;63:750–760. doi: 10.1016/j.neuropharm.2012.05.042. [DOI] [PubMed] [Google Scholar]
- Gao Z, Lee P, Stafford JM, von Schimmelmann M, Schaefer A, Reinberg D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature. 2014;516:349–354. doi: 10.1038/nature13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgi B, Voight BF, Bucan M. From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 2013;9:e1003484. doi: 10.1371/journal.pgen.1003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelkruys A, Lagger S, Krahmer J, Leopoldi A, Artaker M, Pusch O, Zezula J, Weissmann S, Xie Y, Schofer C, et al. A single allele of Hdac2 but not Hdac1 is sufficient for normal mouse brain development in the absence of its paralog. Development. 2014;141:604–616. doi: 10.1242/dev.100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al. A census of human soluble protein complexes. Cell. 2012;150:1068–1081. doi: 10.1016/j.cell.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F, Penn O, Borenstein E, Eichler EE. The discovery of integrated gene networks for autism and related disorders. Genome Research. 2014 doi: 10.1101/gr.178855.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet G, Ey E, Bourgeron T. The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet. 2013;14:191–213. doi: 10.1146/annurev-genom-091212-153431. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Guilmette E, Gosink MM, Fischer JE, Fitzgerald LW, Stephenson DT, Pletcher MT. Transcriptomic analysis of genetically defined autism candidate genes reveals common mechanisms of action. Mol Autism. 2013;4:45. doi: 10.1186/2040-2392-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J, Urban A, Hallmayer J, Snyder M. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol. 2014;10:774. doi: 10.15252/msb.20145487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Sabo A, Neale BM, Nagaswamy U, Stevens C, Lim E, Bodea CA, Muzny D, Reid JG, Banks E, et al. Analysis of Rare, Exonic Variation amongst Subjects with Autism Spectrum Disorders and Population Controls. PLoS genetics. 2013;9:e1003443. doi: 10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nature Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh HJ, Ponting CP, Boulding HC, Meader S, Betancur C, Buxbaum JD, Pinto D, Marshall CR, Lionel AC, Scherer SW, et al. Network Topologies and Convergent Aetiologies Arising from Deletions and Duplications Observed in Individuals with Autism. PLoS Genetics. 2013;9:e1003523. doi: 10.1371/journal.pgen.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power RA, Kyaga S, Uher R, MacCabe JH, Langstrom N, Landen M, McGuffin P, Lewis CM, Lichtenstein P, Svensson AC. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry. 2013;70:22–30. doi: 10.1001/jamapsychiatry.2013.268. [DOI] [PubMed] [Google Scholar]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai Y, Shaw CA, Dawson BC, Dugas DV, Al-Mohtaseb Z, Hill DE, Zoghbi HY. Protein interactome reveals converging molecular pathways among autism disorders. Science translational medicine. 2011;3:86ra49. doi: 10.1126/scitranslmed.3002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature. 2012;493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A. MicroRNA-Mediated Feedback and Feedforward Loops Are Recurrent Network Motifs in Mammals. Molecular Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, Stoykova A. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev Cell. 2013;25:256–269. doi: 10.1016/j.devcel.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Turinsky AL, Razick S, Turner B, Donaldson IM, Wodak SJ. Interaction databases on the same page. Nature Biotechnology. 2011;29:391–393. doi: 10.1038/nbt.1867. [DOI] [PubMed] [Google Scholar]
- van de Leemput J, Boles Nathan C, Kiehl Thomas R, Corneo B, Lederman P, Menon V, Lee C, Martinez Refugio A, Levi Boaz P, Thompson Carol L, et al. CORTECON: A Temporal Transcriptome Analysis of In Vitro Human Cerebral Cortex Development from Human Embryonic Stem Cells. Neuron. 2014;83:51–68. doi: 10.1016/j.neuron.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmar CH, Wahlestedt C. Histone deacetylases (HDACs) and brain function. Neuroepigenetics. 2015;1:20–27. [Google Scholar]
- Wang T, Xie Y, Xiao G. dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biol. 2014;15:R11. doi: 10.1186/gb-2014-15-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wong J, Moreno GT, Young MK, Côté J, Wang W. NURD, a Novel Complex with Both ATP-Dependent Chromatin-Remodeling and Histone Deacetylase Activities. Molecular Cell. 1998;2:851–861. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K, et al. Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron. 2014;83:122–134. doi: 10.1016/j.neuron.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.