

Significance
DNA double-strand breaks (DSBs) occur in all cells, including neural stem/progenitor cells (NSPCs) that give rise to the brain. We previously found that developing neural cells lacking a major DSB end-joining pathway are subject to widespread death. Because DSBs may result from gene transcription, we assayed for DSBs near active transcription start sites (TSSs) genome-wide in NSPCs. DSBs occur near TSSs of highly transcribed genes involved in general cellular processes but occur less often near neural-specific TSSs. These TSS-associated DSBs can translocate to other DSBs by both the normal and alternative DSB repair pathways. We report similar findings in B lymphocytes, suggesting that highly transcribed genes involved in general cellular processes are subject to TSS-associated DSBs in divergent cell types.
Keywords: nonhomologous end-joining, neural stem cells, transcription, translocation, alternative end-joining
Abstract
High-throughput, genome-wide translocation sequencing (HTGTS) studies of activated B cells have revealed that DNA double-strand breaks (DSBs) capable of translocating to defined bait DSBs are enriched around the transcription start sites (TSSs) of active genes. We used the HTGTS approach to investigate whether a similar phenomenon occurs in primary neural stem/progenitor cells (NSPCs). We report that breakpoint junctions indeed are enriched around TSSs that were determined to be active by global run-on sequencing analyses of NSPCs. Comparative analyses of transcription profiles in NSPCs and B cells revealed that the great majority of TSS-proximal junctions occurred in genes commonly expressed in both cell types, possibly because this common set has higher transcription levels on average than genes transcribed in only one or the other cell type. In the latter context, among all actively transcribed genes containing translocation junctions in NSPCs, those with junctions located within 2 kb of the TSS show a significantly higher transcription rate on average than genes with junctions in the gene body located at distances greater than 2 kb from the TSS. Finally, analysis of repair junction signatures of TSS-associated translocations in wild-type versus classical nonhomologous end-joining (C-NHEJ)–deficient NSPCs reveals that both C-NHEJ and alternative end-joining pathways can generate translocations by joining TSS-proximal DSBs to DSBs on other chromosomes. Our studies show that the generation of transcription-associated DSBs is conserved across divergent cell types.
The integrity of mammalian genomes is constantly challenged by DNA double-strand breaks (DSBs) that are generated by cell-intrinsic processes such as DNA replication, transcription, and oxidative stress (1, 2). In addition, during early lymphocyte development the RAG1/2 endonuclease specifically generates DSBs at antigen receptor variable region gene segments; these segments then are assembled to generate variable region exons (3). Likewise, in activated, mature B lymphocytes, activation-induced cytidine deaminases (AID) initiate DSB formation in the noncoding “switch” regions of the Ig heavy chain (IgH) locus to exchange IgH constant region loci to effect IgH class switch recombination (CSR) (3). Recently, our studies of neural stem/progenitor cells (NSPCs) further revealed that recurrent DSB clusters (RDCs) occur within the bodies of a set of at least 27 long genes that mostly are involved in neural functions (4). DSBs around transcription start sites (TSSs) have been implicated in the transcriptional activation of nuclear hormone receptor genes in tumor cell lines (5, 6) and are required for the transcription of a set of early-response genes in activated neurons in culture (7). We also have found that activated B cells in culture generate genomic DSBs at active TSSs, as measured by global nuclear run-on sequencing (GRO-seq) (8) and that these TSS-associated DSBs can translocate to recurrent “bait” DSBs introduced in the body of the c-Myc gene (9).
One of the two major, well-characterized DSB repair pathways in mammalian cells is classical nonhomologous end-joining (C-NHEJ). C-NHEJ is active throughout the cell cycle but is particularly critical in noncycling cells in which the other major DSB repair pathway, homologous recombination, is not available (10). C-NHEJ joins blunt DSB ends directly and also can join ends with several base pairs of short microhomology (MH) to form MH-mediated joins (11). C-NHEJ is required for V(D)J recombination joining and plays a major role in CSR joining (11). C-NHEJ also appears to play a significant role in joining NSPC RDC DSBs, because their translocation frequency increases in its absence (4). In addition to blocking lymphocyte development, C-NHEJ deficiency results in widespread apoptotic death of newly generated postmitotic neurons in mice (12–16). However, because neuronal death in the absence of C-NHEJ results from a p53 tumor-suppressor response to DSBs, such death can be obviated in vivo by p53 deficiency (14, 17). Likewise, p53 deficiency rescues the viability of cultured C-NHEJ–deficient NSPCs (4). In the absence of C-NHEJ (and p53), genomic DSBs can persist unjoined and can be substrates for more frequent translocation (3). In WT NSPCs, RDC-associated DSBs can be fused to other DSBs in the genome by C-NHEJ. In C-NHEJ–deficient NSPCs, such RDC translocations are mediated by an alternative end-joining (A-EJ) pathway that is biased toward the use of longer MHs (4).
C-NHEJ also requires the ataxia telangiectasia-mutated (ATM)-dependent DNA damage response to generate fully efficient DSB repair via end-joining (3, 18). ATM has been shown to have a direct role in DSB end-joining (3, 19, 20) and also is required for the p53-dependent G1 check point response to unrepaired DSBs (21, 22). Because of its dual role in C-NHEJ and DSB checkpoint responses, ATM deficiency can lead to long-term DSB persistence, leading, for example, to V(D)J recombination breaks in early B-cell development that persist developmentally into mature B cells (23–25). Although these ATM roles likely contribute to immunodeficiency and lymphoid cancers associated with ATM deficiency, whether such ATM functions are involved with the neurodegenerative phenotype associated with ATM deficiency in humans remains unknown (21, 22).
We previously developed an unbiased, high-throughput, genome-wide, translocation sequencing (HTGTS) approach to identify DSBs via their translocation to bait DSBs at defined genomic sites in primary progenitor and activated mature B cells, NSPCs, and human cell lines (4, 9, 26–28). Because of cellular heterogeneity in 3D genome organization (3, 27, 29), HTGTS detects, with great sensitivity, recurrent DSBs in any given genomic location (4, 9, 26–29) or recurrent classes of DSBs, such as DSBs at TSSs, that may occur at much lower frequency in any given location (ref. 9 and this study). Because of increased contact frequency, translocation frequency is increased further between sequences on the same cis chromosome (3, 28, 29). HTGTS detects only those DSBs that translocate. Given that most DSBs are rejoined or repaired locally to other DSBs within the same topological domain, HTGTS from DSBs on a different chromosome or even in a different topological domain on the same chromosome provides only a minimal estimate of the actual frequency of DSBs at a given site or region (3, 4).
In the current study, we used GRO-seq and HTGTS to identify TSS-associated DSBs in NSPCs, to quantify the frequency of such DSBs relative to that of RDC-associated DSBs, and to elucidate the class of genes in which most such DSBs occur and compare them with the class of genes that incurs TSS-associated DSBs in B lymphocytes.
Results
DSBs and Translocations Are Enriched Around Active TSSs in NSPCs.
To assess whether transcription-associated DSBs occur and form translocations in NSPCs, we first identified actively transcribed genes by performing GRO-seq (8, 30, 31) in Xrcc4−/−p53−/− NSPCs. In these analyses, a Pearson correlation coefficient analysis showed high reproducibility among three independent experiments (Fig. S1A, Left). Aggregate patterns of transcription profiles showed enrichment of reads within 1.5 kb of TSSs genome-wide (Fig. S1B, Right), consistent with previously published GRO-seq results obtained in other cell types (30, 31). To perform HTGTS for DSB identification, we used a Cas9:sgRNA-based approach to introduce HTGTS bait DSBs into primary NSPCs isolated from Xrcc4−/−p53−/− mice, as previously described (4). More specifically, we used single-guide RNAs (sgRNAs) targeting Cas9-mediated DSBs to a nontranscribed intergenic region ∼50 kb telomeric of N-myc on chromosome 12 (Chr12-sgRNA-1) (Fig. 1A) or to the c-Myc gene on chromosome 15 (Chr15-Myc-sgRNA) (Fig. S2). Primary NSPCs were collected 4.5 d after the introduction of Cas9:sgRNA expression constructs, and genomic DNA was isolated and processed for HTGTS. For both Cas9:sgRNA bait DSBs used, we used HTGTS primers that allowed genome-wide identification of endogenous “prey” DSBs that joined to centromeric broken ends of the bait DSB (Fig. 1A, Upper).
Fig. S1.

Analyses of NSPCs GRO-seq libraries. (A) Reproducibility of GRO-seq biological replicates from Xrcc4−/−p53−/− (Left) or ATM−/− R26I-SceI-GR c-Myc25xI-SceI (Right) NSPCs. The GRO-seq signal per 10-kb genomic bin per replicate is plotted. The Pearson correlation coefficient (r) among biological replicates (n = 3) is stated and indicated by red lines. (B) Combined Metagene profiles of Xrcc4−/−p53−/− (Left) or ATM−/− R26I-SceI-GR c-Myc25xI-SceI (Right) GRO-seq libraries within 1.5 kb of TSSs; antisense (red trace) and sense (blue trace) transcriptions are indicated.
Fig. 1.

DSBs and translocations are enriched around active TSSs in NSPCs. (A, Upper) Diagram of the N-myc locus and sgRNA target site (Chr12-sgRNA-1; vertical black arrowhead) and the location and orientation of HTGTS primer (horizontal blue arrowhead). Cen, centromere; E, exon; Tel, telomere. (Lower) Distribution of HTGTS junctions relative to TSSs genome-wide. Junctions from Chr12-sgRNA-1–expressing Xrcc4−/−p53−/− NSPCs (n = 12,236) are plotted in relation to the nearest RefGene (42) TSS (0 on the x axis). Negative numbers on the x axis indicate locations upstream of TSS; positive numbers on the x axis indicate locations downstream of TSS. The translocation density per 10,000 junctions (bin size 300 bp) relative to the TSS of active (red) or inactive (blue) genes, as determined by GRO-seq. (4), is shown. (B, Upper) Diagram of the 25×I-SceI c-Myc locus; the HTGTS primer and orientation are indicated by the horizontal blue arrowhead. (Lower) Distribution of HTGTS junctions relative to TSSs genome-wide. Junctions from TA-treated ATM−/− R26I-SceI-GR c-Myc25xI-SceI NSPCs (n = 9,788) are plotted in relation to the nearest RefGene (42) TSS; details are as in A.
Fig. S2.

Regions proximal to active TSSs are enriched with DSBs and translocations in NSPCs. (Upper) Diagram of the c-Myc locus. The sgRNA target site (Chr15-Myc-sgRNA) and HTGTS primer are indicated as in Fig. 1A. (Lower) Distribution of HTGTS junctions relative to TSSs genome-wide. Junctions from Chr15-Myc-sgRNA–expressing Xrcc4−/−p53−/− NSPCs (n = 8,640) are plotted in relation to the nearest RefGene (42) TSS; details are as in Fig. 1A.
To examine potential relationships between transcriptionally active NSPC genes and DSBs, we calculated the distance between genome-wide HTGTS junctions generated from either Chr12-sgRNA-1 DSBs or Chr15-Myc-sgRNA DSBs and the nearest TSS of active or inactive genes (lower panels in Fig. 1A and Fig. S2), as described (9). For these analyses, we excluded junctions within 1 Mb on each side of the break site to eliminate junctions that occur via bait DSB rejoining following resection (9) and also eliminated Cas9:sgRNA-mediated off-target hotspot DSBs defined in our separate studies (Fig. 1A and Fig. S2) (4). Although Chr12-sgRNA-1 targets a nontranscribed intergenic region, and Chr15-Myc-sgRNA targets the transcribed c-Myc gene, we found that translocation junctions for both were significantly enriched in active genes, with a clear peak spanning the TSS (Fig. 1A and Fig. S2). We estimate that translocations between a DSB located within 2 kb of an active TSS and a bait Chr12-sgRNA-1 DSB occur in about 1 in 167 XRCC4/p53-deficient NSPCs (i.e., in about 0.4% of cells, based on the observed translocation rate of bait DSBs to DSBs located within 2 kb of an active TSS on the break-site chromosome, multiplied by the total number of chromosomes) (Table S1). We found similar levels of DSBs located within 2 kb of active TSSs for the Chr15-Myc-sgRNA bait DSBs (Table S1).
Table S1.
Frequency estimation of TSS-associated DSBs and translocations in NSPCs
| NSPC genotype | Bait DSB | Input, μg | Total unique junctions | Junctions within 2 kb of the active TSS on the break-site chromosome | Translocation, % of cells | DSBs per cell |
| ATM−/− | c-Myc | 160 | 16,476 | 113 (Chr15) | 1.37 | 0.55 |
| 25xI-SceI | ||||||
| Xrcc4−/−p53−/− | Chr12-sgRNA-1 | 160 | 32,144 | 71 (Chr12) | 0.44 | 0.18 |
| Xrcc4−/−p53−/− | Chr15-Myc-sgRNA | 160 | 21,930 | 45 (Chr15) | 0.41 | 0.16 |
The translocation frequency, indicated as the percentage of cells with HTGTS junctions located within 2 kb of TSSs on the break-site chromosome (Chr12 or Chr15), was determined, and the rate of DSBs per cell was calculated by multiplying the translocation frequency by the total number of chromosomes (n = 40). See main text and ref. 4 for details.
To test whether this DSB/active TSS correlation occurs for a different type of bait DSB end and in a different DNA repair-deficient background, we performed HTGTS and GRO-seq analyses in ATM-deficient (ATM−/−) NSPCs, in which I-SceI–mediated bait DSBs can be induced in a 25×I-SceI knock-in cassette located in c-Myc (c-Myc25×I-SceI) (Fig. 1B, Upper) (9). GRO-seq experiments in the ATM−/− NSPCs again gave high reproducibility across three independent experiments, and the resulting aggregate patterns of transcription profiles indicated enrichment of reads within 1.5 kb of TSSs genome-wide (Fig. S1 A and B, Right). Similarly, as in the two different Cas9:sgRNA-based bait DSB analyses described above, I-SceI–based HTGTS revealed a significant enrichment of translocation junctions around the TSSs of active genes (Fig. 1B, Lower). Based on the observed translocation rate of bait DSBs to DSBs located within 2 kb of active TSSs on the break-site chromosome, multiplied by the total number of chromosomes, we estimate that, genome-wide, TSS-associated DSBs that translocate to bait DSBs occur in about 1 in 73 (1.37%) ATM-deficient NSPCs, consistent with higher bait DSB translocation frequency in the ATM−/− background (Table S1) (4).
To investigate further the set of transcribed genes that contained TSS-proximal junctions in NSPCs, we performed a comparative analysis of GRO-seq transcription profiles of NSPCs and those previously generated for activated B cells (31). This analysis yielded sets of genes transcribed in NSPCs but not in B cells (class 1), genes commonly transcribed in both NSPCs and B cells (class 2), and genes transcribed in B cells but not in NSPCs (class 3). Notably, examination of transcription rates among these classes revealed that the transcription rate of class 2 genes was significantly higher than that of either class 1 or class 3 genes, in all datasets examined (Fig. 2 A and B and Fig. S3). We then evaluated how active genes with translocation junctions located within 2 kb of their TSS in NSPCs were distributed among the different classes of genes. Although ∼2.6% of total genes in each class contained TSS-proximal junctions, we found that the majority of active genes with TSS-proximal junctions (345/440; 78.4%) in experiments based on the Chr12-sgRNA-1 bait DSB represented class 2 genes (Fig. 2C). Similar results were obtained in experiments based on Chr15-Myc-sgRNA bait (Fig. S3A). Moreover, in experiments based on I-SceI-bait DSB 85.4% of active genes with TSS-proximal junctions (303/355) were class 2 genes (Fig. 2D). Notably, the class 2 genes that contained TSS-proximal junctions showed significantly higher average transcription levels than all class 2 genes on average, irrespective of genotype, in both Cas9:sgRNA- and I-SceI–based experiments (Fig. 2 E and F and Fig. S3B) in NSPCs as well as in I-SceI–based experiments in CSR-activated B cells (Fig. S3C). These results suggest an association between transcription rate and TSS-proximal DSBs. Consistent with their common expression in NSPCs and B cells, most class 2 genes with HTGTS junctions are involved in general biological processes (Table S2).
Fig. 2.

TSS-proximal junctions occur in genes that are highly and commonly transcribed in NSPCs and B lymphocytes. (A) Venn diagram illustrating the three classes (1–3) of genes transcribed in WT CSR-activated B lymphocytes and Xrcc4−/−p53−/− NSPCs. (B) Venn diagram showing class 1–3 genes transcribed in WT CSR-activated B lymphocytes and ATM−/−R26I-SceI-GR c-Myc25x I-SceI NSPCs. Class 1 genes are actively transcribed in NSPCs but are inactive in B cells. Class 2 genes are actively transcribed in both NSPCs and B cells. Class 3 genes are transcribed in B cells but not in NSPCs. Gene activity [reads per kilobase of transcript per million reads mapped (RPKM) ≥ 0.05] was determined by GRO-seq analysis. Numbers of genes per category are listed. (C and D) Plots showing the relative distance of junctions from Chr12-sgRNA-1–expressing Xrcc4−/−p53−/− NSPCs (C) or ATM−/−R26I-SceI-GR c-Myc25x I-SceI NSPCs (D) to the closest TSS of class 1 (blue trace), class 2 (red trace), and class 3 (black trace) genes. Figures are organized as described in Fig. 1A. (E) Box-and-whiskers plots of the transcription activity (GRO-seq density) of class 1 (Left) and class 2 (Center) genes and genes in class 2 containing HTGTS junctions located within 2 kb of the TSS from Chr12-sgRNA-1–expressing Xrcc4−/−p53−/− NSPCs or ATM−/−R26I-SceI-GR c-Myc25× I-SceI NSPCs (Right). (F) The number of genes in each group in E is indicated. The upper and lower box edges represent the 25th and 75th percentile, respectively; the horizontal line denotes the median. ****P < 0.0001; **P < 0.01 (Mann–Whitney u test).
Fig. S3.

Distribution of HTGTS junctions in relation to the closest TSS of genes within class 1, 2, or 3. (A) The plot shows the relative distance of junctions from Chr15-Myc-sgRNA–expressing Xrcc4−/−p53−/− NSPCs to the closest TSS of class 1 (blue trace), class 2 (red trace), and class 3 (black trace) genes. Categories are as described in Fig. 2 A and B. Other details are as described for Fig. 2C. (B) Box-and-whiskers plot of transcription activity (GRO-seq density) of Xrcc4−/−p53−/− NSPCs genes in class 1 (Left) and class 2 (Center) genes and genes in class 2 that contain HTGTS junctions located within 2 kb of the TSS (Right) from Chr15-Myc-sgRNA–expressing Xrcc4−/−p53−/− NSPCs. The figure is organized as in Fig. 2E. ****P < 0.0001; *P < 0.05 (Mann–Whitney u test). (C) Box-and-whiskers plot of the transcription activity of WT CSR-activated B-cell genes in class 3 (Left) and class 2 (Center) and of genes in class 2 that contain HTGTS junctions located within 2 kb of the TSS (Right) from ATM−/− c-Myc25×I-SceI B cells; other details are as described in B. ****P < 0.0001 (Mann–Whitney u test).
Table S2.
Gene ontology analysis of class 2 genes harboring TSS-proximal translocation junctions
| GO biological process complete | REFLIST n = 22,320 | Class 2 genes with junctions within 2 kb of the TSS | Expected, n | P value |
| Xrcc4−/− p53−/− Chr15-Myc-sgRNA NSPC experiments (n = 221 class 2 genes with junctions located within 2 kb of the TSS) | ||||
| Metabolic process (GO:0008152)* | 7,858 | 118 | 77.81 | 1.85E-04 |
| Cellular metabolic process (GO:0044237)* | 6,833 | 106 | 67.66 | 3.76E-04 |
| Primary metabolic process (GO:0044238)* | 7,079 | 108 | 70.09 | 6.28E-04 |
| Organic substance metabolic process (GO:0071704)* | 7,367 | 110 | 72.94 | 1.41E-03 |
| Macromolecule metabolic process (GO:0043170)* | 5,892 | 90 | 58.34 | 1.93E-02 |
| Cellular macromolecule metabolic process (GO:0044260)* | 5,282 | 83 | 52.3 | 2.02E-02 |
| ATM−/− R26I-SceI-GR c-Myc25xI-SceI NSPC experiments (n = 277 class 2 genes with junctions located within 2 kb of the TSS) | ||||
| Metabolic process (GO:0008152)* | 7,858 | 153 | 97.52 | 6.12E-08 |
| Cellular metabolic process (GO:0044237)* | 6,833 | 137 | 84.8 | 3.40E-07 |
| Organic substance metabolic process (GO:0071704)* | 7,367 | 144 | 91.43 | 4.35E-07 |
| Primary metabolic process (GO:0044238)* | 7,079 | 140 | 87.85 | 4.64E-07 |
| Cellular macromolecule metabolic process (GO:0044260)* | 5,282 | 112 | 65.55 | 3.43E-06 |
| Macromolecule metabolic process (GO:0043170)* | 5892 | 120 | 73.12 | 6.62E-06 |
| Cellular process (GO:0009987)* | 13,249 | 208 | 164.43 | 2.12E-04 |
| Organelle organization (GO:0006996)* | 2,494 | 58 | 30.95 | 1.46E-02 |
| Heterocycle metabolic process (GO:0046483)* | 3,526 | 74 | 43.76 | 1.83E-02 |
| Organic cyclic compound metabolic process (GO:1901360)* | 3,751 | 77 | 46.55 | 2.44E-02 |
| Nucleobase-containing compound metabolic process (GO:0006139)* | 3,388 | 71 | 42.05 | 3.34E-02 |
| Cellular component organization (GO:0016043)* | 4,121 | 82 | 51.14 | 3.47E-02 |
| Nitrogen compound metabolic process (GO:0006807)* | 4,189 | 83 | 51.99 | 3.48E-02 |
| Cellular component organization or biogenesis (GO:0071840)* | 4,260 | 84 | 52.87 | 3.59E-02 |
| Cellular protein metabolic process (GO:0044267)* | 2,718 | 60 | 33.73 | 4.83E-02 |
| Xrcc4−/− p53−/− Chr12-sgRNA-1 NSPC experiments (n = 306 class 2 genes with junctions located within 2 kb of the TSS | ||||
| Cellular metabolic process (GO:0044237)* | 6,833 | 138 | 93.68 | 5.26E-04 |
| Cellular macromolecule metabolic process (GO:0044260)* | 5,282 | 109 | 72.41 | 1.28E-02 |
| Phosphate-containing compound metabolic process (GO:0006796) | 1,612 | 46 | 22.1 | 1.58E-02 |
| Primary metabolic process (GO:0044238)* | 7,079 | 135 | 97.05 | 2.74E-02 |
| Organic substance metabolic process (GO:0071704)* | 7,367 | 139 | 101 | 3.13E-02 |
| Phosphorus metabolic process (GO:0006793) | 1,656 | 46 | 22.7 | 3.20E-02 |
| Metabolic process (GO:0008152)* | 7,858 | 146 | 107.73 | 3.43E-02 |
| Regulation of organelle organization (GO:0033043) | 1,021 | 33 | 14 | 4.25E-02 |
| ATM−/− c-Myc25xI-SceI B-cell experiments (n = 416 class 2 genes with junctions located within 2 kb of the TSS (31) | ||||
| Cellular metabolic process (GO:0044237)* | 6,833 | 224 | 127.35 | 5.38E-19 |
| Metabolic process (GO:0008152)* | 7,858 | 244 | 146.46 | 1.39E-18 |
| Organic substance metabolic process (GO:0071704)* | 7,367 | 228 | 137.31 | 4.11E-16 |
| Cellular macromolecule metabolic process (GO:0044260)* | 5,282 | 183 | 98.45 | 5.45E-16 |
| Primary metabolic process (GO:0044238)* | 7,079 | 220 | 131.94 | 2.67E-15 |
| Macromolecule metabolic process (GO:0043170)* | 5,892 | 192 | 109.82 | 3.26E-14 |
| Cellular nitrogen compound metabolic process (GO:0034641) | 3,900 | 141 | 72.69 | 4.34E-12 |
| Cellular process (GO:0009987)* | 13,249 | 323 | 246.93 | 1.76E-11 |
| Nitrogen compound metabolic process (GO:0006807)* | 4,189 | 145 | 78.07 | 5.19E-11 |
| Nucleobase-containing compound metabolic process (GO:0006139)* | 3,388 | 121 | 63.15 | 3.27E-09 |
| Heterocycle metabolic process (GO:0046483)* | 3,526 | 122 | 65.72 | 2.21E-08 |
| Cellular aromatic compound metabolic process (GO:0006725) | 3,559 | 122 | 66.33 | 4.14E-08 |
| Cellular macromolecule Biosynthetic process (GO:0034645) | 2,591 | 97 | 48.29 | 1.20E-07 |
| Macromolecule biosynthetic process (GO:0009059) | 2,615 | 97 | 48.74 | 2.01E-07 |
| Nucleic acid metabolic process (GO:0090304) | 2,979 | 106 | 55.52 | 2.07E-07 |
| Cellular protein metabolic process (GO:0044267)* | 2,718 | 99 | 50.66 | 3.25E-07 |
| Gene expression (GO:0010467) | 2,885 | 103 | 53.77 | 3.63E-07 |
| Cellular component organization or biogenesis (GO:0071840)* | 4,260 | 135 | 79.4 | 4.89E-07 |
| Organic cyclic compound metabolic process (GO:1901360)* | 3,751 | 123 | 69.91 | 6.17E-07 |
| Cellular biosynthetic process (GO:0044249) | 3,270 | 111 | 60.95 | 1.03E-06 |
| Negative regulation of biological process (GO:0048519) | 4,133 | 131 | 77.03 | 1.09E-06 |
| Cellular nitrogen compound biosynthetic process (GO:0044271) | 2,508 | 92 | 46.74 | 1.37E-06 |
| Negative regulation of cellular process (GO:0048523) | 3,811 | 123 | 71.03 | 1.71E-06 |
| Biosynthetic process (GO:0009058) | 3,446 | 114 | 64.23 | 2.47E-06 |
| Biological process (GO:0008150) | 20,668 | 412 | 385.21 | 5.40E-06 |
| Organic substance biosynthetic process (GO:1901576) | 3,371 | 111 | 62.83 | 6.13E-06 |
| RNA metabolic process (GO:0016070) | 2,572 | 90 | 47.94 | 2.37E-05 |
| Organelle organization (GO:0006996)* | 2,494 | 88 | 46.48 | 2.52E-05 |
| Cellular component organization (GO:0016043)* | 4,121 | 126 | 76.81 | 2.87E-05 |
| Protein metabolic process (GO:0019538) | 3,362 | 108 | 62.66 | 4.48E-05 |
| Regulation of primary metabolic process (GO:0080090) | 4,897 | 141 | 91.27 | 1.10E-04 |
| Positive regulation of macromolecule metabolic process (GO:0010604) | 2,537 | 87 | 47.28 | 1.19E-04 |
| Regulation of cellular metabolic process (GO:0031323) | 5,062 | 144 | 94.35 | 1.54E-04 |
| Regulation of metabolic process (GO:0019222) | 5,686 | 157 | 105.98 | 1.80E-04 |
| Regulation of macromolecule metabolic process (GO:0060255) | 4,893 | 140 | 91.2 | 1.91E-04 |
| Macromolecular complex subunit organization (GO:0043933) | 1,737 | 66 | 32.37 | 2.36E-04 |
| Negative regulation of cellular metabolic process (GO:0031324) | 2,058 | 74 | 38.36 | 2.82E-04 |
| Negative regulation of metabolic process (GO:0009892) | 2,285 | 79 | 42.59 | 4.61E-04 |
| Translation (GO:0006412) | 271 | 21 | 5.05 | 5.26E-04 |
| Negative regulation of macromolecule metabolic process (GO:0010605) | 2,048 | 72 | 38.17 | 1.07E-03 |
| Regulation of nitrogen compound metabolic process (GO:0051171) | 3,507 | 106 | 65.36 | 1.51E-03 |
| Peptide biosynthetic process (GO:0043043) | 291 | 21 | 5.42 | 1.67E-03 |
| Ribonucleoprotein complex biogenesis (GO:0022613) | 295 | 21 | 5.5 | 2.08E-03 |
| Regulation of macromolecule Biosynthetic process (GO:0010556) | 3,269 | 100 | 60.93 | 2.18E-03 |
| Chromatin organization (GO:0006325) | 554 | 30 | 10.33 | 2.23E-03 |
| Positive regulation of metabolic process (GO:0009893) | 3,172 | 97 | 59.12 | 3.56E-03 |
| Positive regulation of cellular metabolic process (GO:0031325) | 2,700 | 86 | 50.32 | 3.67E-03 |
| Peptide metabolic process (GO:0006518) | 369 | 23 | 6.88 | 5.52E-03 |
| Cellular component biogenesis (GO:0044085) | 1,748 | 62 | 32.58 | 6.81E-03 |
| Negative regulation of macromolecule biosynthetic process (GO:0010558) | 1,228 | 48 | 22.89 | 1.03E-02 |
| Regulation of biosynthetic process (GO:0009889) | 3,497 | 102 | 65.18 | 1.46E-02 |
| Heterocycle biosynthetic process (GO:0018130) | 2,175 | 71 | 40.54 | 1.95E-02 |
| Negative regulation of biosynthetic process (GO:0009890) | 1,303 | 49 | 24.29 | 2.29E-02 |
| Response to stress (GO:0006950) | 2,525 | 79 | 47.06 | 2.37E-02 |
| Amide biosynthetic process (GO:0043604) | 347 | 21 | 6.47 | 2.62E-02 |
| Nucleobase-containing compound biosynthetic process (GO:0034654) | 2,113 | 69 | 39.38 | 2.74E-02 |
| Positive regulation of macromolecule biosynthetic process (GO:0010557) | 1,546 | 55 | 28.81 | 2.79E-02 |
| Homeostasis of number of cells (GO:0048872) | 215 | 16 | 4.01 | 3.14E-02 |
| Regulation of nucleobase-containing compound metabolic process (GO:0019219) | 3,289 | 96 | 61.3 | 3.16E-02 |
| Protein transport (GO:0015031) | 983 | 40 | 18.32 | 3.26E-02 |
| Regulation of cellular macromolecule biosynthetic process (GO:2000112) | 3,171 | 93 | 59.1 | 3.86E-02 |
| Immune system process (GO:0002376) | 1,529 | 54 | 28.5 | 4.25E-02 |
| Aromatic compound biosynthetic process (GO:0019438) | 2,183 | 70 | 40.69 | 4.28E-02 |
| Establishment of protein localization (GO:0045184) | 1,071 | 42 | 19.96 | 4.55E-02 |
Gene ontology (GO) analyses were performed by the Protein Analysis Through Evolutionary Relationships (PANTHER) classification system (43, 44). Gene numbers analyzed for each experiment are indicated. Analysis Type used: PANTHER Overrepresentation Test (release 20150430); annotation version and release date: GO Ontology database, 2015-08-06; Bonferroni correction: true; reference list (REFLIST): Mus musculus (all genes in database).
Shared GO terms across experiments.
To evaluate further the potential relationship between transcription and the generation of DSBs that translocate to HTGTS bait DSBs, we compared the transcription rates of active genes with junctions located within 2 kb of an active TSS (class A) (Fig. 3A) with those of active genes with junctions only in the gene body located at distances greater than 2 kb from the TSS (class B) (Fig. 3A). Analysis of GRO-seq densities of class A and class B genes in Chr12-sgRNA-1– or Chr15-Myc-sgRNA–expressing Xrcc4−/−p53−/− NSPCs (Fig. 3B and Fig. S4) or triamcinolone acetonide (TA)-treated ATM−/− R26I-SceI-GR c-Myc25×I-SceI NSPCs (Fig. 3B) revealed a significantly higher transcriptional activity of class A genes (P < 0.0001; Mann–Whitney u test). Together, these findings are consistent with the notion that increased overall rates of transcription initiation may positively influence the frequency of DSB formation in the vicinity of TSSs.
Fig. 3.

Genes with TSS-proximal junctions show higher transcriptional activity than genes with junctions in the gene body located more than 2 kb from the TSS. (A) Active genes were determined by GRO-seq and were classified as class A (active genes with junctions located within 2 kb of the TSS) or class B (active genes with junctions within the gene body located more than 2 kb from the TSS). TTS, transcription termination site. (B) Box-and-whiskers plots of transcriptional activity (GRO-seq density) of class A and class B genes in Chr12-sgRNA-1–expressing Xrcc4−/−p53−/− NSPCs from four independent experiments or in TA-treated ATM−/− R26I-SceI-GR c-Myc25× I-SceI NSPCs from four independent experiments. Numbers of genes within each group: Chr12-sgRNA-1 Xrcc4−/−p53−/−: class A = 440 genes; class B = 2,377 genes; ATM−/− R26I-SceI-GR c-Myc25× I-SceI: class A = 311 genes; class B = 1,807 genes. The upper and lower box edges represent the 25th and 75th percentile, respectively; the horizontal line denotes the median. ****P < 0.0001 (Mann–Whitney u test).
Fig. S4.
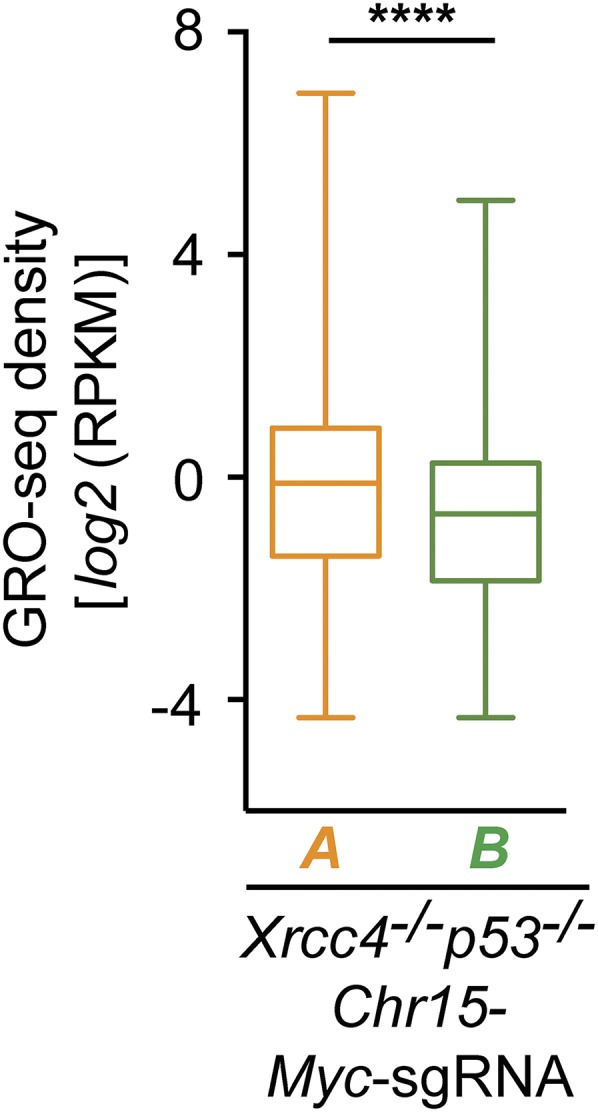
Active genes with TSS-proximal junctions show higher transcriptional activity on average than active genes with junctions in the gene body located more than 2 kb from the TSS. Box-and-whiskers plots of transcriptional activity (GRO-seq density) of class A and class B genes in Chr15-Myc-sgRNA–expressing Xrcc4−/−p53−/− NSPCs from four independent experiments. Numbers of genes within each group: Chr15-Myc-sgRNA Xrcc4−/−p53−/−: class A = 338 genes; class B = 1,915 genes; details are as described in Fig. 3B. ****P < 0.0001 (Mann–Whitney u test).
Both C-NHEJ and A-EJ Processes Can Mediate TSS-Associated Translocations in NSPCs.
HTGTS also provides a powerful DSB end-joining assay. In this regard, junctional repair signatures in HTGTS libraries can be analyzed at the nucleotide level by calculating the difference between the end coordinate of the bait alignment and the start coordinate of the prey alignment. In this calculation, a value of 0 corresponds to a “direct” junction, whereas negative values represent short nucleotide homologies or MHs. To investigate the repair junction structures of translocations between bait DSBs and prey DSBs located within 2 kb of TSSs (either annotated TSSs or “active” TSSs identified by GRO-seq; see Materials and Methods), we examined HTGTS data from Chr12-sgRNA-1–based experiments or Chr15-Myc-sgRNA–based experiments in WT or Xrcc4−/−p53−/− NSPCs (Fig. 4). Four independent experiments in Xrcc4−/−p53−/− NSPCs expressing Chr12-sgRNA-1 yielded 509 junctions located within 2 kb of an annotated TSS (Fig. 4A). Seven independent experiments in WT cells expressing Chr12-sgRNA-1 yielded a total of 366 junctions located within 2 kb of an annotated TSS (Fig. 4A). Notably, ∼35% of TSS-proximal translocation junctions in WT cells were direct, whereas only ∼10% of such junctions were direct in Xrcc4−/−p53−/− NSPCs (Fig. 4A). Moreover, the TSS-proximal translocation junctions in Chr12-sgRNA-1–expressing Xrcc4−/−p53−/− NSPCs displayed significantly longer junctional homologies (mean MH length of 2.71 ± 0.07 bp in Xrcc4−/−p53−/− NSPCs versus 1.79 ± 0.16 bp in WT NSPCs) (Fig. 4B). Examination of junction structures in Chr15-Myc-sgRNA–based experiments in WT or Xrcc4−/−p53−/− NSPCs yielded similar results (Fig. S5). Together, these findings indicate that both the C-NHEJ and A-EJ pathways can mediate translocations arising from transcription-associated DSBs.
Fig. 4.

Repair junction signatures of TSS-proximal HTGTS junctions. (A) TSS-proximal HTGTS junction profiles (located within 2 kb of all TSSs) from Chr12-sgRNA-1–expressing NSPCs of the indicated genotypes. Joins with 0 (direct or “blunt” joins) to 10 bp of junctional MH were identified and plotted as a percentage of total junctions; insertions were excluded from the dataset. The number of independent experiments is indicated. Total junction numbers analyzed: n = 366 WT and n = 509 Xrcc4−/−p53−/− NSPCs. (B) The mean MH length of TSS-proximal HTGTS junctions indicated in bar graphs was determined in the indicated number of independent experiments. Data represent mean ± SEM; ***P < 0.001; **P < 0.01; *P < 0.05 (unpaired two-tailed t test).
Fig. S5.

Repair junction profiles of TSS-proximal HTGTS junctions. (A) TSS-proximal (located within 2 kb of annotated TSSs) HTGTS junction profiles from Chr15-Myc-sgRNA–expressing NSPCs of the indicated genotypes; details are as in Fig. 4A. Total junction numbers analyzed: 421 WT and 374 Xrcc4−/−p53−/− NSPCs. (B) Mean MH length; details are as in Fig. 4B. Data represent mean ± SEM; ***P < 0.001; **P < 0.01; *P < 0.05 (unpaired two-tailed t test).
Discussion
This study, along with our additional recent study (4), identifies various classes of recurrent DSBs in NSPCs. Beyond the TSS-associated DSBs we define here, these other DSB classes in NSPCs include widespread, low-level DSBs that may be caused by cell-intrinsic processes such as replication or oxidative stress (1, 2) and RDCs in long neural genes that mostly are associated with mild replication stress in NSPCs (4). The estimated overall frequency of TSS-associated DSBs corresponds to about one in five NSPCs having a TSS-proximal DSB that translocates to a bait DSB (Table S1). This frequency is about 30-fold and 60-fold lower, respectively, than the frequency per NSPC of low-level widespread and RDC-associated DSBs that translocate to the same bait in the same Xrcc4−/−p53−/− background. Again, these numbers represent the minimal numbers of DSBs per NSPC in each class, because most DSBs are repaired locally, even in an XRCC4-deficient background (4). Based on these estimated numbers, the overall impact of TSS-associated DSBs on genomic stability in NSPCs is likely less than that of the other two classes. In this regard, consistent with these estimates, the great majority of DSBs in long transcribed neural RDC genes are in gene bodies and are not associated with TSSs, also indicating that these two classes of DSBs are independent (4). Finally, also consistent with this notion, TSS-associated DSBs tend to occur in a class of genes (class 2) involved in more general cellular processes that also are expressed and translocated in activated B cells, as opposed to cell-type–specific genes.
We find that DSBs are enriched in transcribed versus nontranscribed genes in NSPCs and are further enriched near the TSSs of generally expressed genes with transcription rates that are higher on average than those of most neural- or B-cell–specific genes. Transcription may contribute to this class of HTGTS-detectable DSBs mainly by promoting their occurrence (2, 5, 7, 9, 32, 33). Formation of transcription-associated DSBs may be related to torsional stress associated with transcription initiation, for example, that is caused by the binding of transcription factors to promoters (32). DSBs at gene promoters have been proposed to induce topological alterations and subsequent changes in chromatin organization that facilitate transcription initiation (32). In this regard, DSBs generated by DNA topoisomerase IIβ (TopoIIβ) during transcription initiation have been mechanistically implicated in the transcriptional activation of genes in various cell types, including postmitotic neurons (5, 7). Another proposed cause of TSS-associated DSBs is the formation of R-loops (2). We note, however, that our findings do not rule out a role for increased proximity of these generally expressed genes resulting from their localization to transcription factories (34). Enhanced interaction in transcription factories also could facilitate the formation of specific translocations of genes that break recurrently because of transcription-associated DSBs in specific cell types (6, 35).
Our analysis of repair junction profiles of TSS-proximal translocations in WT and C-NHEJ–deficient NSPCs shows that both the C-NHEJ and A-EJ pathways can mediate such translocations, as we recently have shown for DSB translocation junctions in RDC genes and in junctions involving translocations of more widespread low-level DSBs (4). A-EJ has been speculated to be a major contributor to oncogenic translocations (36), possibly because of some specific aspect of the A-EJ pathway or simply reflecting increased DSB persistence (and availability for translocation) in the absence of C-NHEJ (37). Although our studies do not rule out a more dominant role for A-EJ in contributing to oncogenic or other translocation junctions, they clearly suggest that C-NHEJ also can participate in such events.
Materials and Methods
Primary NSPC Isolation, Culture, and DSB Induction.
Primary NSPCs were prepared from postnatal day (P) 8–14 frontal brains and were cultured as described (38). All work involving mice was approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital (Protocol 14-10-2790R). Passage 0 dissociated cells at 0 d in vitro (DIV) were seeded into ultra-low-attachment six-well plates (Corning) at 4 × 105 cells/mL. DIV 4.5 cultures were dissociated into single cells, and 5 × 106 NSPCs were nucleofected with 5 μg of Cas9:sgRNA expression vector pSpCas9(BB) (Addgene plasmid 42230) (39) by using the Mouse Neural Stem Cell Nucleofector reagent (VPG-1004; Lonza). Cas9:sgRNA expression vectors were constructed by ligating annealed oligonucleotides into BbsI-digested pSpCas9(BB) (4). GR-I-SceI–mediated bait DSBs (9) were induced by the addition of 10 μM TA (Sigma) on DIV 5.5. Cells were collected and processed for GRO-seq or HTGTS analyses on DIV 9.
Global Run-On Sequencing.
Three biological replicate GRO-seq libraries per genotype (ATM−/−R26I-SceI-GRc-myc25xI-SceI or Xrcc4−/−p53−/−) were prepared and analyzed as previously described (4, 30, 31). In brief, Bowtie2 (40) was used to align GRO-seq data to the mouse genome (mm9/NCBI37), HOMER (41) was used for de novo identification of transcripts, and gene transcription rates were estimated as described (31).
HTGTS and Bioinformatic Analyses.
HTGTS was performed and data were analyzed as described (4, 9, 27). In brief, libraries (500–1,000 bp fragment size) were purified and sequenced on the Illumina MiSeq platform. After demultiplexing of FASTQ output files, unique reads were aligned to the mouse genome (mm9/NCBI37) by Bowtie2 (40) and were processed further through a custom HTGTS pipeline (27). The distances of HTGTS junctions to the closest TSS were calculated by a custom script. Translocation rates and DSB frequency were estimated (4), and Cas9:sgRNA off-target sites were identified (4, 27) as described. Nucleotide-level analysis of HTGTS junctional repair signatures was performed by calculating the difference between the end coordinate of the bait alignment and the start coordinate of the prey alignment (4). In this calculation, a value of 0 corresponds to a direct junction, and negative values represent MHs.
Acknowledgments
We thank members of the F.W.A. laboratory for stimulating discussions, Drs. Chunguang Guo, Monica Gostissa, and Jiazhi Hu for experimental advice; and Drs. Yi Zhang, Li Shen, and Fei-Long Meng for assistance with high-throughput sequencing. This work in the F.W.A. laboratory was supported by the Porter Anderson Fund from Boston Children's Hospital and the Howard Hughes Medical Institute. B.S. is a Martin D. Abeloff Scholar of The V Foundation for Cancer Research and is supported by National Institute on Aging/NIH Grant K01AG043630. P.-C.W. is supported by a National Cancer Center postdoctoral fellowship.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the Gene Expression Omnibus database (accession no. GSE74356).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1525564113/-/DCSupplemental.
References
- 1.Aguilera A, García-Muse T. Causes of genome instability. Annu Rev Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 2.Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet. 2012;13(3):204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152(3):417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei PC, et al. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. doi: 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ju BG, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312(5781):1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 6.Lin C, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139(6):1069–1083. doi: 10.1016/j.cell.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madabhushi R, et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161(7):1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322(5909):1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiarle R, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147(1):107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 12.Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8(25):1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 13.Gao Y, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95(7):891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 14.Frank KM, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5(6):993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, et al. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci USA. 2000;97(6):2668–2673. doi: 10.1073/pnas.97.6.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKinnon PJ. Maintaining genome stability in the nervous system. Nat Neurosci. 2013;16(11):1523–1529. doi: 10.1038/nn.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y, et al. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404(6780):897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- 18.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141(1):27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zha S, et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469(7329):250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar V, Alt FW, Oksenych V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst) 2014;16:11–22. doi: 10.1016/j.dnarep.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 22.Shiloh Y, Ziv Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210. [PubMed] [Google Scholar]
- 23.Callén E, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130(1):63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 24.Hu J, Tepsuporn S, Meyers RM, Gostissa M, Alt FW. Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc Natl Acad Sci USA. 2014;111(28):10269–10274. doi: 10.1073/pnas.1410112111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tepsuporn S, Hu J, Gostissa M, Alt FW. Mechanisms that can promote peripheral B-cell lymphoma in ATM-deficient mice. Cancer Immunol Res. 2014;2(9):857–866. doi: 10.1158/2326-6066.CIR-14-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong J, et al. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature. 2015;525(7567):134–139. doi: 10.1038/nature14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frock RL, et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33(2):179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, et al. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell. 2015;163(4):947–959. doi: 10.1016/j.cell.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148(5):908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min IM, et al. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011;25(7):742–754. doi: 10.1101/gad.2005511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meng FL, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159(7):1538–1548. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju BG, Rosenfeld MG. A breaking strategy for topoisomerase IIbeta/PARP-1-dependent regulated transcription. Cell Cycle. 2006;5(22):2557–2560. doi: 10.4161/cc.5.22.3497. [DOI] [PubMed] [Google Scholar]
- 33.Klein IA, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutherland H, Bickmore WA. Transcription factories: Gene expression in unions? Nat Rev Genet. 2009;10(7):457–466. doi: 10.1038/nrg2592. [DOI] [PubMed] [Google Scholar]
- 35.Mani RS, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326(5957):1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17(4):410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. The role of mechanistic factors in promoting chromosomal translocations found in lymphoid and other cancers. Adv Immunol. 2010;106:93–133. doi: 10.1016/S0065-2776(10)06004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nat Protoc. 2007;2(6):1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- 39.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruitt KD, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41(Database issue):D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]