

Significance
Membrane proteins perform diverse functions in the cell while being embedded in lipid bilayers, but the presence of the anisotropic, nonpolar membrane environment has slowed progress in understanding how these proteins fold and function. Herein, we study GlpG, an intramembrane protease, using computationally efficient models to fill in structural details that are currently invisible to experimental techniques and inaccessible to atomistic simulations. We find that GlpG’s modular functional architecture leaves an imprint throughout its folding and functional landscape, leading to multiple possible folding pathways and the population of near-native states with functional significance. We propose a mechanism by which destabilizing mutations can accelerate folding in detergent micelles, a previously puzzling experimental observation.
Keywords: membrane proteins, micelle folding, bilayer folding, folding mechanism, intramembrane proteolysis
Abstract
We investigate the folding of GlpG, an intramembrane protease, using perfectly funneled structure-based models that implicitly account for the absence or presence of the membrane. These two models are used to describe, respectively, folding in detergent micelles and folding within a bilayer, which effectively constrains GlpG's topology in unfolded and partially folded states. Structural free-energy landscape analysis shows that although the presence of multiple folding pathways is an intrinsic property of GlpG’s modular functional architecture, the large entropic cost of organizing helical bundles in the absence of the constraining bilayer leads to pathways that backtrack (i.e., local unfolding of previously folded substructures is required when moving from the unfolded to the folded state along the minimum free-energy pathway). This backtracking explains the experimental observation of thermodynamically destabilizing mutations that accelerate GlpG’s folding in detergent micelles. In contrast, backtracking is absent from the model when folding is constrained within a bilayer, the environment in which GlpG has evolved to fold. We also characterize a near-native state with a highly mobile transmembrane helix 5 (TM5) that is significantly populated under folding conditions when GlpG is embedded in a bilayer. Unbinding of TM5 from the rest of the structure exposes GlpG’s active site, consistent with studies of the catalytic mechanism of GlpG that suggest that TM5 serves as a substrate gate to the active site.
GlpG is a rhomboid protease that sits and functions in the cell membrane. GlpG’s homologs are found across all kingdoms of life. GlpG has been the subject of several biophysical experimental studies aimed toward understanding membrane protein folding and the relationships among protein structure, dynamics, and function (1–5). An extensive experimental φ-value analysis found φ-values significantly different from zero, indicative of structural changes during the rate-limiting step of folding, in transmembrane helices 1 through 5 (TM1-5) and the intervening loops (4). Most of the nonzero φ-values, particularly in TM3-5 and in the large loop L1, were negative, meaning that although the corresponding mutation destabilizes the native state, the mutation nonetheless accelerates folding. The preponderance of negative φ-values was puzzling and unprecedented, and at the time, these effects were tentatively ascribed to nonnative interactions in the transition state ensemble. In this work, we show that, in fact, simple models with perfectly funneled energy landscapes that lack nonnative interactions are able to explain the origin of these negative φ-values and how the values arise when folding in detergent micelles rather than bilayers.
α-Helical membrane protein folding is thought to occur in two stages in vivo (6). The first stage, setting up the proper topology of transmembrane helices, is handled by the translocon (7, 8). In the present context, topology refers to specifying the directions in which a membrane protein’s constituent transmembrane helices traverse the bilayer. The second stage, converting from properly inserted but dissociated helices into a functional folded structure, occurs spontaneously and is, in some ways, analogous to soluble protein folding. However, we know, ranging from the hydrophobic effect (9, 10) to water-mediated (11) and screened electrostatic interactions (12), the solvent plays a role in determining what types of noncovalent interactions are stabilizing and destabilizing. Whereas soluble proteins fold in polar and isotropic aqueous solutions, membrane proteins fold in largely apolar and anisotropic environments. These environmental differences complicate applying directly methods developed for studying soluble protein folding to the study of membrane protein folding. Nonetheless, experimentalists have been able to apply a variety of methods to study the kinetics and thermodynamics of membrane protein folding through the use of detergent micelles as a membrane-mimicking environment. Experiments that probe the folding mechanisms of membrane proteins have used micelles composed of a mixture of anionic and nonionic detergents (4, 13, 14), which not only keep membrane proteins soluble but also, through use of mixed micelles, allow the equilibrium between folded and unfolded states to be tuned. Micelles predominantly composed of nonionic detergents, such as n-dodecyl-β-d-maltopyranoside (DDM), preferentially stabilize a folded state that has been shown to be functional and is therefore likely to be structurally similar to the folded state in vivo. Micelles predominantly composed of anionic detergents, on the other hand, preferentially stabilize an unfolded state that contains significant amounts of secondary structure. This ability to tune the equilibrium means that stopped-flow kinetic experiments can be combined with protein-engineering techniques to determine folding mechanisms at the single-residue level (4, 13, 15), in analogy to what has been done for soluble proteins (16–18). Because carrying out these types of experiments in bilayers is still difficult, it is presently unknown how folding mechanisms determined in micelles compare with those in membranes. Confining proteins to a 2D membrane is expected to constrain unfolded and partially folded ensembles to having structures with helices that are largely properly aligned and embedded in the membrane; such topological restrictions would be relaxed in a micellar environment.
Theoretical (19, 20) and experimental (3, 4) work suggests that at least some membrane proteins can reversibly fold and unfold without the aid of the translocon or chaperones in vitro. It is therefore likely that membrane protein folding landscapes are funneled, much like globular protein landscapes (21, 22). Structure-based models with perfectly funneled energy landscapes have proven useful for investigating the folding and binding of proteins (23, 24). In this study, we use a structure-based model to investigate folding of a membrane protein in two different situations: in the absence and presence of an implicit membrane energy term that biases conformations to have the correct topology with respect to the membrane. Simulations with the implicit membrane term are thus taken to model folding in a bilayer, whereas simulations without the implicit membrane energy are taken to model folding in detergent micelles. Although this way of modeling micelles and bilayers is an oversimplification, it captures the significantly increased topological freedom of membrane proteins in micellar environments compared with lipid bilayer. Fig. 1 shows schematic representations of the corresponding denatured states of membrane proteins in bilayers and micelles.
Fig. 1.

Schematic diagrams of the unfolded state of α-helical membrane proteins in bilayers (Left) and detergent micelles (Right). The transmembrane helices (cylinders) are connected by loops. Transmembrane helices are either embedded in a membrane (rectangular prism) or are surrounded by detergent micelles (transparent gray spheres). In this work, we use an implicit membrane model to simulate folding within a bilayer and assume that folding in detergent micelles corresponds to folding without constraints on the alignment of helices. In both cases, we assume that the unfolded state has near-native levels of secondary structure, as has been observed in experiments on the SDS-denatured state of membrane proteins.
The same energy landscape that dictates folding routes also encodes functional motions. It has been suggested that the modularity in the structure of GlpG supports functional motions (1, 25). The N-terminal domain, which contains transmembrane helices 1 and 2 (TM1-2) as well as the intervening L1 loop, functions as a structural scaffold (25), whereas the C-terminal domain with its four transmembrane helices (TM3-6) includes the catalytic site (25). The C-terminal domain is apparently more flexible than the N-terminal domain; both the loop L5 (5) and the transmembrane helix TM5 (25) have been crystallized in multiple conformations. Because of this flexibility, it has been suggested that either L5 alone (5) or L5 and TM5 (25) may serve as a substrate gate for access to the catalytic site. Using free-energy landscape analysis and perturbation methods along with structural analysis, we show that there is a near-native state significantly populated under folding conditions and elucidate the state’s connections to GlpG’s folding mechanism and function.
Methods
Simulation and Analysis Methodology.
We performed molecular dynamics simulations of a coarse-grained structure-based model (26) of GlpG based on the crystal structure with Protein Data Bank (PDB) ID code 2XOV (27). We carried out two parallel sets of simulations: one with an implicit membrane present and one without a membrane. The implicit membrane model is described in ref. 19, and the assignment of residues into the intramembrane and extramembrane residues is described in Fig. S1. We sampled at multiple temperatures above and below the corresponding folding temperatures and used umbrella sampling at each of these temperatures to sample a wide range of folded, partially folded and unfolded structures. We then used the Multistate Bennett Acceptance Ratio (MBAR) method (28) to reconstruct unbiased free-energy profiles, compute expectation values of structural order parameters, and perform perturbative calculations to test the effect of small changes to the Hamiltonian. We infer folding mechanisms by looking for low free-energy routes between the unfolded and folded states in the unbiased free-energy profiles and then performing analysis on structures sampled in the basins and saddle points along these routes. Whereas the appropriateness of various reaction coordinates for describing protein folding kinetics is vigorously discussed (29–31), here we take the pragmatic approach of comparing our inferred mechanisms to experimental data and find highly nontrivial agreement based on reaction coordinates that measure the degree of nativeness of different parts of the molecule. See the Supporting Information for a complete explanation of the methods.
Fig. S1.

Proper topology of the native structure of GlpG used in the implicit membrane model. Residues in the transmembrane region are colored in red. Periplasmic and cytoplasmic residues are colored in yellow. L1 is large and contains two interfacial helices, of which residues 137–143 were assigned to be in the transmembrane region.
Structure-Based Model of GlpG.
The crystal structure used to define the stabilizing native interactions in our structure-based model is shown in Fig. 2. GlpG has six transmembrane helices connected by five loops. The first loop, L1, is notable because it is large and contains several small interfacial helices. Our definition of the N- and C-terminal domains of GlpG was arrived at based on the analysis of our simulation results and is therefore not imposed on the model beforehand; these two domains are found to fold semi-independently (Results and Discussion) using our structure-based model. Therefore, this definition arises as a direct consequence of the structure of GlpG given our way of defining its contact map. Structural bioinformatics studies have indicated that membrane proteins are stabilized by tight helix–helix interactions that are mediated by small and polar residues (32). We therefore used a 6.5-Å Cβ–Cβ cutoff to define stabilizing native interactions, which is somewhat shorter than the cutoffs that have been applied to simulations of soluble proteins in the past. We have also selectively strengthened the local-in-sequence interactions to decouple secondary and tertiary structure formation. This modification of the model is motivated by the observation of native-like levels of secondary structure in the SDS-unfolded state of GlpG (4). See the Supporting Information for a precise description of the parameters used in the model.
Fig. 2.

Crystal structure of GlpG (PDB ID code 2XOV). A black sphere demarcates the boundary between the N- and C-terminal domains. The catalytic dyad in the active site (AS), shown in yellow and located on TM4 and TM6, is buried by TM5 and L5. The large loop L1 is made up of several interfacial helices whose axes run parallel to the membrane surface. The color of the backbone varies smoothly from red (N terminus) to white and then to blue (C terminus).
Results and Discussion
Unfolding Always Corresponds to Loss of Tertiary Structure with Retention of Secondary Structure but Leads to a More Expanded Ensemble in the Absence of the Implicit Membrane.
Experimental circular dichroism and tryptophan fluorescence measurements indicate that unfolding of GlpG in micelles corresponds to loss of tertiary structure but retention of native levels of secondary structure (4). In the simulations, the expectation values of secondary and tertiary structure formation order parameters (see the Supporting Information for precise descriptions) as a function of temperature indicate that likewise, both in the absence and the presence of the implicit membrane, unfolding corresponds to loss of tertiary structure and retention of secondary structure (Fig. S2). When the implicit membrane is present, the unfolded structures largely retain native-like topologies with respect to the membrane (Fig. 3E), although excursions to the extramembrane regions are possible. The simulated unfolded ensemble thus resembles what is commonly understood to be the starting point for the “second stage” of membrane protein folding (6), which takes place once the helices have been inserted into the membrane by the translocon in their native orientations. The simulated unfolded ensemble in the absence of the bilayer is significantly more expanded (Fig. 3B). In the Supporting Information, we discuss a more detailed comparison of these two ensembles and experiment (Fig. S3).
Fig. S2.

Expectation value of tertiary and secondary structure formation order parameters both with and without the implicit membrane model. Temperature is normalized by the folding temperature of each model independently. Precise definitions of short- and long-range foldedness are given in Order Parameters.
Fig. 3.

Free-energy analysis and structural characterizations of GlpG without (A–C) and with (D–F) the implicit membrane. (A and D) Two-dimensional free-energy profiles above (Left), at (Center), and below (Right) the folding temperature (Tf) with respect to QN and QC. QN and QC measure the degree of folding within the N- and C-terminal domains, respectively. Precise definitions are given in the Supporting Information. Key structural states are labeled, and the inferred folding pathways are indicated with arrows. Areas shown in white are high in free energy. (B and E) Structural ensembles made up of 10 representative structures selected from low free-energy basins and transition states; folded regions in each ensemble have been aligned for clarity. (C and F) Schematic representations of the structural ensembles. Transmembrane helices and the large loop L1 are shown as fully folded (full color), partially folded (half color), or unfolded (black). The colors used in B, C, E, and F are the same as those established in Fig. 2.
Fig. S3.

Comparison of interhelical (Upper) and intrahelical (Lower) distances in the simulated denatured states of GlpG. The mean interhelical distances are plotted as a function of the sequence separation between the probed residues for both the model without (blue) and with (green) the implicit membrane present. Experimentally measured interhelical and intrahelical distances in the SDS-denatured state of bR (red) are plotted for the sake of comparison. In all cases, SDs are indicated with error bars.
Folding Can Be Initiated in Either the N- or C-Terminal Domain of GlpG.
Both in the absence and presence of the implicit membrane, free-energy profiles plotted as a function of and (see the Supporting Information for precise descriptions), which quantify how native-like the structures are for the N- and C-terminal parts of the molecule, respectively, suggest that folding can be initiated by moving either along or [i.e., by forming native-like structure within either the N-terminal or C-terminal parts of the molecule (Fig. 3)]. Above but near the folding temperature and in the presence of the implicit membrane, the molecule populates both the fully unfolded state (U) and the C-terminal folded state (C) with TM3-6 folded. An orthogonal folding route toward the N-terminal folded state (N) is also present, although less favorable. In the absence of the implicit membrane and above the folding temperature, the molecule prefers the fully unfolded state (U) and a partially formed N-terminal structure with L1 folding onto TM1 (N1). Slightly higher in free energy in the same direction is another state with both TM1 and TM2, as well as the intervening L1, being well folded (N2). As in the case of the model with the implicit membrane, another folding route is available at higher free energy. There are also two intermediates along this route, the first with TM4-6 folded (C1) and a second which also includes folding TM2, TM3, and part of L1 onto the C-terminal part of the molecule (C2).
Optimal Energy–Entropy Compensation for the Modular Structure Results in a Multistep Folding Pathway That Backtracks During the Rate-Limiting Step Without the Implicit Membrane but Does Not Backtrack in the Membrane with Its Accompanying Topological Constraints.
After initiating folding through either the N- or C-terminal domains, GlpG must fold the other half of the molecule to arrive at the folded state. In the membrane, this completion of folding occurs in a straightforward manner, with both pathways (U→C→TS1→F and U→N→TS2→F) being approximately equal in free energy (Fig. 3D). Without the implicit membrane energy term to constrain the topology, however, folding becomes more complex. Although initiating folding via the N-terminal domain (U→N1→N2) is more favorable than initiating folding via the C-terminal (U→C1→C2), starting along this route is ultimately not productive as the molecule later encounters a relatively high free-energy barrier (TS2) associated with organizing the large and unconstrained C-terminal domain. Folding does not proceed by propagating the folding “front” through the interface between the N- and C-terminal domains because there are relatively few contacts on the interface. Instead, the high free-energy barrier to folding is lowered somewhat through simultaneous organization of TM4-6 (a decrease in energy) at the same time as breaking the interface between L1/TM2 and TM3 (an increase in entropy), which was formed in N2. Breaking the interface between L1/TM2 and TM3 is an example of “backtracking” (i.e., the required unfolding of natively folded substructures while proceeding from the unfolded state to the folded state). By making optimal use of energy-entropy compensation, GlpG is able to reduce the free-energy barrier between a partially folded state and the completely folded state because there are multiple sites for nucleating folding. Once both domains are independently folded in TS2, a saddle point in the free-energy surface is reached and folding can proceed downhill to the folded state (F). This effect is also operative when folding is initiated in the C-terminal direction (U→C1→C2). Proceeding initially uphill in free energy, GlpG arrives at C2 where TM2-6 and parts of the loop L1 are folded. Because L1 is quite large, however, there exists a high entropic barrier to consolidate folding of TM1. Again, a compromise is made by simultaneously forming the interface TM1-TM2 and contacts within L1 along with releasing of L1 from its position docked against L3 and breaking the interface between TM2 and the C-terminal domain (TM3/L3/TM4). Finally, folding can proceed downhill toward the folded state by reforming the interface between TM2 and the C-terminal domain and reinserting L1. Note that the presence of high-energy intermediates and multiple folding pathways are compatible with the apparent two-state behavior observed in the micelle-mediated folding experiments. Folding is cooperative in experiments and in our simulations, but free-energy landscape analysis allows us to resolve high free-energy intermediate states and multiple pathways that would not necessarily be apparent from the initial experimental data alone. With the simulation-derived structural model for the parallel pathways, it should be possible to design experiments that probe this aspect of GlpG folding.
Of the two putative folding pathways, the latter one, initiated through folding the C-terminal domain, has the lower free-energy transition state (TS1) and should be dominant. TS1 differs somewhat from the transition state ensemble with an unfolded C-terminal domain and N-terminal folding nucleus, which was inferred without the aid of modeling and based on the distribution of experimentally measured canonical (0 < φ < 1) φ-values in GlpG (4). However, in that study, thermodynamically destabilizing mutations that accelerated folding and unfolding were found throughout TM3-5 and in L1. The resulting φ-values are negative. Destabilizing mutations that slow folding, leading to positive φ-values, were found largely on the interface TM1-TM2 but also in L1. Fig. 4 shows the difference between the average contact maps of TS1 and C2 as well as the connections between the contacts that are gained and lost in going from C2 to TS1 and the experimentally measured φ-values. Mutations that destabilize the interface between L1/TM2 and the C-terminal domain accelerate folding because formation of TS1 involves breaking those contacts. Mutations that destabilize the interface TM1-TM2 will slow folding because formation of TS1 also involves forming that interface. Mutations that primarily affect contacts within the C-terminal domain result in near-zero φ-values, because those contacts are largely preserved in the C2→TS1 transition. Thus, we see that the dominant mechanism predicted by simulations in the absence of the membrane (U→C1→C2→TS1→F) provides a detailed structural explanation of the previously puzzling preponderance of negative φ-values measured in the C-terminal domain of GlpG. On a topologically unconstrained but perfectly funneled landscape, folding is complicated by GlpG’s modular structure and the high entropic cost of organizing helical bundles from their unconstrained partially folded states. Nonnative frustrated interactions need not be invoked to explain the presence of a large number of negative φ-values in GlpG.
Fig. 4.

Contact map of GlpG showing the C2→TS1 structural transition. The axes are labeled with residue indices. Contacts that change their occupancy by more than 20% when going from C2 to TS1 are shown in blue filled circles (gained in TS1; upper diagonal) and red filled circles (lost in TS1; lower diagonal). All other native contacts satisfying |i − j| > 4 are shown as empty circles. Positive (blue) and negative (red) experimental φ-values satisfying | φ | > 0.2 are plotted along the diagonal as filled diamonds. Arrows illustrate the proposed connections between the experimental φ-values and the contacts that are either lost or gained in the simulated structural ensembles. Text labels indicate the interfaces that are either formed or broken during the transition. Note that the positive φ-value at position 219 (the only significantly positive φ-value in the C-terminal domain) is derived from a mutation that actually accelerates folding and unfolding, like those that lead to the negative φ-values, but is formally positive because the mutation slightly stabilizes (rather than destabilizes) the native state.
A recent single-molecule force spectroscopy study in bicelles and micelles also found evidence for structural modularity in GlpG unfolding (3). The authors found that the unfolding of GlpG at high force was cooperative. The authors were also able to characterize two transiently populated metastable states. The authors’ structural interpretation of the unfolding via intermediates closely corresponds to the reverse of one of our folding pathways (F→TS2→N→U) in the presence of the bilayer, whereas the structural decomposition of GlpG into domains given in the supplementary information of the study’s article corresponds more or less exactly to the reverse of one of our dominant folding pathways (F→C2→C1→U) in the absence of the bilayer. These encouraging correspondences (see the Supporting Information for a more detailed discussion) suggest that further computational and experimental work should allow us to create a unified picture of SDS- and force-induced unfolding of GlpG in micelles and bilayers.
TM5 Is Loosely Bound Even Under Folding Conditions.
GlpG is an intramembrane protease of the rhomboid serine protease class (33). GlpG cleaves specific transmembrane substrates using a catalytic dyad that is buried within the lipid bilayer (25). Fig. S4 shows two crystal structures of GlpG, one in a “closed” conformation, the one used to construct our structure-based energy landscape, and the other in an “open” conformation, where L5 and TM5 have bent away from the rest of the structure to expose partially the catalytic dyad. It has been suggested that TM5 functions as a substrate gate that opens for full-sized substrates to gain access to the catalytic site (25).
Fig. S4.
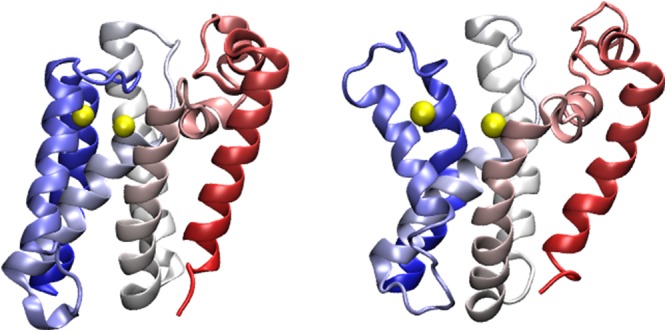
Comparison of the closed (PDB ID code 2XOV) (Left) and open (PDB ID code 2NRF chain A) (Right) crystal structures of GlpG. The catalytic dyad (shown in yellow) is buried in the closed state and exposed in the open state. The largest differences between the open and closed states are in TM5 and L5. This observation led to the suggestion that TM5 serves as a gate for access to the catalytic dyad.
Preferential stabilization of the contacts within the N-terminal domain by 10% suffices to populate a near-native state (F*) under folding conditions in the presence of the implicit membrane (Fig. 5), according to our perturbation calculations. Structural analysis of this state revealed a heterogeneous ensemble of near-native conformations with a common feature: TM5 was unbound from TM4 and TM6, thereby exposing the catalytic dyad. In this state, deviations from the closed crystal structure occur most significantly in TM5 and the connecting loops L4 and L5 (Fig. 6). Whether or not TM5 must undergo significant conformational rearrangements in order for full-sized substrates to access the proteolytic site is a matter of some controversy (5, 25, 34). Our model suggests that the conformation of TM5 is highly dynamic even under folding conditions, which is consistent with the experimental observation that tethering TM5 to TM2 eliminates enzymatic activity (1). The fact that stabilizing the N-terminal part of the molecule increases the population of this state agrees with the experimental observation that destabilizing L1 reduces enzymatic activity (1, 25), highlighting the role of the N-terminal part of the molecule as a structural scaffold. Whereas TM5 is mobile in F*, F* differs, crucially, from TS2 in the implicit membrane (Fig. 3 D–F) by TM6 remaining bound to TM4. The tight association between TM4 and TM6 is mediated by GXXXAXXG and GXXXGXXXA motifs, which stabilize the C-terminal domain and protect against unfolding during GlpG’s functional motions.
Fig. 5.

Two-dimensional free-energy profiles of GlpG without (Upper) and with (Lower) the implicit membrane below the folding temperature, and with an N-terminal domain destabilized (Left) and stabilized (Right) by 10%. A near-native state (F*) is highly populated and accessible from the folded state (F) when the N-terminal is stabilized and the implicit membrane is present.
Fig. 6.

Representative structures from a near-native state (F*) (Fig. 5) sampled while simulating with the implicit membrane present. The structures were all aligned to the closed crystal structure (PDB ID code 2XOV) and colored according to the individual residue rmsd values. Blue indicates low rmsd (high similarity to the crystal structure), and red indicates high rmsd. The catalytic dyad is shown using yellow spheres. High rmsd values are localized to the C-terminal half of the molecule and to TM5 in particular. Movement of TM5 exposes the catalytic dyad, thereby allowing substrate access. This state is highly populated under folding conditions when strengthening the contacts in the N-terminal half of the molecule by 10% relative to the contacts in the C-terminal half of the molecule.
Conclusions
Experiments that probe membrane protein folding on the single-residue (4, 35) and the single-molecule (3) levels begin to allow us to determine the mechanisms by which membrane proteins fold and function. Nevertheless, many details of these processes remain hidden to even the most sensitive experiments. Using mixed micelles provides powerful tools for investigating membrane protein biophysics because of the relative simplicity and general applicability of these micelles, but the structure of the denatured state and its effect on folding mechanisms needs to be better understood. Thus far, studies of how residual structure in the denatured state affects folding have focused on soluble proteins and have used atomistic simulations (36), NMR and other types of spectroscopy (37), or combinations of the two (38). The question of residual structure is certainly no less important for membrane proteins, but the membrane environment poses challenges to both NMR and atomistic simulations. In this work, we used a coarse-grained energy landscape model to explore two limiting models of the folding of an intramembrane protease, GlpG: one limit in which the helices largely remain embedded in the membrane with their proper orientations, as is expected for the denatured state in lipid bilayers, and another limit where no constraints are placed on the alignment of helices in the unfolded state, this being taken as a model for the SDS-denatured state in micelles. Despite the simplicity of these models, on their basis, we have been able to propose a solution to the major puzzle in the experimental study of GlpG’s folding mechanism, characterize a near-native state with potential functional significance, and show how these phenomena are related to GlpG’s modular structure and topological constraints on the motions of partially folded states. The modular architecture of GlpG supports functional motions, including a highly mobile TM5, and leads to backtracking during the rate-limiting step of folding when the entropic cost of organizing helical bundles is high, as is the case in the absence of a bilayer. By providing a structurally detailed resolution of the φ-value puzzle, our analysis gives strong support to the notion that GlpG folding in mixed micelles proceeds by assembling helices with native levels of secondary structure from a state with few other constraints, as guided by a funneled, minimally frustrated landscape.
Simulation Methodology
Simulations were performed using the associative-memory, water-mediated, structure and energy model (AWSEM-MD) (39) simulation package, which is implemented in the large-scale atomic/molecular massively parallel simulator (LAMMPS) molecular dynamics simulation package (40). We used a modified version of the structure-based model described in (26). The value of “p”, which determines the degree of nonadditivity in the structure-based model, was set to 1, yielding a pairwise additive model. The local-in-sequence contacts () were given a strength of 1, whereas long-range contacts were given a strength of 0.5. For soluble proteins, typical values for short- and long-range interactions are 0.25 and 0.5, respectively. A 6.5-Å cutoff between Cβ atoms (Cα for glycine) was used to define native contacts based on the crystal structure with PDB ID code 2XOV. The implicit membrane model used is described in (19), and the assignment of residues for GlpG is explained below. We performed two sets of simulations, with and without the implicit membrane present. For each model, umbrella sampling simulations using the potential given in Eq. S1 were performed at four temperatures separated evenly by 25-K intervals:
| [S1] |
The definition of all order parameters including Q is given below. The temperature range in the case of the simulations with the implicit membrane was 150 to 225 K in the case of the simulations with the implicit membrane and 135–210 K in the case of the simulations without the implicit membrane. The folding temperature was determined empirically using the peak of the heat capacity curves, and all temperatures referenced within the body of the paper were normalized to units of the folding temperature of each model independently. Twenty umbrella sampling simulations were run at each temperature with bias centers ranging from Q0 = 0.00 to Q0 = 0.95, spaced evenly. Each simulation was run for 20,000,000 time steps of 2 fs each. Structures and energies were saved every 1,000 steps.
Order Parameters
Several order parameters were calculated for all structures. Global Q (used for umbrella sampling), QN, and QC as well as the secondary and tertiary structure foldedness parameters were calculated using Eq. 2, varying only the pairs of residues that were summed over and the corresponding normalization constant such that all order parameters had a maximum range of 0–1:
| [S2] |
Np is the normalization constant equal to the number of pairs included in the sum. rij is the instantaneous distance between the Cβ atoms of residues i and j. is the corresponding distance in the crystal structure. σij is a weakly sequence separation dependent well width, For global Q, all unique pairs of residues are included. For QN and QC, the sum runs over all unique pairs within the N- and C-terminal domains, residues 91–171 and 172–271, respectively. The short-range foldedness is calculated by summing over all unique pairs satisfying the long-range foldedness by those pairs of residues satisfying Intra- and interhelical distances were calculated using the Cα atoms of select residues as described in Comparison of Simulated Unfolded Ensembles in the Absence and Presence of the Implicit Membrane to Double Electron–Electron Resonance Experiments.
Free Energy and Expectation Value Calculations
All free-energy profiles and expectation values were calculated using MBAR as implemented in the pyMBAR package (28).
Visualization of Structures
Visualization of structures was performed using VMD (41) and PyMOL (42). Representative structures were picked based on the range of QN and QC of the low free-energy basins and transition states found in the free-energy profiles. For each state, 10 structures were visualized, chosen evenly from throughout all samples that belong to that state, and aligned according to which parts of the molecule are folded in that state.
Implicit Membrane Energy Term and Topological Assignment
The implicit membrane force field is a function of the z coordinates of the Cα atoms. Residues were assigned to be either intramembrane or extramembrane based on the z coordinate of their Cβ atom: |z| < 15 Å, intramembrane, otherwise extramembrane. The proper topology of GlpG within the membrane was obtained directly from the 3D experimentally determined structure using the TMDET webserver (43). Residues are assigned to be in periplasmic, transmembrane, or cytoplasmic regions in the simulation, in which periplasmic and cytoplasmic environments are treated equally. Residues 135–143 (those residues in L1 that are below the membrane plane) were reassigned to be in the transmembrane region. Proper topology of GlpG used in the implicit membrane model is shown in Fig. S1.
Closed and Open Conformations of GlpG
Two crystal structures of GlpG, “closed” and “open” conformations, are shown in Fig. S4. The “closed” conformation (PDB ID code 2XOV) was used to construct our perfectly funneled structure based model. The “open” conformation (PDB ID code 2NRF chain A) has L5 and TM5 bent away from the rest of the structure to make the catalytic site partially accessible.
Comparison with Single-Molecule Force Spectroscopy Study in Bicelles
We noted a close structural correspondence between states described in ref. 3 and those that we found during our simulations. The other study’s structural interpretation of the unfolding via intermediates given in the main text (in their notation) (N→I1→I2→U) closely corresponds to the reverse of one of our folding pathways (F→TS2→N→U) in the presence of the bilayer. Note that, in the other authors’ notation, N refers to “native,” whereas in our notation, N refers to N-terminal. Using mutational perturbations and by examining unfolding rip lengths, the other authors infer a unidirectional unfolding pathway that starts at the C-terminal and proceeds roughly two helices at a time. I1 therefore corresponds to unfolding of TM5 and TM6. In our TS2, TM6 is unfolded and TM4 and TM5 are in the process of being folded onto the N-terminal domain. I2 corresponds to the unfolding of two more helices, leaving only TM1 and TM2 folded. The N-terminal folded domain in our simulations (N) in the presence of the bilayer consists of a folded TM1 and TM2 and a partially folded TM3. The structural decomposition of GlpG into domains given in the other study’s supplementary information (into N, M, and C domains) corresponds more or less exactly to the reverse of one of our dominant folding pathways (F→C2→C1→U) in the absence of the bilayer. The N domain consists of TM1 and L1, which is the unfolded part of the molecule in our C2. The M domain consists of TM2 and TM3, which are the two helices that unfold when going from C2 to C1 in our analysis. Finally, the C domain consists of TM4-6, which is the minimal folding unit for the C-terminal domain in our simulations and makes up the folded region in C1.
Comparison of Simulated Unfolded Ensembles in the Absence and Presence of the Implicit Membrane to Double Electron–Electron Resonance Experiments
To more precisely compare the degree of compaction between the two simulated unfolded ensembles and to expose these unfolded ensembles to potential experimental falsification, we calculated the expected value of several intrahelical and interhelical distances as a function of temperature. These distances were chosen in analogy to distances that were measured in the SDS-unfolded state of bacteriorhodopsin (bR) using double electron–electron resonance (DEER) experiments (44). To measure these distances during the simulations, we virtually labeled the Cα atoms of residues D116 (TM1), F146 (TM2), P195 (TM3), G199 (TM4), F245 (TM5), and A250 (TM6) on the periplasmic side of the protein and A93 (TM1), S171 (TM2 and TM3), P219 (TM4), L225 (TM5), and N271 (TM6) on the cytoplasmic side. Note that the cytoplasmic loop between TM2 and TM3 is very short, so we use S171 to represent the cytoplasmic ends of both TM2 and TM3. Based on the location of these atoms, we calculated six intrahelical distances (A93-D116, TM1; F146-S171, TM2; S171-P195, TM3; G199-P219, TM4; L225-F245, TM5; A250-N271, TM6) corresponding to the length of each helix and 12 interhelical distances (TM1–TM2, TM1–TM4, TM1–TM6, TM2–TM4, TM2–TM6, and TM4–TM6 on both the cytoplasmic and periplasmic sides).
The expected value of the intrahelical and interhelical distances above the folding temperature (i.e., in the unfolded ensembles) are plotted as a function of sequence separation in Fig. S3. For comparison, and because these distances have not yet been measured in experiment for GlpG, experimental measurements of analogous distances in bacteriorhodopsin (44) are plotted alongside the simulation results for GlpG. Whether or not the implicit membrane is present, the intrahelical distances in GlpG show good agreement with those measured for bR, attributable to the fact that, in all cases, the helices present in the folded state remain formed in the denatured state. The interhelical distances measured for the simulated ensemble of GlpG in the presence of the implicit membrane are also in approximate agreement with those measured in experiment for bR. However, whereas the distances in GlpG increase nearly monotonically with sequence separation, the measurements on bR indicate that the interhelical distances are nearly independent of sequence separation. The interhelical distances measured in the simulated ensemble of GlpG in the absence of the implicit membrane are considerably higher than those in the presence of the implicit membrane but show the same increasing trend with sequence separation as the distances in the presence of the implicit membrane. Most of the distances measured in the absence of the implicit membrane exceed the stated maximum range of the DEER experiments that were used to measure the distances for bR (44). Further experimental work on GlpG and computational study of unfolded ensembles of bR will be required to fully understand what constraints, if any, are imposed on the SDS-unfolded ensembles of membrane proteins and how the ensembles might differ from protein to protein.
Acknowledgments
We thank Sin Urban for ongoing constructive discussions about GlpG. K.L.L. and N.P.S. acknowledge support from the Novo Nordisk Foundation. K.L.-L., N.P.S., and D.E.O. were supported by Danish Research Council Grant DFF-4090-00220 and Carlsberg Foundation Grant CF14-0287. H.H.T. and P.G.W. were supported by National Institute of General Medical Sciences Grant R01 GM44557 and the D. R. Bullard-Welch Chair at Rice University (Grant C-0016). Computational resources were supported in part by the Data Analysis and Visualization Cyberinfrastructure funded by the National Science Foundation Grant OCI-0959097.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1524027113/-/DCSupplemental.
References
- 1.Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci USA. 2007;104(20):8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker RP, Urban S. Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol. 2012;8(9):759–768. doi: 10.1038/nchembio.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min D, Jefferson RE, Bowie JU, Yoon TY. Mapping the energy landscape for second-stage folding of a single membrane protein. Nat Chem Biol. 2015;11(12):981–987. doi: 10.1038/nchembio.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paslawski W, et al. Cooperative folding of a polytopic α-helical membrane protein involves a compact N-terminal nucleus and nonnative loops. Proc Natl Acad Sci USA. 2015;112(26):7978–7983. doi: 10.1073/pnas.1424751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zoll S, et al. Substrate binding and specificity of rhomboid intramembrane protease revealed by substrate-peptide complex structures. EMBO J. 2014;33(20):2408–2421. doi: 10.15252/embj.201489367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popot JL, Engelman DM. Membrane protein folding and oligomerization: The two-stage model. Biochemistry. 1990;29(17):4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 7.Pohlschröder M, Prinz WA, Hartmann E, Beckwith J. Protein translocation in the three domains of life: Variations on a theme. Cell. 1997;91(5):563–566. doi: 10.1016/s0092-8674(00)80443-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Miller TF., 3rd Long-timescale dynamics and regulation of Sec-facilitated protein translocation. Cell Reports. 2012;2(4):927–937. doi: 10.1016/j.celrep.2012.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29(31):7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 10.Kauzmann W. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 11.Papoian GA, Ulander J, Wolynes PG. Role of water mediated interactions in protein-protein recognition landscapes. J Am Chem Soc. 2003;125(30):9170–9178. doi: 10.1021/ja034729u. [DOI] [PubMed] [Google Scholar]
- 12.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268(5214):1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 13.Schlebach JP, Woodall NB, Bowie JU, Park C. Bacteriorhodopsin folds through a poorly organized transition state. J Am Chem Soc. 2014;136(47):16574–16581. doi: 10.1021/ja508359n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curnow P, et al. Stable folding core in the folding transition state of an alpha-helical integral membrane protein. Proc Natl Acad Sci USA. 2011;108(34):14133–14138. doi: 10.1073/pnas.1012594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otzen DE. Mapping the folding pathway of the transmembrane protein DsbB by protein engineering. Protein Eng Des Sel. 2011;24(1-2):139–149. doi: 10.1093/protein/gzq079. [DOI] [PubMed] [Google Scholar]
- 16.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38(3):245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 17.Fersht AR, Sato S. Phi-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101(21):7976–7981. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: Evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254(2):260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 19.Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding α-helical transmembrane proteins. Proc Natl Acad Sci USA. 2014;111(30):11031–11036. doi: 10.1073/pnas.1410529111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truong HH, Kim BL, Schafer NP, Wolynes PG. Predictive energy landscapes for folding membrane protein assemblies. J Chem Phys. 2015;143(24):243101. doi: 10.1063/1.4929598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol. 2004;14(1):70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Bryngelson JD, Wolynes PG. Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci USA. 1987;84(21):7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Go N. Theoretical studies of protein folding. Annu Rev Biophys Bioeng. 1983;12:183–210. doi: 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- 24.Levy Y, Wolynes PG, Onuchic JN. Protein topology determines binding mechanism. Proc Natl Acad Sci USA. 2004;101(2):511–516. doi: 10.1073/pnas.2534828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Z, et al. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13(12):1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 26.Eastwood MP, Wolynes PG. Role of explicitly cooperative interactions in protein folding funnels: A simulation study. J Chem Phys. 2001;114(10):4702–4716. [Google Scholar]
- 27.Vinothkumar KR, et al. The structural basis for catalysis and substrate specificity of a rhomboid protease. EMBO J. 2010;29(22):3797–3809. doi: 10.1038/emboj.2010.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129(12):124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho SS, Levy Y, Wolynes PG. P versus Q: Structural reaction coordinates capture protein folding on smooth landscapes. Proc Natl Acad Sci USA. 2006;103(3):586–591. doi: 10.1073/pnas.0509768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du R, Pande VS, Grosberg AY, Tanaka T, Shakhnovich ES. On the t ransition coordinate for protein folding. J Chem Phys. 1998;108(1):334–350. [Google Scholar]
- 31.Zheng W, Best RB. Reduction of All-Atom Protein Folding Dynamics to One-Dimensional Diffusion. J Phys Chem B. 2015;119(49):15247–15255. doi: 10.1021/acs.jpcb.5b09741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eilers M, Patel AB, Liu W, Smith SO. Comparison of helix interactions in membrane and soluble alpha-bundle proteins. Biophys J. 2002;82(5):2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinothkumar KR, Freeman M. Intramembrane proteolysis by rhomboids: Catalytic mechanisms and regulatory principles. Curr Opin Struct Biol. 2013;23(6):851–858. doi: 10.1016/j.sbi.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444(7116):179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 35.Hong H, Blois TM, Cao Z, Bowie JU. Method to measure strong protein-protein interactions in lipid bilayers using a steric trap. Proc Natl Acad Sci USA. 2010;107(46):19802–19807. doi: 10.1073/pnas.1010348107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE. How fast-folding proteins fold. Science. 2011;334(6055):517–520. doi: 10.1126/science.1208351. [DOI] [PubMed] [Google Scholar]
- 37.Mayor U, Grossmann JG, Foster NW, Freund SMV, Fersht AR. The denatured state of Engrailed Homeodomain under denaturing and native conditions. J Mol Biol. 2003;333(5):977–991. doi: 10.1016/j.jmb.2003.08.062. [DOI] [PubMed] [Google Scholar]
- 38.Lindorff-Larsen K, et al. Determination of an ensemble of structures representing the denatured state of the bovine acyl-coenzyme a binding protein. J Am Chem Soc. 2004;126(10):3291–3299. doi: 10.1021/ja039250g. [DOI] [PubMed] [Google Scholar]
- 39.Davtyan A, et al. AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J Phys Chem B. 2012;116(29):8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plimpton S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J Comput Phys. 1995;117(1):1–19. [Google Scholar]
- 41.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL, Lam JW. PyMOL: A communications tool for computational models. Abstr Pap Am Chem S. 2005;230:U1371–U1372. [Google Scholar]
- 43.Tusnády GE, Dosztányi Z, Simon I. TMDET: Web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005;21(7):1276–1277. doi: 10.1093/bioinformatics/bti121. [DOI] [PubMed] [Google Scholar]
- 44.Krishnamani V, Hegde BG, Langen R, Lanyi JK. Secondary and tertiary structure of bacteriorhodopsin in the SDS denatured state. Biochemistry. 2012;51(6):1051–1060. doi: 10.1021/bi201769z. [DOI] [PubMed] [Google Scholar]