

Abstract
Transposable elements (TEs) are genomic repeated sequences that display complex evolutionary patterns. They are usually inherited vertically, but can occasionally be transmitted between sexually independent species, through so-called horizontal transposon transfers (HTTs). Recurrent HTTs are supposed to be essential in life cycle of TEs, which are otherwise destined for eventual decay. HTTs also impact the host genome evolution. However, the extent of HTTs in eukaryotes is largely unknown, due to the lack of efficient, statistically supported methods that can be applied to multiple species sequence data sets. Here, we developed a new automated method available as a R package “vhica” that discriminates whether a given TE family was vertically or horizontally transferred, and potentially infers donor and receptor species. The method is well suited for TE sequences extracted from complete genomes, and applicable to multiple TEs and species at the same time. We first validated our method using Drosophila TE families with well-known evolutionary histories, displaying both HTTs and vertical transmission. We then tested 26 different lineages of mariner elements recently characterized in 20 Drosophila genomes, and found HTTs in 24 of them. Furthermore, several independent HTT events could often be detected within the same mariner lineage. The VHICA (Vertical and Horizontal Inheritance Consistence Analysis) method thus appears as a valuable tool to analyze the evolutionary history of TEs across a large range of species.
Keywords: codon usage, synonymous substitutions, transposable elements, horizontal transfer, vertical transmission, Drosophila, mariner element
Introduction
Vertical transmission from ancestral to derived species is the primary way explaining the distribution of genetic divergence in phylogenies. In general, horizontal transfers of genetic material between reproductively isolated species are viewed as a rare phenomenon among eukaryotes. However, some specific DNA sequences, such as transposable elements (TEs), often exhibit a higher propensity of being horizontally transferred compared with the rest of the genome (Schaack et al. 2010; Wallau et al. 2012). Both Class I (retroelements) or Class II (DNA transposons) can multiply easily, using genome-free steps, which increases the chance of these “selfish DNA” sequences to effectively colonize new genomes, as compared with nonmobile genome components (Doolittle and Sapienza 1980; Orgel and Crick 1980; Le Rouzic and Capy 2005).
After a successful invasion in a new genome, TEs’ natural fate is inactivation, degradation and loss from the host genome as a consequence of the natural selection and/or genetic drift (Hua-Van et al. 2011; Petrov et al. 2011). Horizontal transposon transfers (HTTs) can be seen as a way to escape this natural process by allowing TE perpetuation through continuous invasion of new genomes (Lohe et al. 1995; Kidwell and Lisch 2001; Schaack et al. 2010; Wallau et al. 2012).
Horizontal transfers are considered as rare events, mainly because we can only detect the successful ones, and because it is difficult to obtain direct experimental evidence in the wild. Nevertheless, past transfers can be inferred from genome sequences and genetic data across populations. Since the discovery of the first HTT of a P element between Drosophila melanogaster and D. willistoni (Daniels et al. 1984), three types of indirect evidence have been proposed to support an HTT hypothesis: 1) Unexpectedly high nucleotide identity between TEs present in divergent species, 2) incongruences between gene and TE phylogenies, and 3) patchy distribution of TEs, when only some species of a monophyletic group of species have a given TE family whereas it is absent in other species (for further review, see Loreto et al. 2008). In the P element case, high identity (only one substitution between the P element of D. willistoni and D. melanogaster), patchy distribution, and its well-cut intrapopulation distribution (absence in old lab strains vs. presence in natural populations) were used as a strong support for the HTT inference (Daniels et al. 1990). Nevertheless, for other HTT cases, conclusive evidence is less straightforward, as, at least in theory, patchy distribution, high sequence identity or phylogenetic incongruences can also be compatible with vertical transmission, involving, for example, differential evolution rates between species, stochastic loss, or ancestral polymorphism (Capy et al. 1994; Cummings 1994; Wallau et al. 2012).
A widely used method to detect HTTs is based on the comparison between host genes and TEs, assuming neutral or nearly neutral changes in the DNA coding sequence among synonymous sites. Rates of neutral evolution in coding genes and TEs can be calculated as the rate of synonymous substitutions (dS) assuming that they accumulate at a constant rate. As a result, if the dS for a given TE between two species is equivalent to that observed for the genes, both kinds of sequences should have diverged for the same time, an expected pattern under vertical transmission. In contrast, if the TE displays a significantly lower dS compared with host genes, divergence times are not compatible, which is a signal of horizontal transmission. This method was originally described by Silva and Kidwell (2000), and used along with dN/dS, codon bias analyses and phylogenetic approach to identify numerous HTTs involving the P element.
Nowadays, the massive sequence data, accumulated thanks to recent sequencing technologies, offer greater investigation fields, but require more precise and sophisticated analysis. For instance, Bartolomé et al. (2009) used a genome-scale method comparing neutral substitution changes between TEs and host genes between the closely related species D. melanogaster, D. simulans, and D. yakuba. More recently, Modolo et al. (2014) proposed a genome-wide alignment method with little prior assumptions to assess for horizontal transfer of any DNA segment. Both methods are based on comparison of neutral divergence between two species, not considering phylogenetic information and other evolutionary estimators.
Silva and Kidwell (2000) previously pinpointed the fact that a careful analysis must be performed to distinguish between selection and HT to explain a lower TE dS. Indeed, synonymous substitutions are not totally neutral for a number of genes, as some genes experience a substantial purifying selection at the mRNA and translational level (Akashi 1994; Cannarozzi et al. 2010; Shah and Gilchrist 2010), which generates a Codon Usage Bias (CUB) all along the coding region. A simple method is to consider the CUB, characterized by a higher frequency of some codons in detriment of synonymous ones that may be different between distantly related species. However, the codon bias index (CBI) is itself quite variable among genes of the same species. Assuming that the codon bias partly reflects genome-wide constraints, the codon bias of a recently transferred TE is expected to be more similar to the donor species than the receptor host species (Lerat et al. 2002; Jia and Xue 2009). However, such an analysis is restricted to distantly related species, as CUB is likely to be phylogenetically correlated, and thus similar between closely related species (Vicario et al. 2007; Behura and Severson 2012).
Interestingly, these two methods rely on paradoxical assumptions: The dS method assumes that the synonymous sites evolve freely, whereas the codon bias method assumes that synonymous positions are constrained. With such a constant selection pressure over the coding sequences with high codon bias, the rate of synonymous substitutions is biased downwards, with the risk of underestimating the real distance between two species (Vidal et al. 2009). As a consequence, using genes with high codon bias and hence low dS as reference “host” genes can compromise the detection method, as the low “pseudosynonymous” divergence between these genes may hide horizontal transfers (Wallau et al. 2012). The most recent installments of the dS methodology (Silva and Kidwell 2000; Loreto et al. 2008; Ludwig et al. 2008) thus suggest that one should first perform a codon-bias analysis of host genes before selecting only those presenting a codon bias similar to TEs. However, arbitrarily discarding genes is not completely satisfactory as it leads to a loss of potentially informative data, and thus of statistical power.
In this work, we present a sophisticated and statistically supported method, hereafter called VHICA (Vertical and Horizontal Inheritance Consistence Analysis), to detect vertical and horizontal transposon transfer among related species, from good-quality genome (or transcriptome) sequences. VHICA was tested and validated on various well-known TE families present in the genome of different Drosophila species, previously tested using different methods, and reported to be horizontally transferred or not. We further applied it to 26 lineages of mariner DNA transposons recently characterized in the 20 Drosophila genomes (Wallau et al. 2014). Indeed, mariner elements present in Drosophila genomes appear as good horizontal transfer candidates, as most TE lineages present a patchy distribution (Robertson 1993; Brunet et al. 1994) and have been suspected in numerous interspecies HTT (Wallau, Hua-Van, et al. 2011; Dotto et al. 2015). The numerous examples presented here illustrate that our method increases the statistical power of horizontal transfer detection (and offer more ways to discriminate between HTT and vertical transmission of transposons [VTT]). The method also proposes original graphical representations of significant HTT signal, allowing the analysis of many species or many transposons at the same time, and making it easier to interpret the inferred transfer events in an evolutionary perspective.
New Approaches
Method Overview
The VHICA method is based on the discrepancies between the evolution rate of synonymous positions (dS) between TEs and a set of vertically transferred reference genes. VHICA is more powerful than traditional tests because it accounts for selection at synonymous sites, estimated by the effective number of codons (ENC), which results in a phenomenon known as CUB.
As dS and CUB are correlated, low dS is not necessarily a sign of horizontal transfer when associated with a high CUB, but the same dS associated with a low CUB is inconsistent with vertical transmission (fig. 1). Therefore, considering together dS and CUB leads to a substantial gain in statistical power.
Fig. 1.
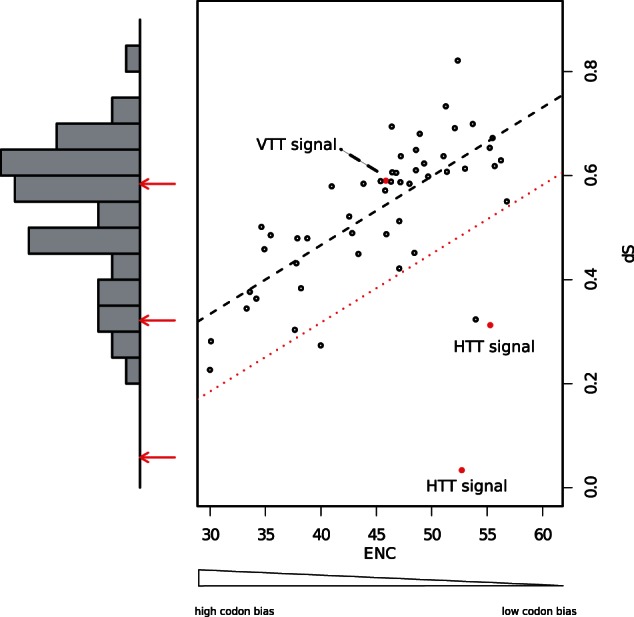
Comparison of a dS-only-based method (left bars chart) and the method proposed in this work (ENC–dS correlation graph in the right side of the figure). White circles represent the 50 host genes used as our control for vertically transmitted genetic information, red circles are the TE ENC–dS plotted against the vertically inherited host genes, the dotted black line represents the predicted distribution of the ENC–dS correlation between host genes derived from the observed data, and the dotted red line represents the variance of the observed measurements. If the TE ENC–dS red circle is plotted inside of the variance of the host data, then it is not significantly different from the host genes and it is considered vertically transmitted. On the contrary, if it is plotted far from the dotted red line it is significantly different from the host genes, hence it will be considered horizontally transferred between the two species. dS, number of synonymous substitutions per synonymous sites; ENC, Effective Number of Codons.
In VHICA, for each pair of species, the correlation between CUB and dS is accounted for by considering the residuals of a linear regression CUB = a dS + b among reference genes assumed to be vertically transmitted. TEs are then mapped on this reference CUB–dS relationship, and statistically significant deviation is interpreted as a signal of horizontal transfer. The corresponding P values (H0 = Vertical transfer, H1= horizontal transfer) can be calculated assuming a Gaussian distribution of residuals.
Evolutionary Interpretation Keys
Simplified theoretical outputs of VHICA, based on the concomitant analysis of four imaginary species are presented in figure 2 and illustrate four simple hypothetical TE evolutionary scenarios that may involve an HTT and a stochastic loss. Each off-diagonal small square represents the statistical signal of horizontal transfers (in practice, the P value is the probability of vertical transmission for a species pairwise comparison). The level of significance is encoded as indicated in the color bar, red squares corresponding to significant statistical signal. In VHICA, the deviation from the vertical transmission scenario is directly used to estimate the strength of statistical signal (P value); for simplicity, both were assimilated when interpreting the results.
Fig. 2.

Interpretation of theoretical patterns for the graphical matrix view with four fictive species (A–D). The green tree represents the host (species) tree and the overlapping black tree corresponds to the TE tree. TE loss is represented by an “X” over the species tree, and HTT event is indicated by a red arrow. The expected divergences between species are plotted on a scale below the tree, and appear in green for host genes (HG), and in black for TEs. The matrix of squares on the right represents all pairwise comparisons. Each square is colored according to the color bar of the P value calculated for the null hypothesis of vertical transmission. For simplicity, we assumed here that the statistical power was identical in all pairwise comparisons, so that the P value reflects directly the divergence difference between genes and TE. (A) TEs are vertically transmitted. (B) Recent HTT from species B to A associated with the loss of ancestral copies in species A. (C) Old horizontal transfer between the ancestor of species D and the ancestor of B and C. (D) Recent HTT from species A to B.
In the scenario depicted in figure 2A, the TE lineage was present in the ancestor of all studied species and was vertically transmitted to the four species. The present-day copies had the same time to diverge than host genes since their common ancestor, and the ENC–dS observed for TEs is not significantly different of the ENC–dS of host genes.
In figure 2B, a recent HTT event occurred from species B to A, with loss of the original A copies. Such a scenario can generate a significant P value between the recipient and the donor species. It is noteworthy that in this case, the species closely related to the donor will also present a (lower) HTT signal against the recipient strain.
Figure 2C illustrates the scenario of an HTT between the ancestor of species D and the ancestor of species B and C. A similar HTT signal is expected for species D versus B and D versus C as elements in species B and C had the same time to diverge since the HTT event.
Finally, in figure 2D, the HTT involves species A and B as in figure 2B, but the direction of the transfer is inverted. Again, the transferred element has replaced the ancestral one. Note that the expected pattern is different from the one in figure 2B. Indeed, in this case, no HTT signal is expected between A and species related to B. Therefore, it is possible to distinguish the direction of the transfer when close species exist in the data set: The horizontal transmission signal is expected to be present in the species close to the donor, and absent in the species close to the recipient.
These examples illustrate simple cases with few species, but empirical patterns can be less straightforward to interpret, in particular when several HTTs and more species are involved. Yet, even when evolutionary scenarios are more difficult to reconstruct, the analysis still provides reliable information about the occurrence and the nonoccurrence of HTTs between clades.
Underlying Assumptions
The VHICA method relies on a series of biological (B) and statistical (S) assumptions, which are listed here and discussed later. HTT inference is valid assuming that B1: Genes from the reference set are transmitted vertically, B2: Molecular evolution of genes and TEs follow the same process, B3: Horizontal transfer scenarios are parsimonious, and B4: The average CUB between pairs of species reflects the average evolutionary selection pressure. On the statistical side, we assume that S1: The relationship between dS and CUB is linear, S2: The residuals of this linear regression are Gaussian, and S3: The uncertainty on the dS versus CUB regression slope can be neglected.
Results and Discussion
The “Vhica” Package
The VHICA analysis is fully automated through the R package “vhica,” available in CRAN (https://cran.r-project.org/). The package uses the seqinr package for the dS calculation (Charif and Lobry 2007).
The analysis is performed in two steps. In the first step, the average CUB (measured by the ENC) and the dS are computed for each species pair and for each reference gene, and the linear regression between ENC and dS is calculated. In the second step, the average ENC and the dS for each TE and for each species pair are used to compute the P value of the departure from the null hypothesis (vertical transmission: TEs follow the gene pattern).
Different outputs can be asked for 1) a graphical matrix (similar to fig. 2) can be drawn for one particular element, P values being displayed according to a color bar. By default, a Bonferroni correction for multiple testing is applied, but alternative methods are available; 2) a graphical plot of the genes ENC–dS and the linear regression line can be drawn for one pair of species, with mapping of all the TE P values (without correction for multiple testing) available in the species comparison, similar to figure 1; and 3) synthetic tables can be generated that compile all the ENC or/and all the dS obtained for all genes and all TEs for all species, or all species pairwise comparison. They can be saved as flat files.
Sequence Data
Gene and TE sequences are processed altogether, and species or TEs need to be named consistently across the data set. The package input consists in a list of files (gene alignment files, and TE alignment files) in a FASTA format. Alignments must be in frame codon alignments, and one file per gene or TE must be provided. Gaps and missing data can be encoded by “N” or by “-.”
For genes, FASTA sequence names must start with the species name, optionally followed by a comment starting with an underscore character. Only one sequence by species must be present, and only the species name information is used. Each file should contain only one gene, the file name being used as the gene identifier.
For TEs, the species name is extracted according to the same rule as for genes. However, several TE copies by species are accepted, provided they are identified by a sublineage name encoded in the sequence name (character string that follows the last period character). Indeed, for a particular TE lineage, several sublineages are sometimes present in some species, and each sublineage could have experienced independent HTTs. We suggest the user to use several sequences only when sequences are distantly related. In this case, several comparisons are done between two species, and the P-value matrix is divided on smaller squares or rectangles accordingly. For closely related TE sequences, a single representative sequence should be selected for the analysis.
HTT Detection in Drosophila
The VHICA method was tested with the 20 sequenced genomes of the Drosophila genus. Fifty single-copy orthologous genes were extracted from the genomes (reference set), as well as various TEs, spanning different classes, including numerous mariner-like elements previously identified in these genomes (Wallau et al. 2014). More details in the Materials and Methods section.
Drosophila Phylogeny
The interpretation of HTT signals requires a solid species phylogenetic tree. All the nodes of the phylogenetic tree of the 20 Drosophila species built from the 50 orthologous genes obtained high statistical support (fig. 3A). The positions of two species D. eugracilis and D. ficusphila in our tree are not congruent with previous work (van der Linde and Houle 2008; Yang et al. 2012) (fig. 3B and C). However, the phylogenetic position of these two species inside of the melanogaster group was already considered as problematic by van der Linde and Houle (2008).
Fig. 3.

Phylogenetic reconstruction for the 20 Drosophila sequenced genomes. (A) Phylogeny built by a Bayesian analysis using the GTR+I+G nucleotide substitution model. (B) and (C) are the phylogenies from the literature. (D) The consensus tree used for HTT analysis.
As our tree is based on a larger amount of sequence data compared with previous phylogenetical analyses, we are confident that it likely reflects the true evolutionary history of these 20 species. However, as the aim of this study is to detect horizontal transfers, we took the precaution to use a consensus polytomic tree merging our phylogenetic hypothesis and the recent trees from the literature (fig. 3D).
Reanalysis of Data from the Literature
First, we explored the properties of the VHICA method by reanalyzing TE lineages that have already been studied. We had three goals: 1) To show that VHICA was able to detect well-documented horizontal transfers, ii) to prove that the method does not generate multiple false positives, and iii) to illustrate the improved statistical power of the VHICA regression by detecting new unpublished HTT events in TE families having no reported HTTs. For this reanalysis, we did not correct P values for multiple testing, in order to make sure to get the same statistical support than the existing literature.
The Confirmed HTT of P Element
The horizontal transfer of a P element between D. willistoni and D. melanogaster is the first documented HTT among eukaryotes (Daniels et al. 1984, 1990). This recent transfer took place around 50–60 years ago between those two species that diverged around 35–40 Ma. Since then various P-like elements have been detected, and classified into different types and groups (Clark and Kidwell 1997). Using the C type canonical P element from D. melanogaster, we retrieved P elements in eight other species (fig. 4A). The phylogenetic analysis of all sequences revealed that the retrieved P copies belonged to various types or groups (Loreto et al. 2012), and that different groups can coexist in the same species. For example four P element groups were found in D. willistoni (fig. 4B and C), and we analyzed representative sequences from each of these groups (also called sublineages). The resulting P-value matrix is shown in figure 4D. No HTT signal could be detected in most comparisons except for D. willistoni. We recovered a highly significant signal for the HTT to D. melanogaster (fig. 4D). This HTT (evidenced by the red rectangle) concerns only elements from the sublineage/group a, although a less significant signal could be also detected between D. melanogaster P element and D. willistoni sublineage b (pale red rectangle). Some other P sublineages of D. willistoni also exhibit HTT signals with various species (e.g., the i sublineage, in D. willistoni and D. bipectinata).
Fig. 4.

(A) Species tree phylogeny presenting the distribution of the P elements in Drosophila. Black branches indicate absence of the element. (B) P elements ML-tree following the species-specific color from part (A), and showing the different group/type of P elements (GTR+I+G). Bootstraps (1,000 replicates) are indicated below branches, and the subfamilies numbering above the branches (a:i). Stars denote representative copies used in the study. (C) Simplified phylogeny obtained with these elements. Names include the species names and the subfamily. (D) Consistency graphical representation for the P element. Each square represents one species comparison. When several elements are analyzed for one species (sublineages), the square is divided into rectangles, each one represents one sublineage. Sublineages are indicated on the sides. The horizontal transfer between Drosophila melanogaster and D. willistoni concerns elements of the subfamily a only, and is then visible as a small red rectangle. Other rectangles from this square correspond to comparisons between the D. melanogaster element (sublineage a) and the other elements (sublineages b,d,i) from D. willistoni. (E) ENC–dS graph between D. melanogaster and D. willistoni showing the P HTT transfer. Open circles represent the ENC–dS measures of the 50 single copy ortholog genes. The dotted black line represents the linear regression of ENC–dS from genes, and the dotted red line corresponds to the cutoff P value of 0.05. TE comparisons are figured as red triangles: P.a is the comparison of sequences from the a sublineage. Other comparisons are between the D. melanogaster P element from sublineage a with D. willistoni elements from sublineages b, d, or i (P.a.b, P.a.d, P.a.i).
The result found here is globally consistent with the existence of an ancestral polymorphism with few HTT for species outside the willistoni group. In this species however HTTs seem more frequent, which is in agreement with previous results that suggested a high rate of HTTs between the willistoni group, and the saltans group (not represented here), and may explain the coexistence of several sublineages (Silva and Kidwell 2000).
Other Elements
We chose to reanalyze a set of lineages previously reported by various methods to be horizontally and vertically transferred: four mariner-like and three Tc1-like elements for DNA transposons, three LINE (non-LTR [long terminal repeat]) elements and four LTR retroelements (table 1 and fig. 5).
Table 1.
Previously Reported Evolutionary History of TEs and Its Reanalysis with VHICA.
| Element | Type | Previous Results | Method | VHICA |
|---|---|---|---|---|
| P | DNA | HTT Dmel–Dwill1 | Sequence similarity | HTT Dmel–Dwill |
| Paris1 | DNA | No HTT2 | dS | HTT signal |
| Paris2 | DNA | 2 Suspected HTT 2 | dS | No HTTHTT signal |
| 17.6 | LTR | 5 Suspected HTT 3 | dS | No HTTHTT signal |
| BS | LINE | No HTT4 | Phylogeny | HTT signal |
| Helena | LINE | No HTT 5 | Phylogeny | HTT signal |
| I | LINE | No HTT6 | Genome-wide dS | HTT signal in new species |
| 1731 | LTR | No HTT6 | Genome-wide dS | HTT signal Dmel–Dsim |
| Chouto | LTR | No HTT6 | Genome-wide dS | HTT signal Dmel–Dsimand new species |
| micropia | DNA | HTT Dmel–Dsim6 | Genome-wide dS | HTT signal Dmel–Dsim |
| Bari1 | DNA | HTT Dmel–Dyak6 | Genome-wide dS | HTT signal Dmel–Dyakand new species |
| Dromar1 | DNA | HTT Dsim–Dyak6 | Genome-wide dS | HTT signal Dsim–Dyak |
| Dromar17 | DNA | HTT in Dmoj | Amplification dynamic | HTT signal Dfic–Dsim |
| Dromar8 | DNA | Suspected HTT7 | Amplification dynamic | HTT signal |
| Dromar6 | DNA | Suspected HTT7 | Amplification dynamic | HTT signal |
Sources: .—
6Bartolome et al. 2009
Fig. 5.

vhica analysis for elements already described in the literature for horizontal transfer and vertical transmission.
Among the three TE lineages (Paris2, Paris1, and 17.6) analyzed by dS comparison to few genes selected for codon bias similar to TEs (Vidal et al. 2009; Wallau, Lima, et al. 2011), HTTs have been suspected for two of them. The VHICA analysis resulted in significant HTTs for all lineages (fig. 5A–C). For Paris1, HTTs were observed between D. ananassae and D. biarmipes, and D. elegans, that were not detected before. For Paris2, three suspected HTTs involved the nonsequenced species D. buzzatii (Wallau, Lima, et al. 2011), but VHICA identified potential HTTs between D. mojavensis, a closely related species and D. persimilis and D. pseudoobscura. However, no signal was detected for the last suspected HTT involving D. rhopaloa. For 17.6, only two of the HTTs (Dmel × Dsim, Dmel × Dsec) among the three involving close species (Vidal et al. 2009) were detected. Both suspected HTTs involving more distantly related species (D. mojavensis, vs. D. melanogaster or D. virilis) were likely false-positives. In most of these cases, the discrepancies are likely due to the low number of reference gene sequences used in the previous analysis, compared with the 50 genes used here.
For the LINE elements, previously considered to be vertically transmitted based on phylogenetic congruence (BS, Helena; Granzotto et al. 2009, 2011), VHICA also gave HTT signal for some comparisons (fig. 5D and E). This result suggests that a mere phylogenetic analysis lacks sensitivity, and might have missed horizontal transfers.
Six elements, present at least in the melanogaster complex and previously analyzed using a genome-wide dS method using more than 10,000 nuclear genes (Bartolomé et al. 2009), were also reanalyzed (fig. 5F–K). For the I element, VHICA confirmed the absence of HTT among species of the melanogaster complex. Supported HTTs were detected only among distant species not analyzed previously (ananassae subgroup × D. eugracilis). For 1731 and Chouto, we detected significant HTTs not considered as significant enough in the previous analysis. The HTTs identified for Dromar1 (mariner), Bari1 and micropia could be confirmed, between D. yakuba or D. melanogaster, and the sister species D. simulans and D. sechellia (this last species was not included in the published analysis). VHICA then appears as much sensitive than this genome-wide method that used more than 10,000 genes but did not include the ENC.
Finally, three mariner-like elements, Dromar17, Dromar8 and Dromar6, have been recently suspected to have underwent HTT, based on the date of amplification burst in the different species, as described in Wallau et al. (2014), although no dS analysis had been performed at this time (table 1). For Dromar17 in D. mojavensis, characterized by a very recent amplification burst, no HTT signal was detected with the copies in D. elegans and D. ficusphila, but we observed an HTT between the two relic sequences present in these last species. This indicated that the putative donor species for D. mojavensis (if HTT) is not among the 20 sequenced species (fig. 5L).
Finally, HTT could be confirmed for Dromar8 and Dromar6, and these examples are described in more details in the next paragraphs, in which we tried to propose evolutionary scenarios (fig. 6).
Fig. 6.

Detailed analysis for two mariner lineages: Dromar8 (A–E) and Dromar6 (F–H). (A) and (F) Distribution of Dromar8 and Dromar6 in Drosophila, with indications of copy number and lineages with potentially active copies (data from Wallau et al. 2014). (B) and (G) TE phylogenies of Dromar8 and Dromar6 with stars denoting the representative copies (Evolutionary models were HKY+I+G and TPM3uf+G, respectively). (D and H) HTT matrices generated by vhica. (E) ENC–dS plot obtained from the comparison between the two species in which Dromar8 has amplified. (C) Amplification dynamics analysis of Dromar8 in the two species with large copy numbers (see Le Rouzic et al. 2013 for a full description of the method).
Dromar8.
The Dromar8 lineage has a patchy distribution (fig. 6A). It is present in moderate copy number in two species belonging to different subgenera (D. ficusphila and D. grimshawi) in which it is potentially functional (Wallau et al. 2014). The element could also be detected in D. erecta and D. ananassae as few relic inactive sequences (fig. 6A). A very high sequence identity was observed between sequences from D. ficusphila and D. grimshawi, as well as a phylogenetic incongruence (D. grimshawi sequences form a sister clade to D. ficusphila; fig. 6B). Combined to the very recent amplification burst in D. grimshawi, detected by the method described in Le Rouzic et al. (2013) (fig. 6C), these observations strongly suggested a recent HTT toward D. grimshawi. The VHICA analysis revealed significant HTT signal for all comparisons involving D. grimshawi sequences (fig. 6D), reflected by the extremely low dS observed relative to gene dS (fig. 6E).
These various arguments strongly support an HTT event from D. ficusphila or a closely related species, to the genome of D. grimshawi, plus one or two other HTT between species of the Sophophora subgenus.
Dromar6.
The Dromar6 case is a little bit more puzzling. This lineage is also featured by a patchy distribution, as it is present in 7 out of the 14 species of the melanogaster group. Potential autonomous elements were found in four species (D. yakuba, D. erecta, D. ananassae, and D. bipectinata), and relic inactive copies in the three others (fig. 6F). This lineage corresponds to the mariner-like element previously described in D. erecta by Lohe et al. (1995), who suspected horizontal transmission from the cat flea to the ancestor of the melanogaster subgroup, and a subsequent HTT between D. erecta and D. ananasse. Several observations could be made from the amplification dynamics in the five species carrying more than ten copies, previously illustrated in Wallau et al. (2014). Recent bursts of transposition have been observed in the closely related species D. ananassae and D. bipectinata, but they are not concomitant. In D. ananassae, the element seems to have also transposed very anciently. Finally, bursts of transposition have occurred at different times in the other species (see Wallau et al. 2014). The phylogenetic analysis of copies reported in figure 6G clearly distinguished one supported clade with old copies from D. ananassae and the copies from D. rhopaola. Old copies of D. elegans and D. ficusphila, and more recent potentially active lineages from D. erecta, D, yakuba, D. ananassae, and D. bipectinata were grouped in another clade but without bootstrap support. Nevertheless, several incongruences are visible when compared with the species tree (fig. 6A).
The VHICA analysis performed on some representative sequences revealed very strong significant HTT signals between the ananassae subgroup and the other subgroups, and between D. rhopaloa and the melanogaster subgroup (fig. 6H). Comparisons involving elements of the clade “a” with long terminal branches (dfic × dele, dele × drho) or elements from different clades in close species (dana b × dbip a) clearly gave no HTT signals. All the other comparisons gave intermediate HTT signals (pink squares).
The global pattern for Dromar6 can be interpreted with at least two independent well-supported horizontal transfer events, if considering only strong HTT signals, and at least five if intermediate signals are considered. Assuming that newly transferred active TEs immediately start to amplify in the new genome, this multiple HTT hypothesis is compatible with the amplification timing previously observed for this element in these different species (Wallau et al. 2014).
Overall, using the same significance threshold than the literature (P = 0.05 without correction for multiple testing), VHICA confirmed most HTTs, rejected some doubtful ones, and detected new candidates. These analyses highlight three properties of the VHICA method: 1) The use of few genes, even selected for comparable codon bias with TE codon bias is not resolutive enough and can lead to false-positives as well as false-negatives. Analyzing 50 genes for both dS and ENC improves both the specificity and the sensitivity; 2) phylogenetic congruence is not enough to discard horizontal transfer hypotheses; and 3) a set of 50 genes, when combined to ENC is enough to detect HTTs, even in relatively close species, with at least the same power than methods that uses the whole gene data set.
Note that several HTT signals faded after Bonferroni correction for multiple testing, especially for widely spread lineages, whereas some other remained highly significant. Hence, some documented cases are not grounded on strong statistical support (data not shown). Most of them corresponded to HTTs that are mildly supported (pink squares). In any case, as our method allows lots of comparisons at the same time, it is advisable to use some correction for multiple testing. Although it may apparently decrease the sensitivity of the method (while potentially increase its specificity), it prevents the accumulation of false positives expected in large-scale studies, an issue sometimes poorly considered.
The Mariner Data Set
The mariner element is known to be involved in numerous horizontal transfers (see Dotto et al. 2015), sometimes on a wide phylogenetic scale trans-order, or even trans-phylum. We have recently characterized 36 different mariner-like elements in the 20 Drosophila genomes (Wallau et al. 2014). Twenty-six lineages were present in more than one species, sometimes distantly related, and they displayed either restricted, patchy or widespread distribution, or phylogenetic discrepancies. In several cases, the amplification age of the lineage was different across species, suggesting that the element appeared in the different genomes at different time, which is the expected pattern in case of horizontal transfers. These suspected HTTs were confirmed in the two previous examples (Dromar6 and Dromar8). However, amplification dynamic is only available for elements that have sufficiently amplified into the genomes and we systematically ran VHICA on all other lineages shared by a least two species (table 2).
Table 2.
HTT Evidence Found for All 20 Mariner Lineages That Presented HTT Signal in the VHICA Test.
| Lineages Names | Species Number | Number of Codons Analyzed Min–Max | VHICA | Incongruences | Patchy Distribution |
|---|---|---|---|---|---|
| Dromar3 | 2 | 345 | − | − | − |
| Dromar19 | 2 | 344 | − | − | − |
| Dromar7 | 2 | 77 | + | − | − |
| Dromar36 | 2 | 131–133 | + | − | + |
| Dromar29 | 2 | 333 | + | − | + |
| Dromar25 | 2 | 218 | + | − | + |
| Dromar33 | 2 | 318 | + | − | + |
| Dromar23 | 2 | 231 | + | − | + |
| Dromar12 | 3 | 108–249 | + | + | + |
| Dromar1 | 3 | 320–344 | + | − | − |
| Dromar9 | 3 | 154–164 | + | − | − |
| Dromar26 | 3 | 345 | + | − | + |
| Dromar17 | 3 | 184–189 | + | − | + |
| Dromar34 | 4 | 198–306 | + | + | + |
| Dromar8 | 4 | 45–331 | + | + | + |
| Dromar30 | 4 | 48–132 | + | + | + |
| Dromar24 | 4 | 163–258 | + | + | + |
| Dromar21 | 5 | 336–349 | + | + | + |
| Dromar22 | 5 | 187–213 | + | + | + |
| Dromar13 | 6 | 277–331 | + | + | + |
| Dromar6 | 7 | 197–356 | + | + | + |
| Dromar16 | 7 | 95–348 | + | − | + |
| Dromar2 | 7 | 41–301 | + | + | + |
| Dromar11 | 7 | 33–340 | + | + | + |
| Dromar5 | 9 | 54–329 | + | + | − |
| Dromar4 | 10 | 90–338 | + | + | + |
| Dromar10 | 11 | 91–335 | + | + | − |
Eight lineages are shared by two species only (table 2). The two cases in which no HTT could be detected involved sister or closely related species. Dromar3, shared by the sister species D. persimilis and D. pseudoobscura, amplified and diverged prior the divergence of the two species, as expected from the existence of several orthologous insertion sites detected by Wallau et al. (2014). Dromar19, present in two species of the melanogaster subgroup, also appeared to have been vertically transmitted. All the other lineages exhibited HTT signals, even between closely related species. However, the absence of the element in other species makes it difficult to draw evolutionary scenarios concerning the direction or the timing of the transfer.
For the remaining 15 other lineages, present in three or four species, or exhibiting a wider distribution which may be compatible with an ancestral presence, HTT signals were consistently detected in numerous pairwise comparisons, especially between species belonging to different groups. Hence, the pattern of multiple HTTs detected for Dromar8 and Dromar6 is also largely observed for all other mariner lineages. In most cases, such a saturated pattern limits the possibility of evolutionary inference. This pattern of very frequent HTTs is unlikely to be due to methodological issues, as it was not observed for other TEs analyzed, including other DNA transposons. Furthermore, the same conclusion was drawn when using the conservative Bonferroni correction for multiple tests: Most mariner lineages still exhibit fully supported HTT during their evolution within the Drosophila genus (fig. 7). Hence, it is clear that this TE family is especially prone to HTT.
Fig. 7.

ENC–dS plot obtained for closely related-species (Drosophila ananassae × D. bipectinata, and D. simulans × D. erecta) and more distantly related species (D. ficusphila × D. bipectinata, D. melanogaster × D. mojavensis). White circles represent host genes and red triangles are TEs. The dotted black line is the linear regression, and the dotted red lines the cutoff P value of 0.05.
Four pairwise comparisons, shown in figure 7, illustrate the apparent propensity of mariner element to undergo horizontal transfer. The first one is the comparison of D. ananassae and D. bipectinata, two species of the ananassae subgroup, that share 15 of all the elements analyzed here, including 13 mariner lineages. Half of these elements, including five mariner lineages, present codon bias and dS similar to genes, the ten others, all mariner elements, exhibiting a significant departure from the ENC/dS correlation. In the D. simulans/D. erecta comparison, two species from the melanogaster subgroup devoid of mariner elements, the shared elements are within the gene cloud, or sometimes separated by larger phylogenetic distance than genes, which is expected when TE lineages have diverged prior to the species. In more distantly related species such as D. ficusphila and D. bipectinata that share 12 elements including 11 mariner, all mariner exhibited strong HTT signals, whereas P was vertically transmitted. Finally, in the comparison involving very distantly related species belonging to different subgenera, such as D. melanogaster and D. mojavensis, the elements 17.6, Helena, and BS displayed the same evolutionary pattern than genes. In comparison, the obvious HTT of Dromar8 has occurred between D. grimshawi and D. ficusphila, which also belong to different subgenera (fig. 6D).
It is widely acknowledged that TEs are generally prone to undergo frequent HT events, but different TE families have different chances of being horizontally transferred depending on the stability of the intermediate state during the transposition process (Silva et al. 2004; Schaack et al. 2010). Essentially, DNA transposons and LTR retrotransposons perform HTTs more frequently than non-LTR retrotransposons, which transposition involves no free step (Loreto et al. 2008). This tendency was observed in three Drosophila genome comparisons, by Bartolomé et al. (2009). However, only five DNA transposons could be analyzed. Here, we could confirm that DNA elements such as the mariner family are very prone to perform recurrent and successful horizontal transfers, in support to the conclusions of previous works (Robertson and Lampe 1995; Wallau, Lima, et al. 2011; Dupeyron et al. 2014; Gilbert et al. 2014)
Robustness of the VHICA Method
The VHICA method is based on a number of biological and statistical assumptions, listed in the “New Approaches” section. Violation of these assumptions might lead to biased or misleading results, and we ran additional tests to assess their potential impact on the HTT inference.
Most of the biological assumptions are realistic and are not likely to raise substantial issues. Assumption B1 (vertical transmission of reference genes) is classical in HT detection methods, and does not imply that all genes are vertically transferred, as only the set of reference genes (well-conserved, single copy, orthologous genes) has to match this assumption. If B1 were violated, the only consequence is the loss of power, as HTTs would be more difficult to detect (inflated residual variance). Assumption B2 implies that the divergence/CUB relationship is identical for TEs and genes. Although it is likely that TE sequence is not subject to the same evolutionary pressures as regular genes, it is reasonable to assume that the correlation between synonymous divergence and codon bias is conserved among all coding sequences. Assumption B3 (parsimony) is classical in phylogenetic models, and consists in choosing the evolutionary scenario with the minimum number of horizontal transfers. The underlying hypothesis is that horizontal transmission is much less frequent than vertical transmission, which seems to be verified for most organisms.
Assumption B4 (evolutionary meaning of divergent CUB between species) deserves deeper investigation. The VHICA method does not assume that CUB is identical between species (which is known to be incorrect), but rather than taking the average CUB between pairs of species measures the average evolutionary pressure during sequence evolution. CUB is evolutionary correlated (supplementary material S3.1, Supplementary Material online), supporting the hypothesis that CUB does not fluctuate rapidly during evolution. We compared the ENC for the 50 genes for all 190 pairwise species comparisons, and obtained a positive correlation for all of them (average correlation: r = 0.64, SD = 0.16). An analysis of variance attributes 56% in ENC variation to the genes, and only 15% to the species. This is in agreement with previous work in the Drosophila genus showing that the CUB is conserved among all Drosophila genomes (Vicario et al. 2007; Behura and Severson 2012). We also performed the same analysis with another CUB measurement, the CBI (Morton 1993), and we obtained very similar results (data not shown). Additional tests show that using alternative ways to measure the CUB (using the CUB of a single species, or alternatively the average CUB of all species) does not drastically change the interpretation, as the resulting P values appear to be very similar (supplementary figs. S3.2 and S3.3, Supplementary Material online). Overall, it seems that VHICA is robust to assumption B4.
The statistical assumptions of the model can also be tested empirically. The first assumption S1, stating that the relationship between dS and ENC is linear, is unproblematic, as 1) a linear regression catches the trend even if the real pattern is not linear, and 2) in case the relationship is clearly nonlinear, it is possible to slightly change the VHICA method and compute the residuals from a nonlinear regression instead. Assumption S2 deals with the Gaussian distribution of residuals, from which P values are calculated. Further investigation showed that residuals from our data set were almost Gaussian, meaning that estimated P values are properly estimated (supplementary material S4, Supplementary Material online). Finally, assumption S3 states that the number of reference genes and the sequence length are large enough to neglect uncertainties on the regression parameters. Supplementary material S5, Supplementary Material online, shows that a sample of 50 genes is likely to be large enough to estimate P values in a robust way, as 1) for most TE sequences, the conclusion (vertical vs. horizontal transfer) is unchanged when resampling the 50 reference genes; and 2) the method accuracy is not improved above 30 genes, suggesting that the stochasticity associated with the gene sample becomes negligible above this limit.
The Choice of the TE Representative Sequence
Although it is theoretically possible to include in the analysis all copies of a TE family within one species, it is much simpler and quicker to use only few representatives. Ideally, one sequence is enough if all the sequences are closely related. In case where divergent groups of sequence coexist (sublineages), one representative of each sublineage should be chosen. However, how to choose the best representative sequence? Obviously the most complete sequences are best suited, and short sequences or sequences with doubtful homologous regions should be discarded. For most TEs, we applied this simple rule and the representative sequence was chosen by eye after a comparison with the consensus sequence (assumed to be complete). However, this artisanal method may bias the choice toward slowly evolved sequence. We tested this risk on some TE families, by running the analysis either with arbitrarily selected sequences, or with the sequences that present an ENC, of a dS value close to the average calculated from all sequences within the species. We also tested the impact of using randomly chosen sequence. Finally, the effect of the sequence length on the ENC and dS value was also evaluated. The results are detailed in supplementary material S6, Supplementary Material online, and show that there is no significant difference between the P-value matrices obtained with arbitrary sequences or with sequences selected for their average dS or ENC, indicating that the arbitrarily choice is suitable. Consistent conclusions were systematically obtained when representative sequences were randomly chosen: P values were always significant for strong HTT signals, always nonsignificant for vertically transmitted sequences, and remained always close to the cutoff (then doubtful) for the doubtful cases. Finally, we could show that the length of the representative sequence does not impact the dS, but can highly bias the ENC when too short. The consequence of using too short sequences (<100 codons) is a decrease in the sensitivity of VHICA (it tends to generate more false negatives), as lower ENC (highly biased) are expected, whereas dS will stay stable.
Inherent Methodological Limitations
Phylogenetic Distance between Species and Taxonomic Range of Application
Obviously, the power of the VHICA method is limited by the information contained in the data. In one hand, as many other sequence-based method, VHICA is not suited to deal with very closely related species such as the pairs D. simulans, and D. sechellia, or D. pseudoobscura and D. persimilis (estimated to have diverged around 500,000 years ago), in which a TE dS of 0 is still nonsignificant due to the lack of divergence among the host genes. Nevertheless, at this phylogenetic scale, horizontal transfers can often be attributed to hybridization and introgression, and not characterized by the transfer of genetic material without sexual reproduction. This issue is not specific to TEs, as similar problems arise with any genetic exchange between not completely isolated species. However, we also noticed that VHICA is at least as sensitive as the method of Bartolomé et al. (2009) for comparison between species as close as D. melanogaster and D. simulans or D. sechellia.
In the other hand, the VHICA method is not designed for very distantly related comparisons. Obviously, genes that are too divergent cannot be aligned with confidence, or have suffered so many changes that the dS is very high, which affect the precision of the linear regression. In such situations, the power of detection of VHICA is likely impaired toward being less sensitive (more false negatives). In the extreme case where dS and ENC would not correlate (because even highly constrained genes have a high dS), the VHICA method becomes equivalent to dS-based methods.
With our Drosophila benchmark, we probably reached both limits with very closely related species, and very distantly related species, separated by about 47 My. However, taxonomic level is arbitrary, and the genus Drosophila is known to be very large. Indeed, the high divergence observed, along with paraphyletic status of some groups have raised a polemic about the splitting of the genus in several genera (Van der Linde et al. 2007). So we are confident that for other groups, comparison between genera can be performed accurately with VHICA.
Detection of Old Transfers
Detecting horizontal transfer signal for very old events can also be challenging, as substitutions accumulated during vertical transmission tend to erase progressively the HTT signal, and will eventually saturate the phylogenetic information. Fading signals are also expected when the assumption that TE evolves at a similar constant rate in the different species is not fulfilled. Indeed, rapidly evolving sequences in one species will increase the dS value. Again in this case, we expect a decrease of the sensitivity, but the specificity should not be affected. More important than the age of the transfer is in fact the time spent between the speciation event and the HTT. Hence, an old transfer will still be detectable if it has occurred between species having diverged for a significantly longer time.
The Necessity for Coding Sequences
The dS and codon bias measurements rely on the assumption that the sequence is coding, and that the inferred protein sequence is meaningful, both for TEs and reference genes. It is thus important to select carefully a single good-quality copy of reasonable length per genome, excluding untranslated region (UTR), promotor, intronic, and terminal UTRs of the element. Noncoding TE families, such as SINES or some MITEs, that lack any remnants of coding sequences cannot be analyzed in this framework. However, as soon as enough coding sequences are present even if nonfunctional or pseudogenized, the VHICA method can be used. Indeed, we were able to distinguish HTT or vertical signal with highly degenerated relic copies, suggesting that our method is powerful enough. Tests carried out on the length of the sequence revealed that the dS is poorly affected by the number of codons available for the comparison, unlike the ENC.
Conclusion
Sequence similarity, tree incongruence, or patchy distribution are the main evidence used for detecting HTTs. Divergence analysis offers a less arbitrary and more sensitive method than sequence similarity, in particular for old events. As illustrated by our results, VHICA, which further takes into account the CUB, is able to detect HTTs independently from phylogenetic incongruence or patchy distribution. The VHICA methodology overcomes existing methods described in the literature, by associating a higher (but reasonable) number of host genes with high statistical robustness. In addition, VHICA does not need fully assembled genomes; in particular, it does not rely on synteny among close species, and can thus be used over a wide phylogenetic range. Overall, given its modest complexity (the method only relies on ordinary regressions), and its good statistical power, VHICA appears as a natural candidate for becoming a standard procedure in the detection of horizontal transfers, when enough gene data and numerous species are available.
Materials and Methods
Drosophila Genomes
We classified the 20 Drosophila genomes into two groups, based on the chronology of their publication: The “first 12 Drosophila genomes” being D. melanogaster, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virilis and D. grimshawi (Drosophila 12 Genomes Consortium 2007), and the “eight new genomes” being D. ficusphila, D. eugracilis, D. biarmipes, D. takahashii, D. elegans, D. rhopaloa, D. kikkawai and D. bipectinata (Chen et al. 2014). A four-letter abbreviation of the names of species was used in the figures and table 1, as for example Dmel for D. melanogaster.
Host Genes Data Set and Host Phylogeny Reconstruction
Based on the data set of orthologous genes obtained from the first 12 Drosophila genomes (Drosophila 12 Genomes Consortium 2007), we arbitrarily picked 150 orthologous genes present in all genomes and used them as query in a BLASTN search against the eight new Drosophila genomes (Chen et al. 2014). We kept only single copy genes that were present in all 20 Drosophila genomes, ending with two data set of 50 single orthologous genes, the list of used genes is available in supplementary table S1, Supplementary Material online.
These gene sequences have been concatenated and used to build a phylogenetic tree of the host species using two independent methods: A maximum-likelihood analysis using the PhyML software (Guindon et al. 2010), and a Bayesian analysis with MrBayes software (Ronquist et al. 2012). These analyses were performed with the nucleotide substitution model GTR+I+G supported by jModeltest (Darriba et al. 2012). The branch support was evaluated with 1,000 bootstraps in the likelihood analysis, and by sampling the most probable tree every 100 steps along 1,000,000 generations for the Bayesian analysis. Both trees displayed exactly the same branching pattern (data not shown). However, this topology was slightly different from previously published Drosophila phylogenies (fig. 3) as described in the Results section.
Transposable Elements
We analyzed TE families of different types: DNA transposons (P, Paris, Bari1, and mariner elements), non-LTR retrotransposons (BS and Helena, and I), and LTR retrotransposons (17.6, Chouto, 1731, and micropia). For mariner and Paris, we used previously published sequence data sets obtained from the analysis of the 20 Drosophila genomes (Wallau et al. 2012, 2014). A lineage here is defined as a functional clade of TE copies, that is, a group of elements that form a monophyletic clade in which any mobile copy is likely to be cross-mobilized by the active transposase of another copy of the clade. Three other elements BS, 17.6, and Helena were previously characterized in the first 12 Drosophila genomes only (Granzotto et al. 2009, 2011; Vidal et al. 2009). A complementary search in the eight new genomes was thus performed for these elements. For the remaining elements (P, I, 1731, micropia, Bari1, and Chouto), for which we had no exhaustive data, we ran the analysis in all 20 genomes using BLASTN, and using as query the consensus sequence provided by RepBase19.01 (Jurka et al. 2005). Note that the P element being absent from the sequenced D. melanogaster genome, we introduced artificially the canonical P element in our data set. For BS and I, we also recovered elements with their flanking sequences to check for orthology. Few orthologous insertions were detected only between the closest species D. persimilis and D. pseudoobscura. For Dromar elements, orthologous insertions had been previously detected only for these two species (Dromar3).
For all TEs, sequence alignment was performed with MAFFT (Katoh and Standley 2013), checked and corrected by hand if needed. Phylogenetic analysis was run with PhyML, with confidence estimated with 500 bootstraps (Guindon et al. 2010). For each TE lineage, the best substitution model suggested by the Bayesian Information Criterion (BIC) in jModeltest (Darriba et al. 2012) was used. Whenever possible we rooted the trees with the closest lineage, and we used midpoint roots only when we found no related sequence that could be aligned at the nucleotide level. These phylogenetic trees included all copies of the lineage and were used for 1) the detection of incongruences when compared with the Drosophila phylogeny, 2) the detection of potential ancestral polymorphism leading to the existence of two or more sublineages, and 3) the choice of the representative sequences used in the following dS–ENC analysis. The longest sequence containing the less deletions, insertions, and nonhomologous sequences relative to the coding consensus was usually selected. When the lineage was present as several sublineages, one sequence from each clade was used for the dS estimate.
Synonymous Substitutions Distance and CUB
The coding regions of these representative sequences were aligned using MACSE (Ranwez et al. 2011). All alignments were further adjusted for in frame codon alignment, using a homemade python script. Codon alignments were finally manually checked using AliView (Larsson 2014).
The dS was calculated using the method of Li (1993) implemented in the seqinR package (Charif and Lobry 2007), with a pairwise deletion option. The CUB was estimated for host genes and TEs as the ENC according to Wright’s method (Wright 1990). The ENC varies from 20 up to 61, ENC = 20 meaning that only one synonymous codon is used for each amino acid high CUB and ENC = 61 means that all synonymous codons are equally used no CUB.
P-Value Calculation
A linear regression ENC = a dS + b is performed for each pair of species s1 and s2, where ENC stands for the average CUB for the gene among both species (fig. 1). The residual variance of this regression is noted VarR. The residual deviation for a transposable element j could then be calculated as r*,j = ENC,j − a dS,j − b. Under the null hypothesis H0: “TE j was vertically transferred,” r*j follows a Gaussian distribution (for the real Gaussian distribution obtained by our genes, see supplementary fig. S4, Supplementary Material online) of mean 0 and of variance VarRs1,s2, and the one-tailed P value is Φ −|r*j| VarR−1/2, Φ being the normal cumulative distribution function. When analyzing new TE lineages in several species, the P values were corrected for multiple testing (by default, we used the Bonferroni correction, known to be conservative).
Supplementary Material
Supplementary materials S1–S6, figure S1, and table S1 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). The first author of this manuscript was a “Bolsista da CAPES—Proc. No. 6145116.” The authors thank the anonymous reviewers for their fruitful comments on the first version of the manuscript.
References
- Akashi H. 1994. Synonymous codon usage in Drosophila melanogaster: Natural selection and translational accuracy. Genetics 136:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomé C, Bello X, Maside X. 2009. Widespread evidence for horizontal transfer of transposable elements across Drosophila genomes. Genome Biol. 10:R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura SK, Severson DW. 2012. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS One 7:e43111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet F, Godin F, David J, Capy P. 1994. The mariner transposable element in the Drosophilidae family. Heredity 73:377–385. [DOI] [PubMed] [Google Scholar]
- Cannarozzi G, Cannarrozzi G, Schraudolph NN, Faty M, von Rohr P, Friberg MT, et al. 2010. A role for codon order in translation dynamics. Cell 141:355–367. [DOI] [PubMed] [Google Scholar]
- Capy P, Anxolabéhère D, Langin T. 1994. The strange phylogenies of transposable elements are horizontal transfers the only explanation? Trends Genet.. 10:7–12. [DOI] [PubMed] [Google Scholar]
- Charif D, Lobry JR. 2007. SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. Structural approaches to sequence evolution: Molecules, networks, populations. New York: Springer; p 207–232. [Google Scholar]
- Chen Z-X, Sturgill D, Qu J, Jiang H, Park S, Boley N, Suzuki AM, Fletcher AR, Plachetzki DC, FitzGerald PC, et al. 2014. Comparative validation of the D melanogaster modENCODE transcriptome annotation. Genome Res. 24:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JB, Kidwell MG. 1997. A phylogenetic perspective on P transposable element evolution in Drosophila. Proc Natl Acad Sci U S A. 94:11428–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings M. 1994. Transmission patterns of eukaryotic transposable elements: arguments for and against horizontal transfer. Trends Ecol Evol. 9:141–145. [DOI] [PubMed] [Google Scholar]
- Daniels SB, Peterson KR, Strausbaugh LD, Kidwell MG, Chovnick A. 1990. Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics 124:339–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels SB, Strausbaugh LD, Ehrman L, Armstrong R. 1984. Sequences homologous to P elements occur in Drosophila paulistorum. Proc Natl Acad Sci U S A. 81:6794–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2 more models new heuristics and parallel computing. Nat Methods. 9:772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle WF, Sapienza C. 1980. Selfish genes, the phenotype paradigm and genome evolution. Nature 284:601–603. [DOI] [PubMed] [Google Scholar]
- Dotto B, Carvalho EL, Silva AF, Silva LFD, Pinto PM, Ortiz MF, Wallau GL. 2015. HTT-DB—horizontally transferred transposable elements database. Bioinformatics 31(17):2915–2917. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. [DOI] [PubMed] [Google Scholar]
- Dupeyron M, Leclercq S, Cerveau N, Bouchon D, Gilbert C. 2014. Horizontal transfer of transposons between and within crustaceans and insects. Mob DNA. 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Chateigner A, Ernenwein L, Barbe V, Bézier A, Herniou EA, et al. 2014. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat Commun. 5:3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzotto A, Lopes FR, Lerat E, Vieira C, Carareto CMA. 2009. The evolutionary dynamics of the Helena retrotransposon revealed by sequenced Drosophila genomes. BMC Evol Biol. 9:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzotto A, Lopes FR, Vieira C, Carareto CMA. 2011. Vertical inheritance and bursts of transposition have shaped the evolution of the BS non-LTR retrotransposon in Drosophila. Mol Genet Genomics. 286:57–66. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 30. Syst Biol. 59:307–321. [DOI] [PubMed] [Google Scholar]
- Hua-Van A, Le Rouzic A, Boutin TS, Filee J, Capy P. 2011. The struggle for life of the genome’s selfish architects. Biol Direct. 6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Xue Q. 2009. Codon usage biases of transposable elements and host nuclear genes in Arabidopsis thaliana and Oryza sativa. Genomics Proteomics Bioinformatics 7:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110:462–467. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:782-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MGMG, Lisch DRDR. 2001. Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution 55:1–24. [DOI] [PubMed] [Google Scholar]
- Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 30:3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E, Capy P, Biémont C. 2002. Codon usage by transposable elements and their host genes in five species. J Mol Evol. 54:625–637. [DOI] [PubMed] [Google Scholar]
- Le Rouzic A, Capy P. 2005. The first steps of transposable elements invasion: parasitic strategy vs genetic drift. Genetics 169:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rouzic A, Payen T, Hua-Van A. 2013. Reconstructing the evolutionary history of transposable elements. Genome Biol Evol. 5:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH. 1993. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 36:96–99. [DOI] [PubMed] [Google Scholar]
- Lohe A, Moriyama E, Lidholm D, Hartl D. 1995. Horizontal transmission vertical inactivation and stochastic loss of Mariner-like transposable elements. Mol Biol Evol. 12:62–72. [DOI] [PubMed] [Google Scholar]
- Loreto ELS, Carareto CMA, Capy P. 2008. Revisiting horizontal transfer of transposable elements in Drosophila. Heredity 100:545–554. [DOI] [PubMed] [Google Scholar]
- Loreto ELS, Zambra FMB, Ortiz MF, Robe LJ. 2012. New Drosophila P-like elements and reclassification of Drosophila Pelements subfamilies. Mol Genet Genomics. 287:531–540. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Valente VL, Loreto EL. 2008. Multiple invasions of Errantivirus in the genus Drosophila. Insect Mol Biol. 17:113–124. [DOI] [PubMed] [Google Scholar]
- Modolo L, Picard F, Lerat E. 2014. A new genome-wide method to track horizontally transferred sequences application to Drosophila. Genome Biol Evol. 6(2): 416–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton BR. 1993. Chloroplast DNA codon use: evidence for selection at the psb A locus based on tRNA availability. J Mol Evol. 37:273–280. [DOI] [PubMed] [Google Scholar]
- Orgel LE, Crick FH. 1980. Selfish DNA: the ultimate parasite. Nature 284:604–607. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Fiston-Lavier A-S, Lipatov M, Lenkov K, González J. 2011. Population genomics of transposable elements in Drosophila melanogaster. Mol Biol Evol. 28:1633–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJP. 2011. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS One 6:e22594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM. 1993. Infiltration of mariner elements. Nature 364:109. [DOI] [PubMed] [Google Scholar]
- Robertson HM, Lampe DJ. 1995. Recent horizontal transfer of a mariner transposable element among and between Diptera and Neuroptera. Mol Biol Evol. 12:850–862. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. 2012. MrBayes 32: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaack S, Gilbert C, Feschotte C. 2010. Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol Evol. 25(9): 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Gilchrist MA. 2010. Effect of correlated tRNA abundances on translation errors and evolution of codon usage bias. PLoS Genet. 6(9): e1001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Kidwell MG. 2000. Horizontal transfer and selection in the evolution of P elements. Mol Biol Evol. 17:1542–1557. [DOI] [PubMed] [Google Scholar]
- Silva J, Loreto ELS, Clark J. 2004. Factors that affect the horizontal transfer of transposable elements. Curr Issues Mol Biol. 6:57–72. [PubMed] [Google Scholar]
- Van der Linde K, Bächli G, Toda MJ, Zhang WX, Katoh T, Hu YG, Spicer GS. 2007. Drosophila Fallén 1823 (Insecta Diptera): proposed conservation of usage 2007. Bull Zool Nomencl. 64:238–242. [Google Scholar]
- Van der Linde K, Houle D. 2008. A supertree analysis and literature review of the genus Drosophila and closely related genera Diptera, Drosophilidae. Insect Syst Evol. 39:241–267. [Google Scholar]
- Vicario S, Moriyama EN, Powell JR. 2007. Codon usage in twelve species of Drosophila. BMC Evol Biol. 7:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal NMM, Ludwig A, Loreto ELS. 2009. Evolution of Tom, 297, 176 and rover retrotransposons in Drosophilidae species. Mol Genet Genomics. 282:351–362. [DOI] [PubMed] [Google Scholar]
- Wallau G, Capy P, Loreto E, Hua-Van A. 2014. Genomic landscape and evolutionary dynamics of mariner transposable elements within the Drosophila genus. BMC Genomics 15:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallau GL, Hua-Van A, Capy P, Loreto ELS. 2011. The evolutionary history of mariner-like elements in Neotropical drosophilids. Genetica 139:327–338. [DOI] [PubMed] [Google Scholar]
- Wallau GL, Lima V, Loreto ELS, Kaminski VL. 2011. The role of vertical and horizontal transfer in the evolution of Paris-like elements in drosophilid species. Genetica 139:1487–1497. [DOI] [PubMed] [Google Scholar]
- Wallau GL, Ortiz MF, Loreto ELS. 2012. Horizontal transposon transfer in eukarya detection, bias, and perspectives. Genome Biol Evol. 4:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. 1990. The “effective number of codons” used in a gene. Gene 87:23–29. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hou Z-C, Qian Y-H, Kang H, Zeng Q-T. 2012. Increasing the data size to accurately reconstruct the phylogenetic relationships between nine subgroups of the Drosophila melanogaster species group Drosophilidae, Diptera. Mol Phylogenet Evol. 62:214–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.