

Abstract
Recent preclinical studies have revealed a functionally important role for the drug efflux pump P‐glycoprotein (P‐gp) at the blood–brain barrier in limiting brain levels and thus antidepressant‐like activity of certain antidepressant drugs. Specifically, acute administration of P‐gp inhibitors, such as verapamil and cyclosporin A (CsA), has been shown to augment brain concentrations and functional activity of the antidepressant escitalopram in rodents. However, depression is a chronic disorder and current treatments require prolonged administration to elicit their full therapeutic effect. Thus, it is important to investigate whether acute findings in relation to P‐gp inhibition translate to chronic paradigms. To this end, the present study investigates whether chronic treatment with the P‐gp inhibitor verapamil and the antidepressant escitalopram results in enhanced brain distribution and antidepressant‐like effects of escitalopram. Verapamil (10 mg·kg−1 i.p.) and escitalopram (0.1 mg·kg−1 i.p.) were administered once daily for 22 days. On the final day of treatment, brain regions and plasma were collected for analysis of cortical and plasma escitalopram concentrations, and to determine the hippocampal expression of genes previously reported to be altered by chronic antidepressant treatment. Verapamil treatment resulted in a greater than twofold increase in brain levels of escitalopram, without altering plasma levels. Neither gene expression analysis nor behavioral testing revealed an augmentation of responses to escitalopram treatment due to verapamil administration. Taken together, these data demonstrate for the first time that P‐gp inhibition can yield elevated brain concentrations of an antidepressant after chronic treatment. The functional relevance of these increased brain levels requires further elaboration.
Keywords: Antidepressant, antidepressant augmentation, blood–brain barrier, escitalopram, P‐glycoprotein, treatment‐resistant depression
Abbreviations
- ANOVA
analysis of variance
- BBB
blood–brain barrier
- BDNF
brain‐derived neurotrophic factor
- CREB
cAMP‐responsive element‐binding protein
- EGR1
early growth response protein 1
- NGFI‐B
nerve growth factor IB
- P‐gp
P‐glycoprotein
- SERT
serotonin transporter
- SSRI
selective serotonin reuptake inhibitor
- TST
tail suspension test
Introduction
Increasing data have revealed that the multidrug efflux transporter P‐glycoprotein (P‐gp), expressed at the blood–brain barrier (BBB), restricts brain levels of several clinically important antidepressant drugs, thereby potentially contributing to the high prevalence of treatment failure (Uhr et al. 2008; O'Brien et al. 2012a,b, 2013a). Moreover, we have recently demonstrated that acute inhibition of P‐gp by verapamil enhances the behavioral effects of the antidepressant escitalopram in the tail suspension test (TST) (O'Brien et al. 2013b), one of the most widely used and well‐validated animal models to assess antidepressant‐like activity (Cryan et al. 2005). Taken together, these findings raise the possibility that adjunctive treatment with a P‐gp inhibitor may represent a potentially beneficial augmentation strategy in treatment‐resistant depression.
Most studies investigating the effect of P‐gp on antidepressant distribution into the brain have focused on acute drug administration (O'Brien et al. 2012b). While a limited number of research articles have reported that brain levels of certain antidepressants are elevated in P‐gp knockout mice relative to wild‐type controls after subchronic (10–11 days) treatment (Grauer and Uhr 2004; Uhr et al. 2008; Karlsson et al. 2011, 2013), no study to date has investigated the effect of chronic P‐gp inhibition on antidepressant distribution into the brain in wild‐type animals, to our knowledge. This is a key consideration, as currently available antidepressants are associated with a delayed response, typically requiring chronic treatment in order to achieve their therapeutic effect in patients (Krishnan and Nestler 2008; O'Leary et al. 2014). Moreover, certain xenobiotics are known to upregulate the expression and activity of P‐gp (Miller 2010). Indeed, the antidepressant venlafaxine, which is known to be a transported P‐gp substrate (O'Brien et al. 2012b), has been reported to increase P‐gp function in vitro and in vivo (Ehret et al. 2007; de Klerk et al. 2010). Thus, even though acute P‐gp inhibition can result in increased brain levels of certain antidepressants, this effect could be negated following chronic exposure due to a hypothesized counteractive upregulation of P‐gp activity.
The primary goal of the present study is to determine whether chronic treatment with the P‐gp inhibitor verapamil and the antidepressant escitalopram results in increased brain distribution of escitalopram. Recent studies, both in P‐gp knockout (acute and subchronic) (Karlsson et al. 2013) and wild‐type (acute only) (O'Brien et al. 2013b) rodents, have identified that escitalopram, a commonly prescribed selective serotonin reuptake inhibitor (SSRI) antidepressant, is a transported P‐gp substrate at the BBB. In addition, putative behavioral and molecular effects of chronic treatment with a P‐gp inhibitor and a P‐gp substrate antidepressant are investigated. In particular, the expression of several genes involved in the regulation of monoaminergic signaling, neurogenesis, responses to stress and gene transcription, and which have been reported to be sensitive to chronic antidepressant treatment, was assessed (Table 1).
Table 1.
Target genes selected for mRNA expression analysis
| Gene name | Protein product | Function | Studies implicating gene in antidepressant response |
|---|---|---|---|
| Nr3c1 | Glucocorticoid receptor | Receptor for glucocorticoids, such as corticosterone in mice | Peiffer et al. (1991), Seckl and Fink (1992), Johansson et al. (1998), Bjartmar et al. (2000), Guidotti et al. (2013) |
| Nr3c2 | Mineralocorticoid receptor | Cytosolic receptor for mineralocorticoids, such as aldosterone, as well as glucocorticoids | Brady et al. (1991), Seckl and Fink (1992), Johansson et al. (1998), Bjartmar et al. (2000) |
| Fkbp5 | FK506‐binding protein | Immunophilin protein involved in immunoregulation and protein folding/trafficking | Guidotti et al. (2013) |
| Egr1 | Early growth response protein 1 (aka Zif268 or NGFI‐A) | Transcription factor | Morinobu et al. (1997), Johansson et al. (1998), Bjartmar et al. (2000), Sillaber et al. (2008) |
| Nr4a1 | Nerve growth factor IB | Transcription factor | Bjartmar et al. (2000) |
| Slc6a4 | Serotonin transporter | Reuptake of 5‐HT from synaptic space | Lesch et al. (1993), Lopez et al. (1994), Benmansour et al. (1999) |
| Tph2 | Tryptophan hydroxylase 2 | Rate limiting enzyme in the synthesis of 5‐HT in CNS | Abumaria et al. (2007), Shishkina et al. (2007), Heydendael and Jacobson (2009) |
| Htr1a | 5‐HT1A receptor | 5‐HT autoreceptor involved in regulation of 5‐HT signaling | Burnet et al. (1994), Abumaria et al. (2007) |
| Kcnk2 | Trek‐1 | Potassium channel | Heurteaux et al. (2006) |
| Bdnf | Brain‐derived neurotrophic factor | Neurotrophin | Nibuya et al. (1995), Martinez‐Turrillas et al. (2005), Sillaber et al. (2008), Alboni et al. (2010) |
| Creb | cAMP response element‐binding protein | Transcription factor | Nibuya et al. (1996), Thome et al. (2000), Blom et al. (2002), Alboni et al. (2010) |
| S100a10 | p11 | Involved in regulation of 5‐HT signaling in brain | Svenningsson et al. (2006), Melas et al. (2012) |
Material and Methods
Drugs and chemicals
Escitalopram oxalate was purchased from Discovery Fine Chemicals (Dorset, UK). Verapamil was obtained from Sigma‐Aldrich (Dublin, Ireland), as were all other chemicals, reagents and materials unless otherwise stated.
Animals
Male C57BL/6JOlaHsd mice (6–8 weeks old at the beginning of the study), purchased from Harlan Laboratories, UK, were used in this study. All animals were group‐housed 4 animals per cage and maintained on a 12 h light/dark cycle (lights on at 08:00 h) with food and water ad libitum. Room temperature was controlled at 22 ± 1°C. All procedures were carried out in accordance with EU directive 2010/63/EU and approved by the Animal Experimentation & Ethics Committee of University College Cork.
Experimental design
Verapamil (10 mg·kg−1 i.p.) or saline were administered at least 30 min before escitalopram (0.1 mg·kg−1 i.p.) or saline each morning for 22 days (n = 9–10 per group). Body weight was measured daily to monitor for potential adverse reactions to treatment. On the penultimate day of treatment (day 21), mice were subjected to the TST to assess the effect of chronic verapamil pretreatment on the antidepressant‐like activity of escitalopram. On the final day of treatment, mice were sacrificed 40 min after escitalopram and 100 min post‐verapamil administration. Timings were based on previous acute experiments (O'Brien et al. 2013b). Brain regions were immediately dissected out in ice‐cold PBS and trunk plasma was collected. All samples were snap frozen in isopentane on dry ice, and stored at −80°C until further processing. The dose of escitalopram was selected based on our previous study, where 0.1 mg·kg−1 of escitalopram was found to elicit a behavioral effect in the TST only when administered in conjunction with verapamil (O'Brien et al. 2013b). However, a lower dose of verapamil was used in the present study (10 mg·kg−1 rather than 20 mg·kg−1) due to concerns about potential systemic side effects that may have arisen with prolonged high dose verapamil treatment.
Tail suspension test
The TST, one of the most widely used models for assessing antidepressant activity in rodents (Cryan et al. 2005), was carried out on the 21st day of treatment, as described previously (O'Brien et al. 2013b). This facilitated investigation of the impact of pretreatment with the P‐gp inhibitor verapamil on the antidepressant‐like activity of escitalopram after chronic administration of each drug. Briefly, on the day of the TST, the P‐gp inhibitor verapamil (10 mg·kg−1 i.p.) or saline was administered one hour before escitalopram (0.1 mg·kg−1 i.p.) or saline treatment. Thirty minutes after the second injection, mice were individually suspended by the tail from a horizontal bar using adhesive tape. Six‐minute test sessions were recorded by video camera and the amount of time spent immobile by each animal was subsequently scored by a trained observer blind to the treatment groups.
Determination of escitalopram and verapamil in brain and plasma samples
Escitalopram and verapamil were extracted from cortical brain tissue and plasma using a liquid–liquid extraction technique described previously (Clarke et al. 2009; O'Brien et al. 2013b). Briefly, 48 μL of plasma was spiked with 2 μL of the internal standard, imipramine, to yield a final concentration of 1 μg·mL−1 imipramine. To this imipramine‐spiked plasma, 1 mL of sodium hydroxide (2 mol/L) and 3 mL of water were added. Extraction was carried out in 7.5 mL of 1.5% isoamyl alcohol in n‐heptane by vortexing for 30 sec, followed by agitation on a mechanical shaker for 15 min and then centrifugation at 4500 g for 15 min at 20°C. The upper solvent layer was transferred to a tube containing 200 μL of 25 mmol/L OPA, vortexed for 30 sec, then agitated on a mechanical shaker for 15 min followed by centrifugation at 5000 rpm for 15 min at room temperature. Twenty microlitres of the lower aqueous phase was injected onto a HPLC system for analysis of escitalopram and verapamil, using a previously described HPLC method (O'Brien et al. 2013b). Brain tissue samples were weighed prior to homogenization in 500 μL of imipramine‐spiked (1 μg·mL−1) homogenization buffer (i.e., HPLC mobile phase). Homogenized brain tissue was centrifuged at 35 280 g at 8°C for 15 min, and escitalopram and verapamil were extracted from 200 μL of the supernatant as described above.
P‐glycoprotein protein expression analysis
The effect of chronic verapamil and/or escitalopram treatment on the relative expression of P‐glycoprotein in hippocampal brain tissue was determined by Western blot (n = 7 per group), as described previously (O'Brien et al. 2013a), with some modifications. Protein was extracted from the hippocampus using a commercially available kit (mirVana™ PARIS™ Kit; Applied Biosystems, Paisley, U.K.), as per the manufacturer's instructions. Protease and phosphatase inhibitor cocktails (Roche, Dublin, Ireland) were included in the tissue lysis buffer used in the extraction procedure. Sixteen micrograms of protein were loaded in a 4–20% gradient gel, according to the manufacturer's instructions (Express PAGE Gels; Genscript, Piscataway, New Jersey, US). After transfer onto a 0.2‐μm nitrocellulose membrane and blocking with 5% skimmed milk and 0.1% Tween 20 in PBS, blots were probed overnight at 4°C with the C219 primary monoclonal P‐gp antibody (1:100 dilution in 2% skimmed milk) (Enzo Life Sciences (UK) Ltd, Exeter, UK). Reprobing was conducted for 1 h at room temperature using a goat anti‐mouse IgG‐HRP conjugate, diluted 1:2000 (Jackson Immunoresearch Europe Ltd, Suffolk, UK.). Images were obtained using a luminescent image analyzer (LAS‐3000; Fujifilm, Dublin, Ireland). For the detection of β‐actin, the membranes were incubated with Monoclonal Anti‐β‐Actin−Peroxidase antibody produced in mouse (1:15,000). Immunoblots were quantified using ImageJ software (http://imagej.nih.gov/ij/, National Institute of Health, Maryland, USA).
Gene expression analysis
The hippocampal expression of twelve genes, which have been shown to be responsive to chronic antidepressant treatment or implicated in the antidepressant response (Table 1), was analyzed using real‐time quantitative polymerase chain reaction (qPCR). To our knowledge, p11, brain‐derived neurotrophic factor (BDNF) and cAMP‐responsive element‐binding protein (CREB) are the only gene targets which have been shown to be altered by chronic escitalopram treatment in the (rat) brain (Jacobsen and Mork 2004; Alboni et al. 2010; Melas et al. 2012). Total RNA was extracted from hippocampal tissue using a commercially available kit (mirVana™ PARIS™ Kit, Applied Biosystems), as per the manufacturer's instructions. RNA purity and quantity was measured by spectrophotometric analysis (NanoDrop® ND‐1000 Spectrophotometer, NanoDrop Technologies, Inc., Wilmington, Delaware, USA). mRNA was reverse transcribed from 1 μg total RNA using a G‐storm thermocycler (G‐storm, Surrey, UK). PCR primers and probes were purchased from Integrated DNA Technologies (Leuven, Belgium). Assays were designed for murine genes, as detailed in Table 1. Analysis of gene expression was performed in triplicate on 384‐well plates using the LightCycler 480 System (Roche), and the expression of each gene was normalized to that of β‐actin. The 2−ΔΔCT method was used to calculate relative changes in gene expression determined from qPCR experiments (Livak and Schmittgen 2001). Gene expression values are expressed relative to the control group.
Data analysis and statistical procedures
Statistical analysis of data was carried out using standard commercial software (SPSS Statistics, version 20.0.0; SPSS, Inc., Chicago, IL). The differences in brain and plasma levels of escitalopram between the two escitalopram‐treated groups were analyzed using an unpaired Student's t‐test. Similarly, verapamil concentrations in brain tissue and plasma were compared by unpaired Student's t‐test. Differences in gene expression between the groups were analyzed by two‐way analysis of variance (ANOVA), with verapamil and escitalopram as factors, and the LSD post hoc was used to elucidate statistically significant differences between treatment groups. Differences in changes in body weight over time were analyzed by two‐way repeated measures ANOVA, with escitalopram and verapamil as factors. The criterion for statistical significance was P < 0.05.
Results
Concomitant chronic treatment with the P‐gp inhibitor verapamil and escitalopram increased the brain levels of escitalopram, without affecting plasma levels
The concentrations of escitalopram in cortical brain tissue were significantly greater in mice pretreated with the P‐gp inhibitor verapamil compared to saline‐pretreated mice, whereas there was no significant difference in plasma escitalopram levels (Fig. 1A and B). Pretreatment with verapamil resulted in a 110% increase in cortical escitalopram levels (t(17) = −5.903, P < 0.001; Fig. 1A), but had no effect on plasma levels of escitalopram (t(17) = −0.98, P = 0.341); Fig. 1B). Thus, the increase in brain concentrations of escitalopram can be attributed to enhanced BBB transport due to P‐gp inhibition, rather than a reflection of elevated plasma concentrations.
Figure 1.
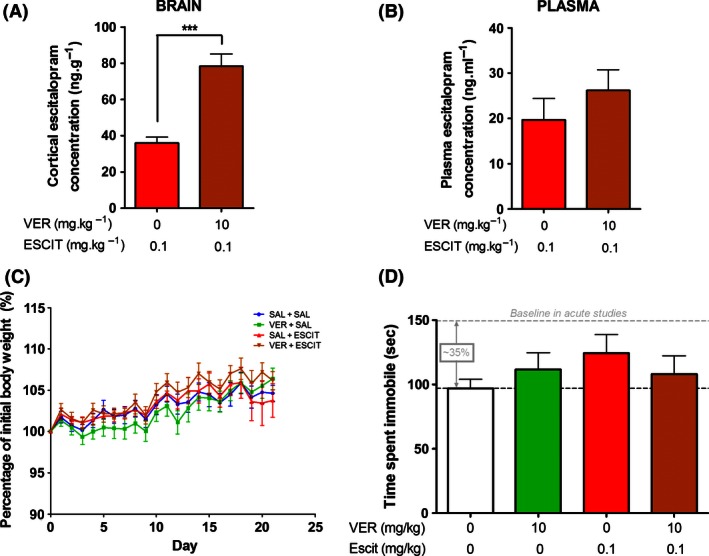
Effect of chronic administration of verapamil and escitalopram on brain concentrations of escitalopram, body weight and antidepressant‐like behavioral effects in the tail suspension test (TST). (A) Pretreatment with verapamil resulted in a 110% increase in concentrations of escitalopram in cortical brain tissue. (B) Pre‐treatment with verapamil did not significantly alter plasma levels of escitalopram. (C) Treatment with verapamil and/or escitalopram had no effect on body weight compared to the saline‐treated control group. (D) There were no statistically significant differences between the treatment groups in terms of immobility in the TST. However, it it worth noting that the baseline immobility of the saline‐treated control group was reduced by ~35% compared to our previous acute work (O'Brien et al. 2013b). (n = 9–10 per group). ***P < 0.001 between groups.
Plasma (t(17) = 0.274, P = 0.787) and brain (t(14) = −0.810, P = 0.937) levels of verapamil were equivalent in both verapamil‐treated groups, with mean (±SEM) concentrations of 390 (±37) ng·mL−1 in plasma and 202 (±15) ng·g−1 in cortical tissue.
Body weight
Neither drug treatment, nor a combination of both drugs, had an effect on body weight during the course of the 22 day of drug administration (Fig. 1C): escitalopram (F(1,35) = 0.376, P = 0.544); verapamil (F(1, 35) = 0.096, P = 0.759); and escitalopram × verapamil (F(1, 35) = 0.195, P = 0.661).
Tail suspension test
Neither escitalopram (F(1, 35) = 0.885, P = 0.353) nor verapamil (F(1, 35) = 0.004, P = 0.950) had a significant impact on the duration of immobility in the TST, nor was there an interaction between the two factors (F(1, 35) = 1.505, P = 0.228) (Fig. 1D). In comparison to previously reported acute experiments performed in our laboratory (O'Brien et al. 2013b), chronic administration reduced the time spent immobile in saline‐treated mice by 35% (97s vs. 150s).
P‐glycoprotein expression analysis
Hippocampal P‐gp protein levels were unaffected by chronic treatment with either verapamil, escitalopram, or a combination of both drugs (Fig. 2). This indicates that P‐gp expression was neither upregulated nor downregulated in response to repeated long‐term administration of this P‐gp inhibitor or P‐gp substrate antidepressant.
Figure 2.
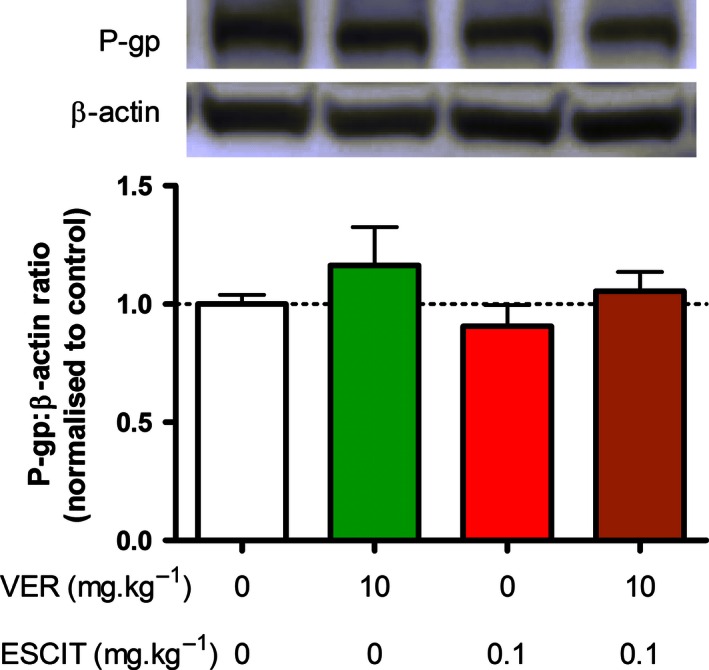
Western blot analysis of P‐gp protein expression in the hippocampus. Chronic administration of verapamil and/or escitalopram elicited no effect on the expression of P‐gp protein in hippocampal tissue (n = 7 per group).
Gene expression analysis
A subset of animals was used for gene expression analysis. The total numbers of samples included for each gene are listed in Table 2. Two‐way ANOVA analysis of PCR results revealed a verapamil, escitalopram or verapamil × escitalopram effect on the expression of four of the twelve genes analyzed (Table 2).
Table 2.
Two‐way ANOVA of the effects of verapamil, escitalopram and verapamil × escitalopram interaction on mRNA expression in the hippocampus (n = 7–10 per group)
| Target | Total n | Verapamil effect | Escitalopram effect | Verapamil × escitalopram interaction |
|---|---|---|---|---|
| GR | 32 | F(1, 28) = 0.15, P = 0.904 | F(1, 28) = 4.132, P = 0.052 | F(1, 28) = 2.337, P = 0.138 |
| MR | 31 | F(1, 27) = 1.530, P = 0.227 | F(1, 27) = 0.004, P = 0.952 | F(1, 27) = 0.059, P = 0.810 |
| FKBP5 | 34 | F(1, 30) = 0.810, P = 0.375 | F(1, 30) = 0.669, P = 0.420 | F(1, 30) = 1.385, P = 0.249 |
| EGR1 | 34 | F(1, 30) = 3.795, P = 0.061 | F (1, 30) = 5.854, P = 0.022 | F(1, 30) = 0.011, P = 0.917 |
| NGFI‐B | 31 | F (1, 27) = 5.336, P = 0.029 | F(1, 27) = 1.838, P = 0.186 | F(1, 27) = 0.389, P = 0.538 |
| SERT | 33 | F(1, 29) = 0.408, P = 0.528 | F(1, 29) = 0.009, P = 0.925 | F (1, 29) = 5.385, P = 0.028 |
| TPH2 | 33 | F(1, 29) = 0.284, P = 0.598 | F(1, 29) = 1.258, P = 0.271 | F(1, 29) = 3.342, P = 0.078 |
| 5‐HT1A | 33 | F(1, 29) = 0.008, P = 0.930 | F(1, 29) = 1.091, P = 0.305 | F(1, 29) = 0.414, P = 0.525 |
| TREK‐1 | 33 | F(1, 29) = 1.568, P = 0.221 | F(1, 29) = 0.091, P = 0.765 | F(1, 29) = 2.216, P = 0.147 |
| BDNF | 34 | F(1, 30) = 0.000, P = 0.990 | F(1, 30) = 0.918, P = 0.346 | F(1, 30) = 0.062, P = 0.805 |
| CREB | 32 | F(1, 28) = 2.163, P = 0.153 | F(1, 28) = 0.008, P = 0.930 | F(1, 28) = 0.477, P = 0.496 |
| p11 | 34 | F (1, 30) = 10.182, P = 0.003 | F(1, 30) = 0.076, P = 0.785 | F(1, 30) = 0.127, P = 0.724 |
Bold font denotes statistically significant effect.
GR, glucocorticoid receptor; MR, mineralocorticoid receptor; FKBP5, FK506‐binding protein; EGR1, early growth response protein 1; NGFI‐B, nerve growth factor IB; SERT, serotonin transporter; TPH2, tryptophan hydroxylase 2; BDNF, brain‐derived neurotrophic factor; CREB, cAMP response element‐binding protein.
The only gene expression target for which a significant escitalopram × verapamil interaction was detected was the serotonin transporter (SERT). Chronic treatment with verapamil or escitalopram individually resulted in a trend toward increased SERT mRNA expression, but this trend was reversed when both verapamil and escitalopram were administered together, resulting in a significant reduction in SERT mRNA levels compared to mice treated with escitalopram only (Fig. 3A).
Figure 3.
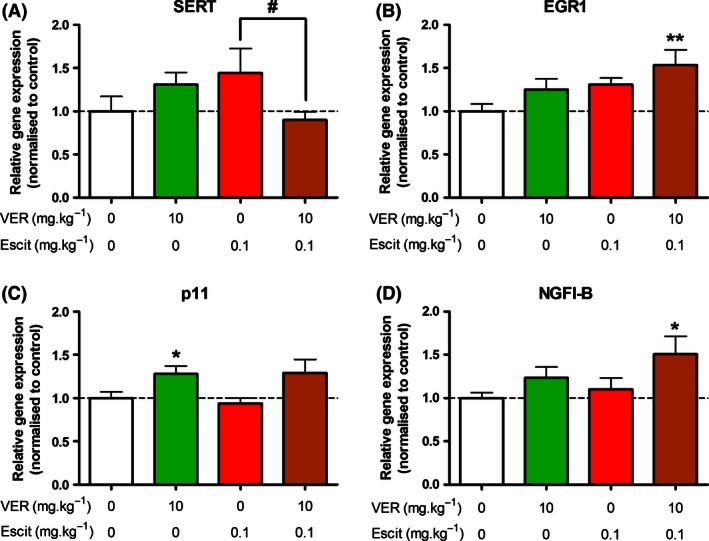
Relative hippocampal expression of genes for which significant effects were observed. (A) A significant interaction between verapamil and escitalopram was observed for SERT expression. SERT mRNA levels were significantly lower in mice treated with both verapamil and escitalopram than those treated with escitalopram only. (B) Escitaloram treatment was found to exert a significant effect on EGR1 mRNA expression. The increase in EGR1 mRNA levels, compared to the control group, was only statistically significant in mice treated with both verapamil and escitalopram. However, there was no significant interaction between the two treatments. (C) Verapamil exerted a significant effect on p11 expression, resulting in a significant increase in mice treated with verapamil only compared to control mice, and a trend toward increased expression in mice treated with both verapamil and escitalopram (P = 0.054). (D) Verapamil also exerted a significant effect on NGFI‐B expression. A statistically significant elevation in NGFI‐B mRNA was only observed in mice treated with both verapamil and escitalopram, but there was no interaction between the two treatments (n = 7–10 per group). *P < 0.05; **P < 0.01 compared to control group. # P < 0.05 between the indicated groups.
Escitalopram treatment had an effect on the expression of early growth response protein 1 (EGR1) mRNA, resulting in increased expression of this gene. Post hoc analysis revealed that this increase was only statistically significant in mice treated with both verapamil and escitalopram. However, there was no significant interaction between the two treatments (Table 2; Fig. 3B).
Verapamil treatment affected the expression of mRNA for p11 and nerve growth factor IB (NGFI‐B), increasing p11 expression to a similar extent with or without escitalopram treatment (Fig. 3C). NGFI‐B mRNA expression was also increased by verapamil treatment. Similar to EGR1, post hoc analysis revealed that this increase was only statistically significant in mice treated with both verapamil and escitalopram, but there was no significant interaction between the two treatments (Table 2; Fig. 3D).
Discussion and Conclusions
Recent evidence from preclinical studies has highlighted an important role for the efflux transporter P‐gp at the BBB in limiting the brain distribution of several antidepressants (O'Brien et al. 2012b). Here, it is shown for the first time, to our knowledge, that chronic treatment with a P‐gp inhibitor and a P‐gp substrate antidepressant results in increased brain levels of the antidepressant drug. We recently demonstrated that acute pre‐treatment with the P‐gp inhibitor verapamil augmented the brain concentrations and functional activity of the SSRI escitalopram in mice (O'Brien et al. 2013b). In addition, in vitro bidirectional transport studies demonstrated that escitalopram is a transported substrate of human P‐gp (O'Brien et al. 2013b), indicating that these pharmacokinetic and pharmacodynamic observations in mice may translate to man. However, prior to the present study, it remained unclear if acute findings would apply to chronic treatment paradigms. Considering that antidepressant drugs are administered chronically in clinical practice, this is an important point, especially given that another P‐gp substrate antidepressant (venlafaxine) has been reported to induce P‐gp in vitro and in vivo (Ehret et al. 2007; de Klerk et al. 2010). Thus, the present results indicate that P‐gp inhibition may represent a promising strategy to augment the brain delivery of certain antidepressants during chronic treatment.
The greater than 2‐fold increase in brain concentrations of escitalopram in mice treated with the P‐gp inhibitor verapamil in the present chronic study is consistent with previously observed effects in an acute setting (O'Brien et al. 2013b), despite using a lower dose of verapamil in the present work (10 mg/kg daily vs. 20 mg/kg previously). However, in contrast with the acute study, no enhancement of behavioral or gene expression responses to chronic escitalopram treatment was detected here. Indeed, chronic treatment with escitalopram elicited no robust effect on the behavioral or molecular parameters investigated in the present study. This is perhaps unsurprising, given that a relatively low dose (0.1 mg·kg−1) of escitalopram was chosen so as to be able to unmask any augmentation effects of verapamil.
In previous acute studies, we had observed a behavioral effect in the TST at this low dose of escitalopram only when mice were pretreated with verapamil (O'Brien et al. 2013b). No such effect was evident here after chronic administration of both drugs. However, when compared to our previous acute experiments (cf. O'Brien et al. 2013b), the baseline immobility in the control group in this chronic study was reduced by 35% (Fig. 1D), indicating that repeated daily handling and injections impacted on the behavioral readout of the TST. This may have resulted in a floor effect, thereby potentially obscuring any putative behavioral impact of the increased brain concentrations of escitalopram due to P‐gp inhibition and rendering the results from the TST inconclusive. Although there have been a limited number of reports of the TST being responsive to chronic antidepressant administration, it is predominantly used to assess behavioral responses to acute antidepressant treatment (Cryan et al. 2005). Interestingly, studies which have yielded positive results in the TST following chronic antidepressant administration have generally involved quite high doses of the antidepressant, for example 20 mg·kg−1 of fluoxetine (Ukai et al. 1998). Hence, increasing the dose of escitalopram may represent a viable strategy to overcome the floor effect seen in the TST here in future. In addition, it will be important to validate the antidepressant potential of esciatlopram in combination with verapamil in alternative antidepressant testing paradigms, such as the social defeat stress or learned helplessness models (O'Leary and Cryan 2013), as these tests are responsive to chronic antidepressant treatment and have the added advantage of being mouse models of depression, as opposed to the present study which involved ‘normal’ mice.
Similar to our behavioral studies, gene expression analysis did not reveal an augmentation of molecular responses to escitalopram administration in verapamil pretreated mice. However, reports of alterations in mRNA expression in response to antidepressant treatment are often inconsistent, and may not be universal among different antidepressants, as evidenced by the variable findings in relation to BDNF expression (Tardito et al. 2006; Groves 2007). Thus, genes which have been reported to be altered by treatment with other antidepressants may not generalize to escitalopram administration. To our knowledge, p11, BDNF and CREB are the only gene targets which have been shown to be affected by chronic escitalopram treatment (Jacobsen and Mork 2004; Alboni et al. 2010; Melas et al. 2012). However, the expression of these genes was not altered by escitalopram treatment in the present study, which may have been due to the 100‐fold lower dose of escitalopram used here than in previous work (0.1 mg/kg per day compared to 10 mg/kg per day). Moreover, our finding that chronic escitalopram treatment exerted a significant effect on mRNA levels of the transcription factor EGR1 has not previously been reported, to our knowledge. Interestingly, the combination of verapamil and escitalopram treatment, but neither in isolation, resulted in a statistically significant increase in EGR1 mRNA expression relative to control mice. Similar findings were observed in relation to another transcription factor, NGFI‐B. While there was not a significant interaction between escitalopram and verapamil treatment for either of these genes, indicating that this could merely reflect an additive effect of the two treatments, these targets warrant further investigation in future studies.
It is also worth noting that antidepressant‐induced changes in gene expression can be dependent on the brain region analyzed and the antidepressant dose used (Tardito et al. 2006). In relation to the dose, the drug administration regimen used in the present study yielded plasma concentrations of escitalopram (23 ng·mL−1) within the therapeutic range (15–80 ng·mL−1) (Baumann et al. 2004), albeit at the lower end of that range. However, most studies which have reported significant alterations in mRNA expression following antidepressant treatment have involved the administration of much higher doses of the antidepressant (10 mg·kg−1 escitalopram daily, for example (Jacobsen and Mork 2004; Alboni et al. 2010)). Thus, the doses of escitalopram used in the present study may have been insufficient to reproduce previously reported changes in mRNA expression. Furthermore, it is possible that there may have been functionally relevant alterations in protein expression, activation or cellular localization in response to antidepressant treatment, without changes in mRNA levels. Future studies should investigate these possibilities.
Even though our analysis did not reveal any augmentation of escitalopram's effects on mRNA expression by verapamil administration, a statistically significant interaction between the two treatments was observed in terms of serotonin transporter (SERT) mRNA levels. Interestingly, this effect constituted an attenuation of a trend toward increased SERT mRNA levels following verapamil or escitalopram on their own. It is important to note that, while studies have shown that SERT mRNA expression can be altered in response to antidepressant treatment, these findings have been somewhat inconsistent. SERT mRNA levels have been variously reported to be increased, decreased or not affected by chronic antidepressant treatment (Lesch et al. 1993; Lopez et al. 1994; Benmansour et al. 1999; Abumaria et al. 2007), indicating that the effect of antidepressant drugs on SERT expression is complex and variable.
Due to the inconclusive nature of our findings in relation to the effect of chronic P‐gp inhibition on behavioral and molecular responses to antidepressant treatment, further studies are required to investigate this question. Such future studies should ideally utilize more specific inhibitors of P‐gp, thereby limiting the contribution of P‐gp independent factors to alterations in gene expression, and helping to specifically elucidate antidepressant‐related changes. For example, two‐way ANOVA analysis revealed that verapamil itself exerted a significant effect on the expression of mRNA for NGFI‐B and p11, thus making it difficult to identify any potential influence of P‐gp inhibition on putative escitalopram effects in relation to these targets. In addition, future studies should use different dosing regimens of escitalopram and investigate other parameters, such as protein expression, rather than focusing on mRNA expression exclusively.
Verapamil, the P‐gp inhibitor used in the present study, is a clinically used calcium channel blocker, indicated for the treatment of several cardiovascular conditions. At higher doses, which it is thought may be required to inhibit P‐gp due to its lack of specificity and relatively low potency, verapamil can elicit toxicity (DeWitt and Waksman 2004). In the present study, in which verapamil was found to inhibit P‐gp, no overt signs of toxicity were detected during daily observations or in terms of alterations in body weight. Furthermore, the plasma concentrations of verapamil measured in the present study (390 ng·mL−1) are within the clinical therapeutic range (50–500 ng·mL−1) (Regenthal et al. 1999). However, it is likely that its other pharmacological actions, including cardiovascular effects and inhibition of cytochrome P450 enzymes, would preclude verapamil's widespread use as a P‐gp inhibitor in clinical practice. Several more specific second and third generation P‐gp inhibitors have been developed which may be more appropriate for this purpose (Colabufo et al. 2010). Nonetheless, verapamil represents a valuable pharmacological tool to investigate the effects of P‐gp inhibition on drug distribution in preclinical studies. Another important consideration in this regard is the possibility that certain antidepressants, such as clomipramine, have been reported to influence P‐gp function in vitro (Pariante et al. 2003; Weiss et al. 2003), even if the in vivo relevance of these findings remains unclear (Mason et al. 2011). Further studies are required to add clarity to this issue.
In conclusion, the present study demonstrates that inhibition of P‐gp results in elevated brain levels of an antidepressant drug in a chronic drug administration paradigm. This finding indicates that P‐gp inhibition may represent a viable strategy to enhance the brain distribution of P‐gp substrate antidepressants after repeated dosing. In addition, these data reveal that chronic escitalopram administration can influence mRNA expression of the transcription factor EGR1. Further studies are required to fully elucidate the functional consequences of increasing the brain levels of an antidepressant drug by P‐gp inhibition on a prolonged basis.
Disclosures
The authors declare no conflict of interest.
Acknowledgements
The authors thank Rachel Moloney, Patrick Fitzgerald, Margherita Bonetti, and the staff at the University College Cork Biological Services Unit for technical assistance with this study. The Alimentary Pharmabiotic Centre is a research center funded by Science Foundation Ireland (SFI), through the Irish Government's National Development Plan. T. G. D. and J. F. C. are supported by SFI (grant nos. 07/CE/B1368 and 12/RC/2273). The SFI‐funded Strategic Research Cluster grant no. 07/SRC/B1154 and the Irish Drug Delivery Network also fund B. T. G. and J. F. C., T. D., J. F. C. and G. C. are also supported by the Irish Health Research Board Health Research Awards (HRA POR/2011/23) and (HRA POR/2012/32). GC is supported by a NARSAD Young Investigator Grant from the Brain and Behavior Research Foundation (Grant number 20771).
O'Brien F. E., Moloney G., Scott K. A., O'Connor R. M., Clarke G., Dinan T. G., Griffin B. T., Cryan J. F.. Chronic P‐glycoprotein Inhibition Increases the brain concentration of escitalopram: potential implications for treating depression, Pharma Res Per, 3 (6), 2015, e00190, doi: 10.1002/prp2.190
References
- Abumaria N, Rygula R, Hiemke C, Fuchs E, Havemann‐Reinecke U, Ruther E, et al. (2007). Effect of chronic citalopram on serotonin‐related and stress‐regulated genes in the dorsal raphe nucleus of the rat. Eur Neuropsychopharmacol 17: 417–429. [DOI] [PubMed] [Google Scholar]
- Alboni S, Benatti C, Capone G, Corsini D, Caggia F, Tascedda F, et al. (2010). Time‐dependent effects of escitalopram on brain derived neurotrophic factor (BDNF) and neuroplasticity related targets in the central nervous system of rats. Eur J Pharmacol 643: 180–187. [DOI] [PubMed] [Google Scholar]
- Baumann P, Hiemke C, Ulrich S, Eckermann G, Gaertner I, Gerlach M, et al. (2004). The AGNP‐TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry 37: 243–265. [DOI] [PubMed] [Google Scholar]
- Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, et al. (1999). Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci 19: 10494–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar L, Johansson IM, Marcusson J, Ross SB, Seckl JR, Olsson T (2000). Selective effects on NGFI‐A, MR, GR and NGFI‐B hippocampal mRNA expression after chronic treatment with different subclasses of antidepressants in the rat. Psychopharmacology 151: 7–12. [DOI] [PubMed] [Google Scholar]
- Blom JM, Tascedda F, Carra S, Ferraguti C, Barden N, Brunello N (2002). Altered regulation of CREB by chronic antidepressant administration in the brain of transgenic mice with impaired glucocorticoid receptor function. Neuropsychopharmacol 26: 605–614. [DOI] [PubMed] [Google Scholar]
- Brady LS, Whitfield HJ Jr, Fox RJ, Gold PW, Herkenham M (1991). Long‐term antidepressant administration alters corticotropin‐releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. Therapeutic implications. J Clin Invest 87: 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnet PW, Michelson D, Smith MA, Gold PW, Sternberg EM (1994). The effect of chronic imipramine administration on the densities of 5‐HT1A and 5‐HT2 receptors and the abundances of 5‐HT receptor and transporter mRNA in the cortex, hippocampus and dorsal raphe of three strains of rat. Brain Res 638: 311–324. [DOI] [PubMed] [Google Scholar]
- Clarke G, O'Mahony SM, Cryan JF, Dinan TG (2009). Verapamil in treatment resistant depression: a role for the P‐glycoprotein transporter? Hum Psychopharmacol 24: 217–223. [DOI] [PubMed] [Google Scholar]
- Colabufo NA, Berardi F, Cantore M, Contino M, Inglese C, Niso M, et al. (2010). Perspectives of P‐glycoprotein modulating agents in oncology and neurodegenerative diseases: pharmaceutical, biological, and diagnostic potentials. J Med Chem 53: 1883–1897. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Mombereau C, Vassout A (2005). The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29: 571–625. [DOI] [PubMed] [Google Scholar]
- DeWitt CR, Waksman JC (2004). Pharmacology, pathophysiology and management of calcium channel blocker and beta‐blocker toxicity. Toxicol Rev 23: 223–238. [DOI] [PubMed] [Google Scholar]
- Ehret MJ, Levin GA, Narasimhan M, Rathinavelu A (2007). Venlafaxine induces P‐glycoprotein in human Caco‐2 cells. Hum Psychopharm Clin 22: 49–53. [DOI] [PubMed] [Google Scholar]
- Grauer MT, Uhr M (2004). P‐glycoprotein reduces the ability of amitriptyline metabolites to cross the blood brain barrier in mice after a 10‐day administration of amitriptyline. J Psychopharmacol 18: 66–74. [DOI] [PubMed] [Google Scholar]
- Groves JO (2007). Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry 12: 1079–1088. [DOI] [PubMed] [Google Scholar]
- Guidotti G, Calabrese F, Anacker C, Racagni G, Pariante CM, Riva MA (2013). Glucocorticoid receptor and FKBP5 expression is altered following exposure to chronic stress: modulation by antidepressant treatment. Neuropsychopharmacol 38: 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thummler S, Peng XD, et al. (2006). Deletion of the background potassium channel TREK‐1 results in a depression‐resistant phenotype. Nat Neurosci 9: 1134–1141. [DOI] [PubMed] [Google Scholar]
- Heydendael W, Jacobson L (2009). Glucocorticoid status affects antidepressant regulation of locus coeruleus tyrosine hydroxylase and dorsal raphe tryptophan hydroxylase gene expression. Brain Res 1288: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JP, Mork A (2004). The effect of escitalopram, desipramine, electroconvulsive seizures and lithium on brain‐derived neurotrophic factor mRNA and protein expression in the rat brain and the correlation to 5‐HT and 5‐HIAA levels. Brain Res 1024: 183–192. [DOI] [PubMed] [Google Scholar]
- Johansson IM, Bjartmar L, Marcusson J, Ross SB, Seckl JR, Olsson T (1998). Chronic amitriptyline treatment induces hippocampal NGFI‐A, glucocorticoid receptor and mineralocorticoid receptor mRNA expression in rats. Brain Res Mol Brain Res 62: 92–95. [DOI] [PubMed] [Google Scholar]
- Karlsson L, Hiemke C, Carlsson B, Josefsson M, Ahlner J, Bengtsson F, et al. (2011). Effects on enantiomeric drug disposition and open‐field behavior after chronic treatment with venlafaxine in the P‐glycoprotein knockout mice model. Psychopharmacology 215: 367–377. [DOI] [PubMed] [Google Scholar]
- Karlsson L, Carlsson B, Hiemke C, Ahlner J, Bengtsson F, Schmitt U, et al. (2013). Altered brain concentrations of citalopram and escitalopram in P‐glycoprotein deficient mice after acute and chronic treatment. Eur Neuropsychopharmacol 23: 1636–1644. [DOI] [PubMed] [Google Scholar]
- de Klerk OL, Bosker FJ, Willemsen AT, Van Waarde A, Visser AK, de Jager T, et al. (2010). Chronic stress and antidepressant treatment have opposite effects on P‐glycoprotein at the blood‐brain barrier: an experimental PET study in rats. J Psychopharmacol 24: 1237–1242. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ (2008). The molecular neurobiology of depression. Nature 455: 894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch KP, Aulakh CS, Wolozin BL, Tolliver TJ, Hill JL, Murphy DL (1993). Regional brain expression of serotonin transporter mRNA and its regulation by reuptake inhibiting antidepressants. Brain Res Mol Brain Res 17: 31–35. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Lopez JF, Chalmers DT, Vazquez DM, Watson SJ, Akil H (1994). Serotonin transporter mRNA in rat brain is regulated by classical antidepressants. Biol Psychiatry 35: 287–290. [DOI] [PubMed] [Google Scholar]
- Martinez‐Turrillas R, Del Rio J, Frechilla D (2005). Sequential changes in BDNF mRNA expression and synaptic levels of AMPA receptor subunits in rat hippocampus after chronic antidepressant treatment. Neuropharmacology 49: 1178–1188. [DOI] [PubMed] [Google Scholar]
- Mason BL, Thomas SA, Lightman SL, Pariante CM (2011). Desipramine treatment has minimal effects on the brain accumulation of glucocorticoids in P‐gp‐deficient and wild‐type mice. Psychoneuroendocrinology 36: 1351–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melas PA, Rogdaki M, Lennartsson A, Bjork K, Qi H, Witasp A, et al. (2012). Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychopharmacol 15: 669–679. [DOI] [PubMed] [Google Scholar]
- Miller DS (2010). Regulation of P‐glycoprotein and other ABC drug transporters at the blood‐brain barrier. Trends Pharmacol Sci 31: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinobu S, Strausbaugh H, Terwilliger R, Duman RS (1997). Regulation of c‐Fos and NGF1‐A by antidepressant treatments. Synapse 25: 313–320. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15: 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS (1996). Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16: 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien FE, Clarke G, Fitzgerald P, Dinan TG, Griffin BT, Cryan JF (2012a). Inhibition of P‐glycoprotein enhances transport of imipramine across the blood‐brain barrier: microdialysis studies in conscious freely moving rats. Br J Pharmacol 166: 1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien FE, Dinan TG, Griffin BT, Cryan JF (2012b). Interactions between antidepressants and P‐glycoprotein at the blood‐brain barrier: clinical significance of in vitro and in vivo findings. Br J Pharmacol 165: 289–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien FE, Clarke G, Dinan TG, Cryan JF, Griffin BT (2013a). Human P‐glycoprotein differentially affects antidepressant drug transport: relevance to blood‐brain barrier permeability. Int J Neuropsychopharmacol 16: 2259–2272. [DOI] [PubMed] [Google Scholar]
- O'Brien FE, O'Connor RM, Clarke G, Dinan TG, Griffin BT, Cryan JF (2013b). P‐glycoprotein inhibition increases the brain distribution and antidepressant‐like activity of escitalopram in rodents. Neuropsychopharmacol 38: 2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary OF, Cryan JF (2013). Towards translational rodent models of depression. Cell Tissue Res 354: 141–153. [DOI] [PubMed] [Google Scholar]
- O'Leary OF, Dinan TG, Cryan JF (2014). Faster, better, stronger: towards new antidepressant therapeutic strategies. Eur J Pharmacol 753: 32–50. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Hye A, Williamson R, Makoff A, Lovestone S, Kerwin RW (2003). The antidepressant clomipramine regulates cortisol intracellular concentrations and glucocorticoid receptor expression in fibroblasts and rat primary neurones. Neuropsychopharmacol 28: 1553–1561. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Veilleux S, Barden N (1991). Antidepressant and other centrally acting drugs regulate glucocorticoid receptor messenger RNA levels in rat brain. Psychoneuroendocrinology 16: 505–515. [DOI] [PubMed] [Google Scholar]
- Regenthal R, Krueger M, Koeppel C, Preiss R (1999). Drug levels: therapeutic and toxic serum/plasma concentrations of common drugs. J Clin Monit Comput 15: 529–544. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Fink G (1992). Antidepressants increase glucocorticoid and mineralocorticoid receptor mRNA expression in rat hippocampus in vivo. Neuroendocrinology 55: 621–626. [DOI] [PubMed] [Google Scholar]
- Shishkina GT, Kalinina TS, Dygalo NN (2007). Up‐regulation of tryptophan hydroxylase‐2 mRNA in the rat brain by chronic fluoxetine treatment correlates with its antidepressant effect. Neuroscience 150: 404–412. [DOI] [PubMed] [Google Scholar]
- Sillaber I, Panhuysen M, Henniger MS, Ohl F, Kuhne C, Putz B, et al. (2008). Profiling of behavioral changes and hippocampal gene expression in mice chronically treated with the SSRI paroxetine. Psychopharmacology 200: 557–572. [DOI] [PubMed] [Google Scholar]
- Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, et al. (2006). Alterations in 5‐HT1B receptor function by p11 in depression‐like states. Science 311: 77–80. [DOI] [PubMed] [Google Scholar]
- Tardito D, Perez J, Tiraboschi E, Musazzi L, Racagni G, Popoli M (2006). Signaling pathways regulating gene expression, neuroplasticity, and neurotrophic mechanisms in the action of antidepressants: a critical overview. Pharmacol Rev 58: 115–134. [DOI] [PubMed] [Google Scholar]
- Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, et al. (2000). cAMP response element‐mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci 20: 4030–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhr M, Tontsch A, Namendorf C, Ripke S, Lucae S, Ising M, et al. (2008). Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron 57: 203–209. [DOI] [PubMed] [Google Scholar]
- Ukai M, Maeda H, Nanya Y, Kameyama T, Matsuno K (1998). Beneficial effects of acute and repeated administrations of sigma receptor agonists on behavioral despair in mice exposed to tail suspension. Pharmacol Biochem Behav 61: 247–252. [DOI] [PubMed] [Google Scholar]
- Weiss J, Dormann G, Martin‐Facklam M, Kerpen CJ, Ketabi‐Kiyanvash N, Haefeli WE (2003). Inhibition of P‐glycoprotein by newer antidepressants. J Pharmacol Exp Ther 305: 197–204. [DOI] [PubMed] [Google Scholar]