

Abstract
The benefits of novel oral anticoagulants are hampered by bleeding. Since coagulation factor IX (fIX) lies upstream of fX in the coagulation cascade, and intermediate levels have been associated with reduced incidence of thrombotic events, we evaluated the viability of fIXa as an antithrombotic target. We applied translational pharmacokinetics/pharmacodynamics (PK/PD) principles to predict the therapeutic window (TW) associated with a selective small molecule inhibitor (SMi) of fIXa, compound 1 (CPD1, rat fIXa inhibition constant (Ki, 21 nmol/L) relative to clinically relevant exposures of apixaban (rat fXa Ki 4.3 nmol/L). Concentrations encompassing the minimal clinical plasma concentration (C min) of the 5 mg twice daily (BID) dose of apixaban were tested in rat arteriovenous shunt (AVS/thrombosis) and cuticle bleeding time (CBT) models. An I max and a linear model were used to fit clot weight (CW) and CBT. The following differences in biology were observed: (1) antithrombotic activity and bleeding increased in parallel for apixaban, but to a lesser extent for CPD1 and (2) antithrombotic activity occurred at high (>99%) enzyme occupancy (EO) for fXa or moderate (>65% EO) for fIXa. translational PK/PD analysis indicated that noninferiority was observed for concentrations of CPD1 that provided between 86% and 96% EO and that superior TW existed between 86% and 90% EO. These findings were confirmed in a study comparing short interfering (si)RNA‐mediated knockdown (KD) modulation of fIX and fX mRNA. In summary, using principles of translational biology to relate preclinical markers of efficacy and safety to clinical doses of apixaban, we found that modulation of fIXa can be superior to apixaban.
Keywords: anticoagulation, factor IX, factor IXa, stroke prevention in atrial fibrillation, thrombosis, translational PK/PD.
Abbreviations
- aPTT
activated partial thromboplastin time
- AVS
arteriovenous shunt
- BID
twice daily dose
- bp
base pair
- Cavg
average plasma concentration
- CBT
cuticle bleeding time
- CD‐IGS
CDR rats bred using the International Genetic Standardization Program
- Cmax
maximal plasma concentration
- Cmin
minimum plasma concentration
- CPD1
compound 1
- CW
clot weight
- DSPC
1,2‐distearoyl‐sn‐glycero‐3‐phosphocholine
- Emax
maximum effect
- EO
enzyme occupancy
- fIX
coagulation factor IX
- fX
coagulation factor X
- Fu
fraction unbound
- hCG
human chorionic gonadotropin
- hrfXIa
recombinant human fXIa
- Imax
maximum inhibitory effect
- Ki
inhibition constant
- LNP
lipid nanoparticle
- KD
knockdown
- mRNA
messenger ribonucleic acid
- PEG
polyethylene glycol
- PEG 2000‐DMG
1‐monomethoxy polyethyleneglycol 2000‐2,3‐dimyristoylglycerol
- PK/PD
pharmacokinetics/pharmacodynamics
- PMSG
pregnant mares’ serum gonadotrophin
- PT
prothrombin time
- RSE
relative standard error
- siRNA
short interfering RNA
- SMi
small molecule inhibitor
- TE
target engagement
- TF
tissue factor
- TGA
thrombin generation assay
- TW
therapeutic window
- WT
wild type
- ZFN
zinc finger nuclease
Introduction
Novel oral anticoagulants blocking the final common pathway of coagulation (dabigatran etexilate targeting fIIa and rivaroxaban–apixaban both targeting fXa) have been approved for the prevention of venous thromboembolism in patients undergoing elective knee or hip replacement (see Rachidi et al. 2013, for review) and for the prevention of stroke and systemic embolism in patients with atrial fibrillation (SPAF, for review see Albert 2014). The substantial benefits are unfortunately accompanied by a high annual incidence of major and nonmajor clinically relevant bleeding (~15% for atrial fibrillation patients) (Connolly et al. 2009; Granger et al. 2011; Patel et al. 2011). There is hope, however, that inhibition of other coagulation targets from the intrinsic pathway such as coagulation factor IXa (fIXa) could provide an improved therapeutic window (TW) relative to fIIa and fXa inhibitors. In one example, it has been reported that approximately 99–50% reduction in levels of fIX (as observed in moderate, mild hemophilia B patients and carriers of hemophilia B) are associated with a reduced risk of thrombosis (Sramek et al. 2003; Darby et al. 2007) for acceptable levels of bleeding. The relative safety of such strategy is reinforced by the fact that severe and spontaneous bleeding appear to be associated with <1% residual activity, an observation recently confirmed in a gene therapy study in severe hemophilia B patients whose bleeding episodes were inhibited by expressing 1–6% of the normal level of fIX (Nathwani et al. 2014). Lower bleeding risks by targeting fIXa are expected from the phenotype of fIX knockout mice in comparison with fX‐deficient mice as deficiency of fX causes partial embryonic and fatal neonatal bleeding (Dewerchin et al. 2000), while fIX‐deficient mice are viable with a high rate of survival at weaning (Lin et al. 1997; Wang et al. 1997). We have further confirmed this hypothesis using a short interfering RNA (siRNA) approach in the rat in which a ~50% reduction in fIX plasma activity provided protection from thrombosis with no prolongation of the bleeding time (Metzger et al. 2015).
Stroke and major clinical bleeding are relatively rare events, therefore, assessment of the TW of novel oral anticoagulants for indications such as SPAF can really only be done in the context of a large phase III trial. This presents considerable risk to the development of drugs against novel targets. Translational pharmacology principles can be leveraged to interrogate novel targets by using quantitative approaches to translate target engagement and efficacy/safety between animals and humans. In order to mitigate this risk, we have employed concepts of translational pharmacology to assess TW associated with modulation of fIXa relative to that of fXa. Because modulation of fXa can be linked to clinical outcomes for both efficacy and bleeding, we have proposed to predict the clinical TW for fIXa inhibitors through preclinical comparison to fXa inhibitors at enzyme occupancy (EO) ranges associated with known clinical efficacy and bleeding rates. In the present work, we aimed at defining the TW associated with modulation of fIXa relative to that of fXa using a head‐to‐head, blinded comparative study using a rat arteriovenous shunt (AVS) and cuticle bleeding time (CBT) model of anticoagulation (the relevance of the rat species and experimental models is presented in the Discussion section). Apixaban was selected as the representative fXa inhibitor for the SPAF indication, achieving statistically significant reduction in stroke and bleeding relative to warfarin at a single dose level (Granger et al. 2011).
A model‐based pharmacokinetics/pharmacodynamics (PK/PD) analysis quantitatively evaluated the range of fIXa occupancies associated with an equivalent or improved TW relative to apixaban. The range of plasma concentrations which resulted in calculated fXa occupancy equivalent to that achieved in humans at the the 5 mg twice daily dose (BID) approved dose of apixaban for SPAF were determined. PK/PD analysis was used to relate clot weight (CW) and CBT to plasma concentrations (kept constant during the duration of the assay). The range of CW and CBT in the AVS/CBT assays associated with the clinically relevant rat plasma concentrations of apixaban were set as targets against which to compare AVS/CBT results using a fIXa small molecule inhibitor (SMi), compound 1 (CPD1). Findings from the apixaban and CPD1 study were compared with those from a comparably designed rat AVS/CBT study in which a siRNA strategy was used to knockdown (KD) fIX or fX mRNA. Results indicate that modulation of fIXa inhibits thrombosis and may represent a novel, promising antithrombotic strategy.
Materials and Methods
FIXa and fXa inhibitors
The optimization to identify selective small molecule inhibitors of fIXa has been described previously (submitted to Bioorganic and Medicinal Chemistry Letters, manuscript number: BMCL‐D‐15‐00383). Preparation of pyrazolo‐pyridine benzamide analog (CPD1) began with acylation of the enolate derived from 2 with acyl chloride 3 followed by pyrazole formation to give 4 by treatment of the intermediate with t‐butyl hydrazine hydrochloride (Fig. 1). Refluxing pyrazole 4 in hydrochloric acid not only effects the removal of both the boc and t‐butyl groups, but also promotes a sequence of furan hydrolysis and subsequent cyclization onto the pyrazole resulting in pyrazolopyridine core 5. Amide coupling with an acid provided CPD1. Apixaban (Fig. 1) was prepared using methods described in the literature (Jian'an and Yafei 2013). The key properties of the compounds are listed in Table 1.
Figure 1.
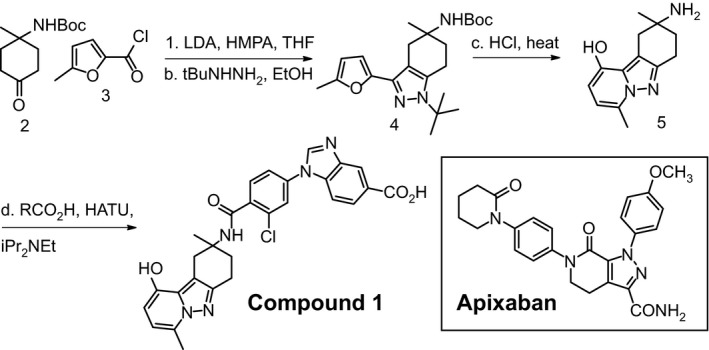
Small molecule inhibitors of fIXa and fXa. (A) Preparation of tricyclic pyrazolo‐pyridine analogs. Reagents and conditions: (a) lithium di‐isopropylamide, hexamethylphosphoramide, tetrahydrofuran, −78°C; (b) t‐butyl hydrazine hydrochloride, ethanol, room temperature; (c) 37% hydrochloric acid, 125°C, sealed tube, 30 min; (d) 2‐chloro‐4‐(5‐(methoxycarbonyl)‐1H‐benzo[d]imidazol‐1‐yl)benzoic acid, O‐(7‐azabenzotriazole‐1‐yl)‐1,1,3,3‐tetramethyluronium hexafluorophosphate, di‐isopropyl ethyl amine, dimethyl formamide, room temperature then sodium hydroxide. (B) Structure of apixaban.
Table 1.
Characteristics of CPD1 and apixaban
| Property | CPD1 | Apixaban |
|---|---|---|
| Human, rat fIXa Ki (nmol/L) | 3.46 ± 0.95, 22 | >30,000, >30,000 |
| Human, rat fXa Ki (nmol/L) | 489 ± 93, >30000 | 0.13 ± 0.025, 2.1 ± 0.39 |
| % unbound (human, rat) | 0.02, 0.6 | 9.3 ± 1.3, 1.9 ± 0.2 |
| Rat Cl (mL/min.kg), Vd (L/kg), T1/2 (h) | 30.4, 0.19, 0.38 | 12, 0.6, 0.86 |
Data are expressed as mean ± standard deviation. CPD1, compound 1; fX, coagulation factor X; Ki, inhibition constant; Cl, clearance; Vd, volume of distribution; T1/2, half‐life.
AVS and CBT models
All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health and the guidelines of Merck Animal Care and Use Committee. Rat protocols of AVS and CBT were based on previously published procedures (Schumacher et al. 2010; Metzger et al. 2015) and performed in the same animal. Male Sprague Dawley rats (350–400 g, Charles River Laboratories, Wilmington, MA, USA) were anesthetized with thiobutabarbital. After a 5‐min stabilization period, compounds or vehicle were administered by an initial intravenous bolus injection (1 mL/kg) followed by an infusion (3 mL kg−1 h−1) that continued for the duration of the experiment. The bolus dose and rate of infusion were selected such that plasma concentrations would be nearly constant during the AVS and CBT assessments, given the rat clearance of CPD1 and apixaban. The dosing regimen was established during a preliminary pharmacokinetics study. The ratio of bolus to infusion dose was deemed appropriate when study exposures across the test interval did not deviate more than 10% from the mean across the measured period (10, 15, 20, 25, and 30 min, data not shown). This preliminary study ensured that constant plasma concentrations would be achieved throughout the duration of the CW and CBT assays and that a single concentration could be paired with each CW and CBT measure. Fifteen minutes after the start of compound administration, an extracorporeal shunt (Tygon tubing containing a silk thread) was connected to carotid artery and jugular vein cannulas and blood allowed to flow through the shunt for a period of 15 min. The weight of the thread at the end of the experiment was recorded and CW calculated via subtracting the weight of the thread prior to the circulation of blood. Five minutes after shunt placement, two cuticles on hind paw toes were cut (where the quick meets the nail) and the paw immersed in 37° lactated ringers solution. CBT was recorded as time for cessation of bleeding up to a maximum of 10 min. In the AVS/CBT study, five doses, with a minimum of n = 10 per group, were evaluated for each compound. To maintain steady‐state exposure throughout the duration of the AVS study period, compounds were dosed using a bolus plus infusion regimen as follows (first value is the bolus dose/second value is the infusion dose): apixaban at 0.1/0.12 mg/kg (A1), 0.3/0.375 mg/kg (A2), 1/1.25 mg/kg (A3), 3/3.75 mg/kg (A4), and 8/10.2 mg/kg (A5); CPD1 at 0.06/0.09 mg/kg (C1), 0.2/0.3 mg/kg (C2), 0.6/0.9 mg/kg (C3), 2/3 mg/kg (C4), and 6.5/9.8 mg/kg (C5). A terminal blood sample was taken in order to determine final plasma concentration, evaluate activity of the compounds on prothrombin time (PT), activated partial thromboplastin time (aPTT), and on fIIa formation (thrombin generation assay, TGA), and relate efficacy and pharmacodynamic activity to the levels of EO. Vehicle control was 35% (w/v) 2‐hydroxypropyl‐β‐cyclodextrin in 10 mmol/L phosphate buffer. The study was conducted in a blinded head‐to‐head fashion.
Model‐based translational PK/PD analysis
A model‐based PK/PD analysis was performed to quantitatively assess the TW of fIXa modulation relative to fXa. The median clinical exposures of apixaban associated with the approved 5 mg BID dose for SPAF were obtained from literature (Leil et al. 2010) and converted to an equivalent range of clinically relevant plasma concentrations in rats. The assumption was made that equivalent fXa occupancies in humans and rats would yield comparable efficacy/safety profiles between the two species. EO (%) was calculated using: EO = (100 × C) / (Ki / fu + C), where Ki is the inhibition constant, fu is the fraction unbound in plasma, and C is the plasma concentration of inhibitor. The fu values were determined via equilibrium dialysis and Ki was measured as described previously. In order to assess the assay‐related uncertainty in the calculation of the rat plasma concentration equivalent to the clinical plasma concentrations of apixaban, Ki and fu were measured in triplicates. Simulations, sampling from the standard error in Ki and fu measurements, were performed to estimate the median and 90% confide‐nce intervals (CIs) of the clinically relevant range of rat plasma concentrations. PK/PD models were fit to the rat CW and CBT data for apixaban and CPD1 using MATLAB R2013a and Monolix. Linear and sigmoidal I max model structures were explored. Final models were selected based on the assessment of goodness‐of‐fit plots, % relative standard error (%RSE) of parameter estimates, and minimum residual variability. The standard error of the model parameter estimates was used to simulate the median and 5th and 95th percentiles of CW and CBT as a function of apixaban or CPD1 rat plasma concentration. The range of CW and CBT in the rat AVS and CBT models associated with the clinically relevant range of rat apixaban concentrations were determined from the model and used as targets against which to compare CPD1 results. The range of CPD1 rat plasma concentrations, and calculated % EO that achieved equivalent, or better, CW and CBT relative to the apixaban targets was assessed.
FIX and fX siRNA lead selection and in vivo qualification
SiRNA lead identification was done as described previously (Metzger et al. 2015). In brief, chemically modified siRNAs were designed against rat fIX (NM 031540) and fX (NM_0143) and synthesized at Merck's oligosynthesis facility (Rahway, NJ, USA). siRNA in vitro screening was done using luciferase reporter constructs derived from psiCHECK2 vector (Promega, Cat# C8021, Madison, WI, USA). The list of lead siRNA sequences for fIX and fX is shown in Table S1 (all in the 5′–3′ direction).
For in vivo studies, siRNAs were encapsulated in lipid nanoparticles (LNPs) as described previously (Chen et al. 2015). In vivo siRNA qualification was done in 8 weeks old male Sprague Dawley rats weighing approximately 144–170 g purchased from Charles River Laboratories. LNP‐encapsulated siRNAs were administered via tail vein. Animals were sacrificed on day 7 postdosing and blood samples were collected. Liver punches were collected for mRNA silencing analysis. RNA isolation and TaqMan reverse transcription polymerase chain reaction (RT‐PCR) analysis were done as described previously (Tadin‐Strapps et al. 2011; Olearczyk et al. 2014). All Taqman probe sets were from Applied Biosystems (Foster City, CA, USA). LNP‐encapsulated siRNAs were administered intravenously to male Sprague Dawley rats weighing approximately 300 g, at a volume of 2 mL/kg. fIX and fX LNP‐siRNA were diluted using 10 mmol/L Tris, 70 mmol/L NaCl, 5 wt% sucrose, pH 7.5 (Thermo Fisher Scientific, Pittsburgh, PA, USA) buffer to achieve doses equivalent to 0.01, 0.03, 0.1, 0.3, and 1 mg/kg (fX siRNA) and 0.003, 0.01, 0.03, 0.1, 0.3, and 0.3 mg/kg (fIX siRNA). A nontargeting control siRNA (nt) was administered at the equivalent of 0.6 or 1 mg/kg to match the high doses of fIX LNP and fX LNP, respectively. All animals weighed 325–400 g at the time of AVS/CBT procedures (7 days after the administration of siRNA).
Endogenous fIXa and fXa enzyme assay
The determination of rat plasma fIX was adapted from Biophen fIX Assay Kit (HYPHEN BioMed, Neuville‐Sur‐Oise, France) as described previously (Metzger et al. 2015). In brief, 25 μL diluted plasma (300X dilution in BSA‐Tris buffer) was incubated with 20 μL activation reagent containing purified human fXIa (Haematologic Technologies Inc., Essex Junction, VT, USA) for 10 min at 37°C, followed by the addition of 50 μL of a mixture containing human fX zymogen and activated fVIII, calcium, synthetic phospholipids, and a fXa‐specific substrate SXa‐11. The plasma fIX was measured based on the release of p‐nitroaniline monitored at 405 nm in a kinetic mode in a SpectraMax 96‐well plate reader (Molecular Devices, Sunnyvale, CA, USA) from SXa‐11 using purified human fIXa as a calibrator. The determination of rat plasma fX was adapted from the Biophen fX Kit (Cat # A221705; Aniara, West Chester, OH, USA). Briefly, plasma samples were diluted 100X with manufacturer‐supplied buffer and mixed with Russell's viper venom (1:1) for 3 min at 37°C, followed by addition of one volume of substrate (R1). The plate was read in a SpectraMax Plus384 (Molecular Devices) in a kinetic mode, that is, the time course of fXa enzymatic reaction was followed and the initial slope of the time course was taken as a measure of the initial velocity of fXa (directly proportional to the amount of fXa present in the plasma). The method was fully validated for rat plasma samples using fXa inhibitors (e.g., apixaban, rivaroxaban) and all fX zymogen were completely converted to fXa with the protocol (data not shown).
aPTT, PT measurement, and TGA
Sodium citrate (11 mmol/L final) blood samples were centrifuged at 2500g at 4°C for 15 min to generate plasma. Both aPTT and PT were determined by standard methods using TriniCLOT aPTT S (Tcoag, Bray, Ireland) and TriniCLOT PT Excel (Tcoag) on a KC4 Delta coagulation analyzer (Tcoag) (Metzger et al. 2015). Ellagic acid was used as a trigger for aPTT, while 100 μL tissue factor (TF) + CaCl2 mix was used to trigger measure of PT. Thrombinoscope (Diagnostica Stago, Parsippany, NJ, USA) was according to the manufacturer's instructions. Plasma (60 μL) was incubated with 15 μL recombinant human fXIa (rhfXIa, 1, 3, 10 pmol/L) or TF (1, 5, 20 pmol/L, Diagnostica Stago, Parsippany, NJ, USA) at 37°C for 5 min. This was followed by automatic injection of 15 μL of FluCa buffer to initiate fIIa. Peak thrombin generation was recorded.
Compliance with design and statistical analysis requirements
Groups have been designed to have at least five per group. There are no exclusion criteria, nor did we use duplicate or triplicate data. Investigators performing the efficacy (thrombosis) and bleeding (CBT) assays were blinded to the treatment or siRNA type.
Results
Rat small molecule inhibitor AVS/CBT studies
Pilot studies were conducted to establish the dosing regimen required to achieve steady‐state plasma concentrations CPD1 and apixaban (data not shown). Accordingly, a range of CPD1 dosing regimens were selected which achieved total concentrations of 0.28–153 μmol/L, which corresponded to a calculated fIXa EO range of 7–98%. Apixaban dosing regimens were selected to achieve steady‐state plasma concentrations ranging from 0.39 nmol/L to 57 μmol/L, which corresponded to a calculated fXa EO 78% to ≥99%. The observed median clinical trough concentration (C min) for apixaban at the 5 mg BID dose approved for SPAF reported in the literature was 107 ng/mL (0.2 μmol/L) (Leil et al. 2010). Accounting for uncertainty in measured rat and human free fraction and enzyme affinity, the equivalent plasma concentration (90% CI) in rats was predicted to be 17 (8.5, 32) μmol/L. Although Leil et al. (2010) reported that C min was more closely related to efficacy (albeit in prevention of venous thromboembolism rather than stroke), and C avg was more closely associated with bleeding risk, only the rat equivalent to clinical C min was considered in this analysis. We found that the interassay variability for Ki and fu when translated from human to rat led to a predicted rat equivalent of C avg (24 μmol/L) within the 90% CI around C min.
Apixaban induced a concentration‐dependent increase in aPTT and PT (Fig. 2A. In contrast, CPD1 only showed an increase in aPTT, as expected for a selective fIXa inhibitor (Fig. 2A), indicating continued selectivity for the intrinsic pathway in the rat species. Dose/concentration‐dependent activity and compound specificity were also confirmed in the TGA assay (Fig. 2B) with CPD1 demonstrating TF concentration‐dependent inhibitory activity, while apixaban activity was constant across the TF concentration tested.
Figure 2.
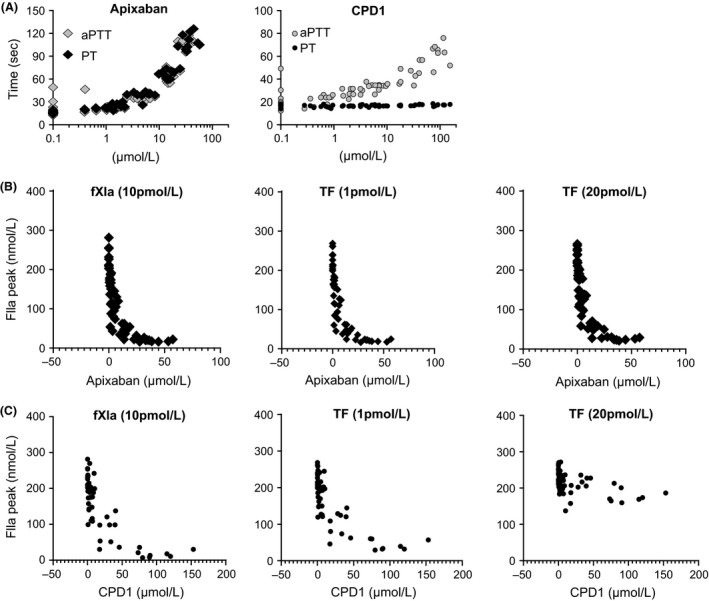
Ex vivo pharmacodynamic activities of CPD1 and Apixaban Blood was collected post‐AVS study and plasma prepared as described in materials and methods. One platelet poor plasma (PPP) sample was utilized to monitor pharmacodynamic activities, the other to determine plasma concentration of the two compounds. A), effects of apixaban and CPD1 on aPTT and PT. B), apixaban and CPD1 effects on aPTT and PT as a function of concentration. C), apixaban and CPD1 effects on thrombin generation in TGA as a function of concentration. Data showed that apixaban inhibited both intrinsic and extrinsic pathways of coagulation, while CPD1 displayed activities in line with known fIXa biology. Apixaban, A1) 0.1 mg/kg bolus followed by 0.12 mg/kg infusion; A2), 0.3 mg/kg followed by 0.375 mg/kg; A3) 1 mg/kg followed by 1.25 mg/kg; A4) 3 mg/kg followed by 3.75 mg/kg; A5) 8 mg/kg followed by 10.2 mg/kg. CPD1, C1) 0.06 mg/kg bolus followed by 0.09 mg/kg infusion; C2) 0.2 mg/kg followed by 0.3 mg/kg; C3) 0.6 mg/kg followed by 0.9 mg/kg; C4) 2 mg/kg followed by 3 mg/kg; C5) 6.5 mg/kg followed by 9.8 mg/kg.
Both apixaban and CPD1 demonstrated dose‐dependent antithrombotic activity with the two highest doses of apixaban (A4, A5, Fig. 3A, top panel) and the three highest doses of CPD1 (C3–C5, Fig. 3A, bottom panel) providing significant CW reduction. The two effective doses of apixaban were associated with significant prolongation of the CBT, whereas only the maximal dose tested for CPD1 led to a significant increase in bleeding. Interestingly, the C3 dose of CPD1 conferred protection from thrombosis with no impairment of the hemostatic function (Fig. 3B, bottom panel), while the C4 dose displayed highly significant protection from thrombosis at a limited cost on bleeding. Figure 3C shows that for a given, effective (antithrombotic) dose, apixaban treatment was associated with prolonged CBT when compared with CPD1.
Figure 3.
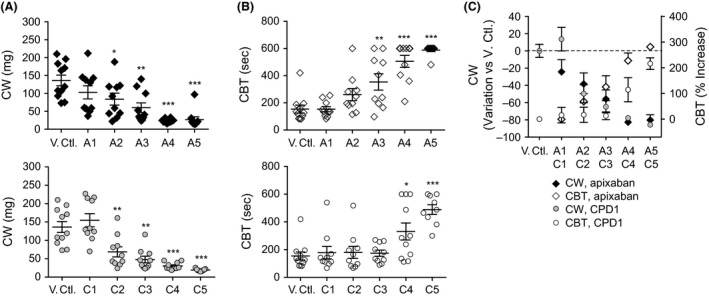
Apixaban and CPD1 effects on thrombosis (AVS) and bleeding (CBT) in the rats. (A) Apixaban and (B) CPD1 effects on thrombosis and primary hemostasis. (C) Separation of bleeding curves for apixaban and CPD1 at equivalent antithrombotic activity. *P < 0.05, **P < 0.01, ***P < 0.0001 versus vehicle control. B, bolus; I, infusion; Apix, apixaban; Apixaban (A1) 0.1 mg/kg bolus followed by 0.12 mg/kg infusion; (A2), 0.3 mg/kg followed by 0.375 mg/kg; (A3) 1 mg/kg followed by 1.25 mg/kg; (A4) 3 mg/kg followed by 3.75 mg/kg; (A5) 8 mg/kg followed by 10.2 mg/kg. CPD1; (C1) 0.06 mg/kg bolus followed by 0.09 mg/kg infusion; (C2) 0.2 mg/kg followed by 0.3 mg/kg; (C3) 0.6 mg/kg followed by 0.9 mg/kg; (C4) 2 mg/kg followed by 3 mg/kg; (C5) 6.5 mg/kg followed by 9.8 mg/kg. CPD1, compound 1; AVS, arteriovenous shunt; CBT, cuticle bleeding time.
We next calculated EO based on the plasma concentrations of CPD1 and apixaban achieved in each animal. The use of apixaban and CPD1 highlighted differences in biology as >99% fXa EO was required to ensure CW <50 mg in all animals, while only >65% EO was required for fIXa (Fig. 4A top and bottom left panels, respectively). Bleeding effects paralleled antithrombotic activity for apixaban (Fig. 4A, top right panel). In contrast, CPD1 was only associated with prolonged CBT at fIXa occupancies >85% (Fig. 4, bottom right panel). Figure 4B shows the CW plotted against CBT for each animal. Although the distribution pattern for the two targets showed considerable overlap, 17 animals treated with the higher doses of apixaban reached the limit of the CBT assay (600 sec), while only five did for CPD1 treatment. This suggested that the upper limit of CBT prevented the ability to fully differentiate between apixaban and CPD1. The apparent right censoring of these data was accounted for in the model‐based PK/PD analysis of these data as described below.
Figure 4.
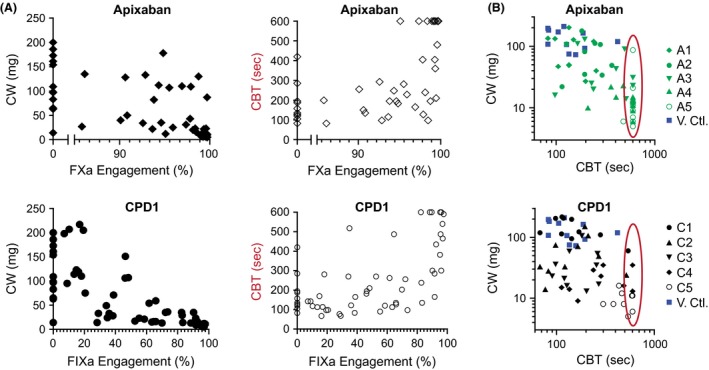
Clot weight and bleeding time as a function of target engagement (EO). (A) High (fXa, apixaban) and moderate (fIXa, CPD1) engagement are required to observe strong antithrombotic activity while >90% EO with both compounds is accompanied by increased bleeding. (B) Clot weight as a function of cuticle bleeding time. The orange ovale focus on animals which reach the upper limit of the CBT measurement. Each point represents measurement for one animal. Enzyme occupancy (EO) was calculated according to the following equation % Occupancy = (100 × [C]) / (Ki / fu + [C]).
Finally, in order to assess the relevance of our pharmacodynamic assays as markers of antithrombotic activity, peak generation of thrombin, aPTT and PT were plotted versus CW (Fig. S1). Decreased CW was correlated with aPTT for both apixaban and CPD1, while decreased CW correlated with PT only for apixaban (Fig. S1A), confirming the anticoagulant profile associated with fIXa and fXa inhibition. Data showed that anticoagulant activity in TGA stimulated with hrfXIa was associated with antithrombotic activity for both inhibitors (Fig. S2). Low TF, but not high TF stimulation, was correlated with antithrombotic activity for CPD1.
Model‐based translational PK/PD analysis
While there was an apparent difference in efficacy and bleeding data by dose of CPD1 and apixaban, we wanted to firmly predict the TW of fIXa relative to fXa inhibitors. To do so, we performed a model‐based PK/PD analysis of the CW and CBT data. A vehicle‐proportional I max relationship of CW with plasma concentration provided the best fit for both apixaban and CPD1: CW = CWvehicle × (1 − I max × C/IC50 + C), where CWvehicle was the clot weight (mg) associated with vehicle‐treated animals, C was the plasma concentration of either apixaban or CPD1, I max was the maximal inhibition of CW, and IC50 was the plasma concentration corresponding to half of the maximal response. A linear relationship with concentration was found to provide the best fit to the CBT data for apixaban and CPD1: CBT = CBTvehicle + m × C, where CBTvehicle was the bleeding time associated with vehicle‐treated animals, m was the slope of the drug effect on bleeding time, and C was the plasma concentration of either apixaban or CPD1. The CBT data appeared to be right censored due to the large number of animals reaching the upper limit of the assay (600 sec) in the apixaban‐treated animals, above which blood loss levels were believed to affect the integrity of the data. Therefore, the data were fit using the likelihood that the predicted data were >600 sec rather than using the likelihood of the data being equal to 600 sec. Parameter estimates for CPD1 and apixaban for the CW and bleeding models can be found in Table 2. Model residuals were found to be approximately normally distributed around zero, and showed no bias with either concentration or predicted CW or CBT. One thousand simulations of CW and CBT versus apixaban or CPD1 concentrations were conducted (Fig. 5). Models showed no evidence of bias from predicted simulation plots. The target CW and CBT corresponding to the apixaban clinical C min (Fig. 5) were 16 (12–24) mg and just above the upper limit of 600 (400 to >600) sec, respectively. The concentrations of CPD1 resulting in <16 mg CW was >23 μmol/L (>86% EO), and in a <600 sec CBT was <80 μmol/L (<96% EO). Occupancy of fIXa between ~86% and ~96% would therefore be expected to result in an equivalent benefit/risk profile relative to a fXa inhibitor. Uncertainty in the model fits was used to assess the upper end of fIXa occupancy which would be expected to achieve reduced bleeding relative to apixaban (i.e., <460 sec, corresponding to a CPD1 concentration of 35 μmol/L or 96% EO, see Fig. 5). Therefore, the window of improved benefit/risk for a fIXa inhibitor (equivalent CW, reduced BT) corresponded to fIXa occupancies of ~86% to ~90%.
Table 2.
Parameter estimates for CW and CBT models for apixaban and CPD1
| Model | Parameter | Estimate | %RSE |
|---|---|---|---|
| Apixaban CW | CWvehicle (mg) | 99.5 | 19 |
| I max | 0.946 | 4 | |
| IC50 (μmol/L) | 1.92 | 53 | |
| Apixaban CBT | CBTvehicle (sec) | 144 | 13 |
| Slope | 29.6 | 26 | |
| CPD1 CW | CWvehicle (mg) | 121 | 17 |
| I max | 0.936 | 2 | |
| IC50 (μmol/L) | 1.69 | 17 | |
| CPD1 CBT | CBTvehicle (sec) | 122 | 15 |
| Slope | 5.93 | 34 |
CW, clot weight; CBT, cuticle bleeding time; CPD1, compound 1; %RSE, % relative standard error; Slope, slope of the drug effect on bleeding time.
Figure 5.
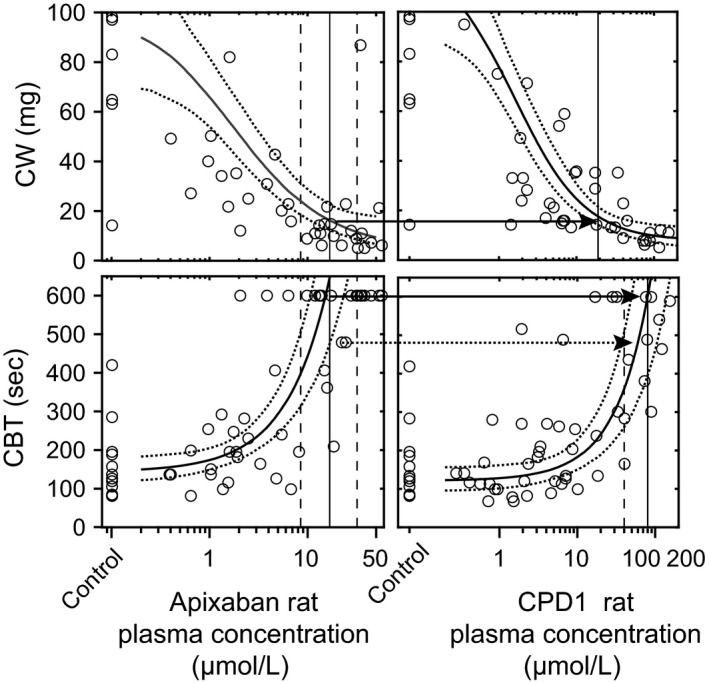
Transational pharmacokinetics/pharmacodynamics (PK/PD) analysis of the efficacy/bleeding study. PK/PD model fits (solid line median, dotted lines are 5th and 95th percentiles incorporating uncertainty of parameter estimates) are overlaid with observed (circles) clot weight (top panels) and bleed time (bottom panels) as a function of apixaban (left panels) and compound 1 (CPD1) (right panels) rat plasma concentrations. The vertical lines on the apixaban figures represent the median (solid) and 90% CI (dotted) range of clinically relevant apixaban C min concentrations. The intersection of the median C min with median model fit were used to set target clot weight (CW) and target cuticle bleeding time (CBT) against which to estimate the upper and lower bounds of the therapeutic window for CPD1 (arrows show the target translated to CPD1). The vertical lines on the CPD1 figures show the rat plasma concentrations that are expected to meet the target CW and CBT. To estimate the CPD1 concentration required to achieve improved CBT relative to apixaban, the target CBT was set at the intersection of median apixaban C min and the 5th percentile of the CBT model; the CPD1 concentration for which the 95th percentile of the CBT model was below this target was then estimated (dotted vertical line, right bottom panel).
siRNA study
Ex vivo characterization and pharmacodynamic monitoring
FIX and fX mRNA levels were decreased by >95% and 97%, respectively (Fig. 6A), for the highest LNP‐siRNA doses. To ensure that the KD of fIX and fX did not affect other coagulation factors, we monitored changes in mRNA levels of fII, fV, fVII, fVIII, fIX, fX, fXI, and fXII. There were no significant changes in coagulation factors beyond those related to fIX and fX siRNA‐treated groups (Figs. S2 and S3) and both fIX and fX activities displayed linear relationship with mRNA KD (Fig. S4). Both aPTT and PT were assessed in all the animals. aPTT increased with increasing doses of fX siRNA to reach a 50% increase at the 1 mg/kg dose (Fig. 6B, top left panel), while a 2.5‐fold increase was noted with the highest dose of fIX siRNA (Fig. 6B, bottom left panel). PT changes for fX siRNAs were more important than their corresponding aPTT values and were dose dependent, reaching 250% increase at 1 mg/kg. There was no variation in PT associated with the administration of fIX siRNA.
Figure 6.
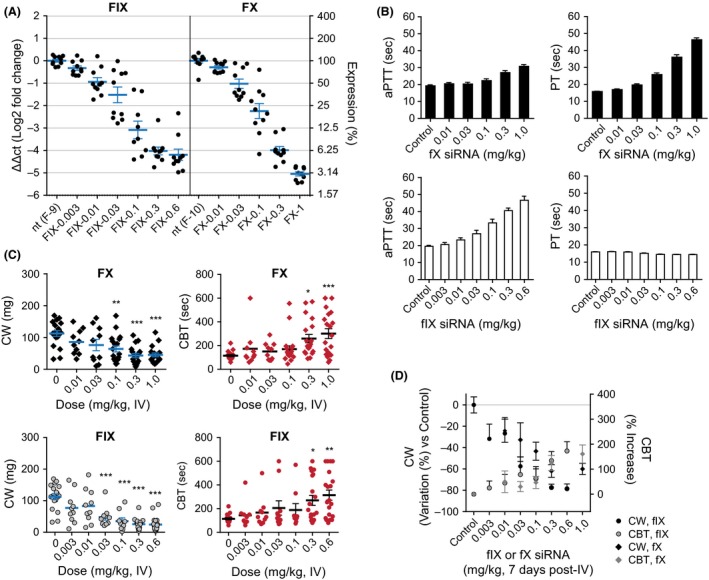
Effects of increasing doses of the fX and fIX lipid nanoparticle (LNP) on mRNA expression, measurement of activated partial thromboplastin time (aPTT) and prothrombin time (PT), clot weight, and bleeding. (A) mRNA expression was calculated relative to vehicle control. Individual animals and group means ± SEM are shown. Animals were dosed with a single dose and sacrificed at day 7 (post efficacy/bleeding study) for determination of hepatic fX and fIX mRNA levels. (B) fX siRNA‐dependent increase in aPTT and PT (top panel). fIX siRNA dose‐dependently increased aPTT but did not affect PT. (C) Effects of fX siRNA (upper panel) and fIX siRNA (lower panel), on CW and CBT showing that intermediate doses of fIX siRNA exist that provide significant inhibition of thrombosis at no cost on bleeding. (D) Separation of efficacy curve for fX and fIX siRNA for similar prolongation of CBT. *P < 0.05, **P < 0.01, ***P < 0.0001 versus 0 (control). FIX–0.003, fIX siRNA‐51797 dosed at 0.003 mg/kg. FIX–0.01, fIX siRNA‐51797 dosed at 0.01 mg/kg. FIX–0.03, fIX siRNA‐51797 dosed at 0.03 mg/kg. FIX–0.1, fIX siRNA‐51797 dosed at 0.1 mg/kg. FIX–0.3, fIX siRNA‐51797 dosed at 0.3 mg/kg. FIX–0.6, fIX siRNA‐51797 dosed at 0.6 mg/kg. FX–0.01, fX siRNA‐53334 dosed at 0.01 mg/kg. FX–0.03, fX siRNA‐53334 dosed at 0.03 mg/kg. FX–0.1, fX siRNA‐53334 dosed at 0.1 mg/kg. FX–0.3, fX siRNA‐53334 dosed at 0.3 mg/kg. FX–1, fX siRNA‐53334 dosed at 1 mg/kg. nt, nontargeting.
Rat siRNA efficacy and bleeding studies
Both fX and fIX siRNA significantly and dose‐dependently inhibited CW (Fig. 6C). Of note, while the two highest doses of fX siRNA procured >97% mRNA and >99% KD of fX activity, they did not confer the same levels of inhibition of CW than that associated with fIX siRNA (CW 43 ± 6.7 and 45 ± 5.8 g for 0.3 and 1 mg/kg fX siRNA vs. 25.5 ± 3.6 and 24.3 ± 5 g for 0.3 and 0.6 mg/kg fIX siRNA, respectively, P < 0.05 for both, Fig. 6C). On the other hand, the CBT prolongation was similar for the two highest fIX and fX siRNA doses (Fig. 6C). Thus, it appeared that greater levels of inhibition of thrombosis could be achieved with fIX siRNA at equivalent levels of bleeding. We then plotted CW and CBT data against KD of fIX and fX activity (Fig. 7) and evaluated the therapeutic index (CW vs. CBT) for each animal. Data indicated greater levels of inhibition of thrombosis with fIX siRNA at high KD levels (Fig. 7A, left panels), however there were no meaningful differences in bleeding profiles (Fig. 7B right panel, C).
Figure 7.
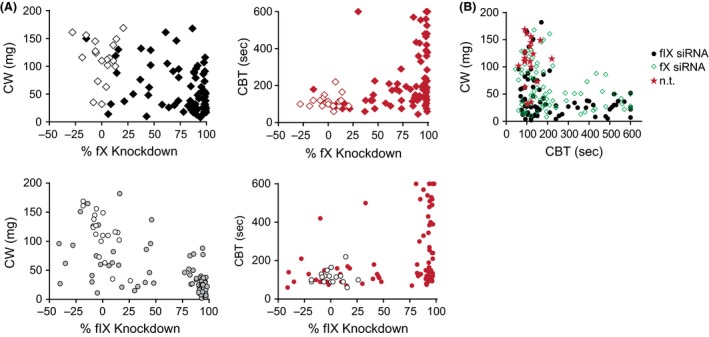
Expression of clot weight (CW) and bleeding (CBT) against mRNA knockdown (KD). (A) High fIX KD levels reach greater levels of inhibition of CW (left panel) than its fX counterpart, for similar bleeding profile. (B) CW expressed as a function of CBT. Data indicate similar L‐shape curve showing that severe inhibition of thrombosis is accompanied by increased bleeding for the two targets. n.t., nontargeting.
Discussion
With the results of this head‐to‐head blinded study we present preclinical evidence that modulation of fIXa (by the use of a selective small molecule antagonist of the catalytic domain) or of fIX (via mRNA KD) inhibited thrombosis. Furthermore, a model‐based translational PK/PD analysis of the TW associated with a SMi of fIXa and fXa demonstrated that a modulator of fIXa is predicted to be at least noninferior to apixaban in the EO range of 86–96% and superior (i.e., reduced bleeding at equivalent efficacy) to apixaban in the range of 86–90%. These findings reinforce the concept that fIXa is a gate keeping enzyme in thrombin generation and a critical player between initiation and propagation phases of coagulation (Butenas et al. 2004; Smiley and Becker 2014).
The relevance of these preclinical findings in predicting clinical outcomes in response to novel oral anticoagulants is dependent on the validity of three key assumptions: (1) the rat AVS/CBT model is a relevant and a sufficiently sensitive model of efficacy and safety to predict the TW for modulation of fIXa and fXa by CPD1 and apixaban, respectively; (2) for a given enzyme, equivalent calculated EO yields comparable efficacy/safety profiles across species; and (3) adequate selectivity for fIXa relative to fXa in our studies across the full range of EO or KD tested was achieved. On the basis of the calculation from concentrations reported in literature, we found that >50% inhibition of thrombosis was observed at >97% fXa EO in a venous and arterial FeCl2 thrombosis model and at >99% fXa EO in stasis and AVS thrombosis models in the rat, thus validating the choice of the model and animal species (Schumacher et al. 2010). These levels were consistent with the 99.2% calculated EO achieved at apixaban median C min (107 ng/mL) at the 5 mg BID SPAF dose. Furthermore, the ranges of fIXa EO associated with off‐scale bleeding in the SMi and siRNA studies were in agreement with reports of severe bleeding in hemophilia B patients with <1% of normal fIX levels, and with those of thromboprotection at intermediate levels. Finally, the third assumption was tested directly both by in vitro assessment of selectivity (CPD1 was >300‐fold selective over fXa in both human and rat plasma), and by use of pharmacodynamic biomarkers of the intrinsic and extrinsic pathways. CPD1, even at the highest doses tested, was not associated with PT prolongation nor did its TGA selectivity profile deviate from that expected for an inhibitor of fIXa. Similarly, apixaban activity and selectivity were conserved across the two species. In conclusion, we believe this evidence supports the validity of the rat AVS/CBT model to predict the clinical benefit/risk profile of fIXa modulation relative to fXa using calculated EO to translate between species.
The quantitative assessment of the TW of our fIXa SMi showed a relatively large range of EOs predicted to yield a similar benefit/risk profile relative to the 5 mg BID dose of apixaban, and a somewhat smaller range of fIXa EOs with the potential for reduced bleeding while achieving efficacy equivalent to apixaban. This is of particular significance as one of the main drawbacks of the fXa inhibitors in the clinic is their dose limitation due to bleeding risks (Albert 2014). Thus, it is possible that fIXa inhibitors could reach greater efficacy at equivalent bleeding risk, or similar efficacy with a safer profile.
While the data indicated noninferiority, and superiority over apixaban for precise set of target engagement, other findings were notable. First, some differences in mechanisms of action and thrombosis/efficacy profiles were observed between mRNA silencing of the zymogen and small molecule inhibition of the catalytic domain of the enzyme. Our results confirmed that as in its human counterpart, high‐target engagement levels (≥99% EO) of fXa are necessary to produce antithrombotic activity. Unfortunately, since minimal fX activity can support thrombosis, almost full silencing of fX mRNA is required to confer antithrombotic activity. In our study, we were unable to achieve doses of fX siRNA high enough to mimic the maximal levels of inhibition of thrombosis provided by apixaban. We were not able to increase the dose of fX siRNA (e.g., 2 mg/kg) nor did we have the option to switch to a different fX siRNA sequence in light of potential nonspecific LNP‐related effects that could have confounded the interpretation of the data (data not shown). On the other hand, there were no major differences between siRNA and pharmacological modalities applied to fIXa as both could mimic the human hemophilia B observations (Sramek et al. 2003; Darby et al. 2007). Thus, differences existed in EO levels that may reflect the key amplification role of fIXa in the coagulation cascade (Eikelboom et al. 2010; Roser‐Jones et al. 2011). A second finding stemmed from the fact that antithrombotic activity was almost paralleled by CBT prolongation upon inhibition of fX and fXa. This narrow TW somewhat contradicted the experimental findings of others who reported extended TWs for apixaban (Wong et al. 2009; Schumacher et al. 2010). It should be noted, however, that the apixaban doses, their pharmacodynamic effects (e.g., PT), and their antithrombotic activities were directly in agreement with published literature (Schumacher et al. 2010). While it is possible that small differences in CBT assay techniques may have accounted for bleeding effects at intermediate levels of inhibition of thrombosis, it seems unlikely that our assay techniques were solely responsible for this discrepancy as the bleeding associated with modulation of fIX and fIXa were in line with the phenotype of hemophilia B patients and the extended bleeding observed in animal models of hemophilia B (Gui et al. 2007; Metzger et al. 2015). Thus, in our studies, apixaban antithrombotic activity was associated with increased bleeding in a “provoked” bleeding model. Third, in our pharmacodynamic monitoring study, we found that ex vivo measurement of thrombin generation in TGA was dose sensitive, could detect antithrombotic activity in vivo, and also distinguished mechanism of action. This suggests that TGA could complement the classical aPTT/PT assays for establishing activity and defining relationship between drug levels and clinical outcomes.
In summary, this multifaceted evaluation of the viability of fIXa as an antithrombotic target, including a model‐based translational PK/PD analysis was the first of its kind, directly comparing fXa and fIXa inhibitory modalities in the context of thrombosis and bleeding using two different approaches, in a head‐to‐head, blinded manner. Results showed that modulation of fIX via mRNA KD or fIXa with a SMi of the catalytic domain of the enzyme can at least match the inhibition provided by apixaban and that fIXa inhibitors could offer superior TW over fXa inhibitors in humans.
Author Contributions
P. A., W. A., T. B., W. G., and H. B. W. designed experiments. H. B. W., J. X., K.‐I. F., W. R. S., and M. T.‐S. contributed reagent tools. H. B. W., M. T.‐S., and D. S. critically reviewed the manuscript. M. S. C. and J. M. M. performed data analysis. P. A and W. A. wrote the manuscript.
Conflict of Interest
All authors are current or past employees of Merck.
Disclosure
All authors have obtained permission to publish from their employers; approvals are held from any persons acknowledged; all authors have seen and approved the final version of the submitted article; content of the manuscript is original and has not been published or accepted for publication, either in whole or in part, and that no part of the manuscript is currently under consideration for publication elsewhere.
Supporting information
Figure S1. Correlation between ex vivo effects in pharmacodynamic assays (aPTT, PT, TGA) and antithrombotic activity. (A) Left panel: apixaban activity on CW expressed against aPTT and PT for each animal. Right panel: CPD1 expressed as a function of aPTT and PT. (B) Apixaban on peak thrombin generation (TGA assay) in response to rhfXIa (top) or TF (bottom) is expressed against CW. (C) CPD1 effects on peak thrombin generation.
Figure S2. mRNA levels of coagulation factors in fIX siRNA‐treated groups (day 7, AVS). Data indicate a dose‐dependent significant decrease in expression of fIX mRNA levels that reached a maximum at 0.6 mg/kg dose. Greater doses were not evaluated in AVS/CBT in order to prevent significant of target effects on other coagulation markers. KLKB1, plasma kallikrein.
Figure S3. mRNA levels of coagulation factors in fX siRNA‐treated groups (day 7, AVS). Data indicate a dose‐dependent significant decrease in expression of fX mRNA levels that reached a maximum at 1 mg/kg dose. Greater doses were not evaluated in AVS/CBT in order to prevent significant off target effects on other coagulation markers (i.e., fVII). KLKB1, plasma kallikrein.
Figure S4. Correlation between enzyme inhibition and mRNA KD upon the administration of fIX and fX siRNA.
Acknowledgements
We thank Duncan Brown for siRNA design, RNA Therapeutics Process Research and Formulation Sciences for synthesizing and formulating siRNAs. We also thank Jillian Dimuzio, Marty Di Pietro, Karen Leander, Anil Thankappan, and Jyoti Disa for in vitro and in vivo siRNA characterization and Laura Sepp‐Lorenzino for her support of the work. We acknowledge the work of Kunal Desai who performed the TGA experiments. Finally, we are thankful to Nina Jochnowitz, Tian‐Quan Cai, Ross Bentley, Michael Calhoun, Lisa Contino, Cesaire L. Gai, Lizbeth Hoos, Harmony Lederman, Alexandra Wickham, and Yuchen Zhou for their contribution to the in vivo studies.
Ankrom W., Wood H. B., Xu J., Geissler W., Bateman T., Chatterjee M. S., Feng K.‐I., Metzger J. M., Strapps W. R., Tadin‐Strapps M., Seiffert D., Andre P., Preclinical and translational evaluation of coagulation factor IXa as a novel therapeutic target, 2015, Pharma Res Per, 4(1), 2016, e00207, doi: 10.1002/prp2.207
References
- Albert NM (2014). Use of novel oral anticoagulants for patients with atrial fibrillation: systematic review and clinical implications. Heart Lung 43: 48–59. [DOI] [PubMed] [Google Scholar]
- Butenas S, Orfeo T, Gissel MT, Brummel KE, Mann KG (2004). The significance of circulating factor IXa in blood. J Biol Chem 279: 22875–22882. [DOI] [PubMed] [Google Scholar]
- Chen Z, Luo B, Cai TQ, Thankappan A, Xu Y, Wu W, et al. (2015). Proof‐of‐concept studies for siRNA‐mediated gene silencing for coagulation factors in rat and rabbit. Mol Ther Nucleic Acids 4: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, et al.; RE‐LY Steering Committee and Investigators (2009). Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 361: 1139–1151. [DOI] [PubMed] [Google Scholar]
- Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, et al. (2007). Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood 110: 815–825. [DOI] [PubMed] [Google Scholar]
- Dewerchin M, Liang Z, Moons L, Carmeliet P, Castellino FJ, Collen D, et al. (2000). Blood coagulation factor X deficiency causes partial embryonic lethality and fatal neonatal bleeding in mice. Thromb Haemost 83: 185–190. [PubMed] [Google Scholar]
- Eikelboom JW, Zelenkofske SL, Rusconi CP (2010). Coagulation factor IXa as a target for treatment and prophylaxis of venous thromboembolism. Arterioscler Thromb Vasc Biol 30: 382–387. [DOI] [PubMed] [Google Scholar]
- Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, et al.; ARISTOTLE Committees and Investigators (2011). Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 365: 981–992. [DOI] [PubMed] [Google Scholar]
- Gui T, Reheman A, Funkhouser WK, Bellinger DA, Hagaman JR, Stafford DW, et al. (2007). In vivo response to vascular injury in the absence of factor IX: examination in factor IX knockout mice. Thromb Res 121: 225–234. [DOI] [PubMed] [Google Scholar]
- Jian'an J, Yafei J (2013). Alternate synthesis of apixaban (BMS‐562247), an inhibitor of blood coagulation factor Xa. Chem Inform 44. [Google Scholar]
- Leil TA, Feng Y, Zhang L, Paccaly A, Mohan P, Pfister M (2010). Quantification of apixaban's therapeutic utility in prevention of venous thromboembolism: selection of phase III trial dose. Clin Pharmacol Ther 88: 375–382. [DOI] [PubMed] [Google Scholar]
- Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW (1997). A coagulation factor IX‐deficient mouse model for human hemophilia B. Blood 90: 3962–3966. [PubMed] [Google Scholar]
- Metzger J, Tadin‐Strapps M, Thankappan A, Strapps WR, DiPietro M, Leander K, et al. (2015). Titrating hemophilia B phenotypes using siRNA strategy: evidence that antithrombotic activity is separated from bleeding liability. Thromb Haemost 113: 1300–1311. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. (2014). Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olearczyk J, Gao S, Eybye M, Yendluri S, Andrews L, Bartz S, et al. (2014). Targeting of hepatic angiotensinogen using chemically modified siRNAs results in significant and sustained blood pressure lowering in a rat model of hypertension. Hypertens Res 37: 405–412. [DOI] [PubMed] [Google Scholar]
- Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, et al. (2011). Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 365: 883–891. [DOI] [PubMed] [Google Scholar]
- Rachidi S, Aldin ES, Greenberg C, Sachs B, Streiff M, Zeidan AM (2013). The use of novel oral anticoagulants for thromboprophylaxis after elective major orthopedic surgery. Expert Rev Hematol 6: 677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser‐Jones C, Chan M, Howard EL, Becker KC, Rusconi CP, Becker RC (2011). Factor IXa as a target for pharmacologic inhibition in acute coronary syndrome. Cardiovasc Ther 29: e22–e35. [DOI] [PubMed] [Google Scholar]
- Schumacher WA, Bostwick JS, Stewart AB, Steinbacher TE, Xin B, Wong PC (2010). Effect of the direct factor Xa inhibitor apixaban in rat models of thrombosis and hemostasis. J Cardiovasc Pharmacol 55: 609–616. [DOI] [PubMed] [Google Scholar]
- Smiley DA, Becker RC (2014). Factor IXa as a target for anticoagulation in thrombotic disorders and conditions. Drug Discov Today 19: 1445–1453. [DOI] [PubMed] [Google Scholar]
- Sramek A, Kriek M, Rosendaal FR (2003). Decreased mortality of ischaemic heart disease among carriers of haemophilia. Lancet 362: 351–354. [DOI] [PubMed] [Google Scholar]
- Tadin‐Strapps M, Peterson LB, Cumiskey AM, Rosa RL, Mendoza VH, Castro‐Perez J, et al. (2011). siRNA‐induced liver ApoB knockdown lowers serum LDL‐cholesterol in a mouse model with human‐like serum lipids. J Lipid Res 52: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zoppe M, Hackeng TM, Griffin JH, Lee KF, Verma IM (1997). A factor IX‐deficient mouse model for hemophilia B gene therapy. Proc Natl Acad Sci USA 94: 11563–11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Crain EJ, Watson CA, Xin B (2009). Favorable therapeutic index of the direct factor Xa inhibitors, apixaban and rivaroxaban, compared with the thrombin inhibitor dabigatran in rabbits. J Thromb Haemost 7: 1313–1320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Correlation between ex vivo effects in pharmacodynamic assays (aPTT, PT, TGA) and antithrombotic activity. (A) Left panel: apixaban activity on CW expressed against aPTT and PT for each animal. Right panel: CPD1 expressed as a function of aPTT and PT. (B) Apixaban on peak thrombin generation (TGA assay) in response to rhfXIa (top) or TF (bottom) is expressed against CW. (C) CPD1 effects on peak thrombin generation.
Figure S2. mRNA levels of coagulation factors in fIX siRNA‐treated groups (day 7, AVS). Data indicate a dose‐dependent significant decrease in expression of fIX mRNA levels that reached a maximum at 0.6 mg/kg dose. Greater doses were not evaluated in AVS/CBT in order to prevent significant of target effects on other coagulation markers. KLKB1, plasma kallikrein.
Figure S3. mRNA levels of coagulation factors in fX siRNA‐treated groups (day 7, AVS). Data indicate a dose‐dependent significant decrease in expression of fX mRNA levels that reached a maximum at 1 mg/kg dose. Greater doses were not evaluated in AVS/CBT in order to prevent significant off target effects on other coagulation markers (i.e., fVII). KLKB1, plasma kallikrein.
Figure S4. Correlation between enzyme inhibition and mRNA KD upon the administration of fIX and fX siRNA.