

Abstract
Resistance to azacitidine is a major issue in the treatments of myelodysplastic syndrome and acute myeloid leukemia, and previous studies suggest that changes in drug metabolism are involved in the resistance. Therefore, drugs with mechanisms resistant or alternative to such metabolic changes have been desired for the treatment of resistant disease. We generated azacitidine‐resistant cells derived from SKM‐1 and MOLM‐13 leukemia cell lines in vitro, analyzed the mechanisms, and examined the impact on the efficacy of other antimetabolic drugs. It appeared that the cell growth‐inhibitory effect of azacitidine, expression levels of uridine–cytidine kinase 2, and the concentrations of azacitidine triphosphate were remarkably decreased in the resistant cells compared with those in parent cells. These results were consistent with previous observations that azacitidine resistance is derived from metabolic changes. Cross‐resistance of greater than 10‐fold (shift in IC50 value) was observed in azacitidine‐resistant cells for decitabine and for cytarabine, but not for gemcitabine or the inosine‐5′‐monophosphate dehydrogenase (IMPDH) inhibitors FF‐10501 and mycophenolate mofetil (cross‐resistance to 5‐fluorouracil was cell line dependent). The IMPDH inhibitors maintained their cell growth‐inhibitory activities in the azacitidine‐resistant cell lines, in which the levels of adenine phosphoribosyltransferase (which converts FF‐10501 to its active form, FF‐10501 ribosylmonophosphate [FF‐10501RMP]), FF‐10501RMP, and the target enzyme, IMPDH, were equivalent to those in the parent cell lines. These results suggest that an IMPDH inhibitor such as FF‐10501 could be an alternative therapeutic treatment for leukemia patients with acquired resistance to azacitidine.
Keywords: Azacitidine, FF‐10501, IMPDH inhibitor, resistance
Abbreviations
- ACN
acetonitrile
- AML
acute myeloid leukemia
- APRT
adenine phosphoribosyl transferase
- aza‐CTP
5‐azacytidine 5′‐triphosphate
- AzaR
azacitidine resistant
- CDA
cytidine deaminase
- 2D2F
2′‐deoxy‐2′‐fluorocytidine hydrate
- DCK
deoxycytidine kinase
- DMSO
dimethyl sulfoxide
- ENT
equilibrative nucleoside transporters
- 5‐FU
5‐fluorouracil
- HMA
hypomethylating agents
- HPLC
high‐performance liquid chromatography
- HPRT
hypoxanthine phosphoribosyltransferase
- IMP
inosine‐5′‐monophosphate
- IMPDH
inosine‐5′‐monophosphate dehydrogenase
- MDS
myelodysplastic syndrome
- MPA
mobile phase A
- PBS
phosphate‐buffered saline
- RMP
ribosylmonophosphate
- UCK
uridine–cytidine kinase
Introduction
Myelodysplastic syndrome (MDS) is characterized by dysplasia of the myeloid cells, in which the number and quality of blood cells decline irreversibly, and has a risk of progression to acute myeloid leukemia (AML) (Tefferi and Vardiman 2009). Therapeutic treatments for MDS and AML patients have been improved with hypomethylating agents (HMAs), azacitidine (5‐azacytidine) and decitabine (5‐aza‐2′‐deoxycytidine), and DNA hypomethylation is believed to be one of the mechanisms of the efficacy (Fenaux et al. 2010; Tefferi 2010). Although azacitidine improves the survival rate in MDS and AML patients (Fenaux et al. 2009, 2010), approximately 40% of the MDS patients fail to respond (Silverman et al. 2002; Fenaux et al. 2009). Prognosis after azacitidine failure is poor both in MDS patients (Prébet et al. 2011), and in AML patients progressed from MDS (Prébet et al. 2012). Although decitabine is another effective HMA for MDS and AML (Lübbert et al. 2011; Kantarjian et al. 2012), the prognosis after decitabine failure is also poor (Jabbour et al. 2010). One common problem of both HMAs is that patients acquire resistance during the treatment (Kadia et al. 2011).
The mechanisms of acquired HMA resistance have been widely studied. There are several reports suggesting that decreased activity or expression of uridine–cytidine kinases (UCK1 and UCK2) (Grant et al. 1984; Sripayap et al. 2014; Valencia et al. 2014) and deoxycytidine kinase (DCK) (Qin et al. 2009, 2011) are responsible for the resistance of azacitidine and decitabine, respectively, as the UCKs and the DCK are required for the HMAs to form their phosphorylated active metabolites. These metabolic changes observed in HMA resistance may have an impact on the efficacies of other antimetabolic agents, such as cytarabine, for the treatment of AML, that is, a possible risk of cross‐resistance in the treatment of MDS and/or AML. Since acquired resistance limits the drug usage in MDS and AML treatments, novel drugs with different mechanisms from hypomethylation are desired.
Inosine‐5′‐monophosphate dehydrogenase (IMPDH) is a key enzyme in de novo purine synthesis (Hedstrom 2009). Human IMPDH includes two isoforms, IMPDH1 and IMPDH2 (Natsumeda et al. 1990). IMPDH1 is expressed ubiquitously in normal cells, whereas IMPDH2 is upregulated in cancer cells, including leukemia cells (Konno et al. 1991; Nagai et al. 1991, 1992; Collart et al. 1992).
Some IMPDH inhibitors, such as tiazofurin (Tricot et al. 1987, 1989; Jayaram et al. 1999), AVN944 (Klisovic et al. 2007; Zuck et al. 2008), and mycophenolate mofetil (Lin et al. 2002; Remacha et al. 2010), have been used in the treatment of patients with hematological malignancies, including MDS and AML, in expectation of repression of proliferation, induction of differentiation, and an immunosuppressive effect through IMPDH inhibition (Chen and Pankiewicz 2007), and the patients showed some response to the IMPDH inhibitors (Jayaram et al. 1999; Klisovic et al. 2007). This implies that IMPDH could be an alternative target for the treatment of MDS and AML, and it would be of interest to investigate whether IMPDH inhibitors are useful in patients with acquired azacitidine resistance.
FF‐10501 (5‐hydroxy‐1H‐imidazole‐4‐carboxamide), formerly known as SM‐108, was investigated in a clinical study of patients with hematological malignancies, and some clinical responses were observed in the MDS and AML patients (Uzuka and Saito 1988; Kimura et al. 1989). FF‐10501 is metabolically transformed to a ribose monophosphate form (FF‐10501RMP), a nucleotide analog, by adenine phosphoribosyl transferase (APRT) intracellularly, and this active form inhibits IMPDH activity (Fukui et al. 1982). The in vitro and in vivo anticancer activities of FF‐10501 have been reported previously (Fukui et al. 1982; Yoshida et al. 1983). It is therefore of particular interest to see whether FF‐10501 is able to inhibit growth of azacitidine‐resistant cells with metabolic changes.
In this study, we analyzed mechanisms of azacitidine resistance using azacitidine‐selected cell lines generated from the leukemia cell lines, SKM‐1 (Kawaguchi et al. 1992; Nakagawa et al. 1993) and MOLM‐13 (Matsuo et al. 1997), both of which were derived from patients with AML following MDS. To determine potential alternative treatments for HMA‐refractory MDS and/or AML, we examined whether the azacitidine resistance would affect the efficacies of other antimetabolic agents. Because azacitidine contains pyrimidine structure, decitabine, cytarabine, gemcitabine, and 5‐fluorouracil (5‐FU) were selected and tested as a pyrimidine analogs, and IMPDH inhibitors, FF‐10501 and mycophenolate mofetil, were also investigated in the resistant cells.
Materials and Methods
Chemicals and reagents
Mycophenolic acid, azacitidine, decitabine, 5‐FU, IMP disodium salt, GTP, high‐performance liquid chromatography (HPLC) grade methanol, acetonitrile (ACN), dimethyl sulfoxide (DMSO), and chloroform were purchased from Wako Pure Chemical Industries (Osaka, Japan). 5‐Azacytidine 5′‐triphosphate (aza‐CTP) ammonium salt was purchased from American Biochemicals (San Diego, CA, USA). Cytarabine and 2′‐deoxy‐2′‐fluorocytidine hydrate (2D2F) were purchased from Tokyo Chemical Industry (Tokyo, Japan). FF‐10501‐01 (3/4 hydrate of FF‐10501) and FF‐10501RMP disodium salt were provided by Toyama Chemical (Tokyo, Japan). Gemcitabine hydrochloride was purchased from Teva Pharmaceutical Industries (Netanya, Israel). Mycophenolate mofetil, ammonium bicarbonate, and ammonium hydroxide solution were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Phosphate‐buffered saline (PBS) was purchased from Life Technologies (Gaithersburg, MD, USA).
Cell lines
SKM‐1 cells were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan), and MOLM‐13 cells were obtained from DSMZ (Braunschweig, Germany). The cells were cultured with culture medium consisting of RPMI 1640 with 10% heat‐inactivated fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. The cultures were incubated in a CO2 incubator at 37°C with 5% CO2 in a humidified atmosphere.
Generation of azacitidine‐resistant cells
SKM‐1 and MOLM‐13 cells were treated with increasing concentrations of azacitidine (from 0.05 to 12.8 μmol/L). The azacitidine concentration was doubled at approximately 2‐ to 4‐week intervals and maintained until cell growth rates were approximately equal to those of the parent cells. About 12 months later, the cells were cultured in the presence of 12.8 μmol/L azacitidine, and the remaining cells were defined as azacitidine‐resistant SKM‐1 (SKM‐1/AzaR) or azacitidine‐resistant MOLM‐13 (MOLM‐13/AzaR). After acquiring the azacitidine‐resistant cells, the cells were cultured without azacitidine and after 1 week the cells were used for several examinations.
Cell growth inhibition assay
Cells were seeded at 5000 cells/well into 96‐well culture plates. FF‐10501‐01, azacitidine, decitabine, cytarabine, gemcitabine hydrochloride, 5‐FU, or mycophenolate mofetil dissolved in PBS, or PBS alone (control) was added to the wells. The cells were incubated in a CO2 incubator for about 72 h. Cell growth inhibition was evaluated using CellTiter‐Glo Luminescent Cell Viability Assay kit (Promega, Madison, WI, USA). Luminescence values were measured using an EnVision plate reader (Perkin Elmer, Waltham, MA, USA).
Measurement of protein expression by western blot analysis
Cells were harvested and washed with PBS. The cells were lysed and protein concentration was determined by BCA assay (Thermo Fisher Scientific, Rockford, IL, USA). Ten micrograms of protein was subjected to SDS‐PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked in Tris‐buffered saline with 0.1% Tween‐20 (TBS‐T) and 5% skim milk (Becton Dickinson and Company, Franklin Lakes, NJ, USA) for 1 h at room temperature. Membranes were incubated with primary antibodies (in a 1:500 dilution of anti‐APRT antibody, 1:500 dilution of anti‐IMPDH2 antibody, 1:1000 dilution of anti‐HPRT antibody [Abcam, Cambridge, UK], 1:100 dilution of anti‐DCK antibody [GenWay, San Diego, CA, USA], 1:1000 dilution of anti‐UCK2 antibody [Proteintech Group, Chicago, IL, USA], 1:10,000 dilution of anti‐β‐actin antibody [Sigma‐Aldrich]), and washed with TBS‐T three times. Reaction with peroxidase‐conjugated secondary antibodies (GE Healthcare Life Science, Piscataway, NJ) was then carried out at room temperature. Reacted proteins were detected with the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) using an LAS‐3000 imaging analyzer (Fujifilm, Tokyo, Japan).
Measurements of intracellular azacitidine, aza‐CTP, FF‐10501RMP, and GTP
Cells (1 × 107) were treated with different concentrations of FF‐10501‐01 or azacitidine, and incubated at 37°C for 6 h (azacitidine) to measure aza‐CTP, 2 h and 24 h (FF‐10501) to measure FF‐10501RMP and GTP, respectively. Cells were harvested and washed with ice‐cold PBS. The cells were deproteinized by the addition of 400 μL methanol. The resulting solution was vortex‐mixed, 200 μL ultrapure water and 400 μL chloroform were added, and the resultant was then mixed well followed by centrifugation at 10,000g for 15 min at 4°C. Four‐hundred microliters of aqueous layer was transferred to an ultrafiltration tube and the tube was centrifuged at 9200g for 120 min at 12°C. The filtrate was separated and concentrated by centrifugation for 120 min at 40°C. The residues were reconstituted with 50 μL ultrapure water containing internal standard, 2D2F (final concentration of 0.4 μg/mL), and vortex‐mixed. Calibration standards were made by spiking various amounts of FF‐10501RMP to the reconstituted residues prepared from PBS‐treated MOLM‐13 or SKM‐1 cells to give final concentrations of 0.3–3.0 μmol/L. A 10‐μL portion of the test drug sample or calibration standard was then injected into an HPLC‐MS/MS system.
HPLC‐MS/MS system and conditions
The HPLC‐MS/MS system used consisted of two LC‐20AD pumps and an SIL‐20AC HT autosampler (Shimadzu, Kyoto, Japan) coupled to a 3200 QTRAP mass spectrometer (AB Sciex, Foster City, CA, USA). The analysis was performed on a ZIC‐pHILIC column, 150 × 4.6 mm, 5 μm polymer (EMD Millipore, Merck Millipore, Darmstadt, Germany), coupled to a ZIC‐pHILIC Guard column, 20 × 2.1 mm (EMD Millipore). The eluents used consisted of mobile phase A (MPA), containing 10 mmol/L ammonium bicarbonate in ultrapure water buffered to pH 9.4 with ammonium hydroxide solution, and mobile phase B, consisting of ACN. A gradient program was used at a flow rate of 0.5 mL/min. The program was initiated with 35–60% MPA from 0 to 5 min, 60–100% MPA from 5 to 7 min, 100% MPA from 7 to 9 min, and 35% MPA from 9 to 12 min. The injection volume was 10 μL. The autosampler temperature was set at 4°C throughout the analysis. The mass spectrometer was operated with an electrospray ionization source in the positive ion mode. The electrospray voltage was set at 5.5 kV and the temperature of the heated capillary was set at 500°C. Semiautomatic tuning was used to optimize all relevant parameters with an infusion of azacitidine, aza‐CTP, FF‐10501RMP, or GTP solution. The ion transitions at m/z 245.1→113.1, 485.0→113.1, 340.1→128.2, 523.9→152.2 for azacitidine, aza‐CTP, FF‐10501RMP, and GTP, respectively, were used in multiple reaction monitoring modes. Collision energy values were optimized to 15–35 kV for these transitions.
IMPDH enzyme inhibition assay
IMPDH activities were measured spectrophotometrically by measuring NADH production at 340 nm. IMPDH1 or IMPDH2 (Abnova, Taipei, Taiwan) recombinant enzyme was added to measurement buffer (50 mmol/L Tris‐HCl, 100 mmol/L KCl, 1 mmol/L dithiothreitol, pH 8.0) to give final IMPDH1 or IMPDH2 concentrations of 7 μg/mL. IMP solution was added to the enzyme solution to give a final concentration of 15 μmol/L for IMPDH1 and 10 μmol/L for IMPDH2. The enzyme solution was mixed with either FF‐10501‐01 or FF‐10501RMP disodium salt (dissolved in distilled water) or mycophenolic acid (dissolved in DMSO) and kept for 5 min. Absorbance at 340 nm was measured immediately after addition of 500 μmol/L NAD+ solution. For each sample, the first measurement was made followed by a second measurement at 30 min later. The measurements were performed using a UV‐2550 spectrophotometer (Shimadzu). All procedures were performed at room temperature.
Statistical analysis
Values are expressed as mean ± standard deviation (SD). The differences between parent and azacitidine‐resistant cells were analyzed by Student's t‐test (equal variance) or Welch's t‐test (unequal variance) using Microsoft Office Excel 2003. P < 0.05 was considered statistically significant.
Results
Generation of azacitidine‐resistant cells, and inhibition of the cell growth by azacitidine, decitabine, other antimetabolic agents, and IMPDH inhibitors
To analyze the mechanisms of azacitidine resistance, we generated two sublines, one each derived from SKM‐1 and MOLM‐13, that were able to grow even in the presence of high concentration of azacitidine (12.8 μmol/L), by gradually increasing concentration of azacitidine, and defined them as SKM‐1/AzaR and MOLM‐13/AzaR. The growth‐inhibitory effect of azacitidine on parent cells (SKM‐1 and MOLM‐13) and the generated cells (SKM‐1/AzaR and MOLM‐13/AzaR) were evaluated by measuring ATP content as an index of viability. We also examined whether the efficacies of decitabine, another HMA, other pyrimidine analogs, cytarabine, gemcitabine, and 5‐FU, and IMPDH inhibitors, FF‐10501 and mycophenolate mofetil, were affected in the azacitidine‐resistant cells. Cytarabine and gemcitabine were selected as these are the known substrates for DCK or cytidine deaminase (CDA), as is decitabine (Heinemann et al. 1988; Eliopoulos et al. 1998). 5‐FU was selected as this is catalyzed by different enzymes, other than UCK or DCK. Mycophenolate mofetil is a prodrug of mycophenolic acid, and both are known to show equivalent activity in cell assays as IMPDH inhibitors (Colic et al. 2003).
The IC50 values and the cell growth inhibition curves for azacitidine, decitabine, cytarabine, FF‐10501, and mycophenolate mofetil against SKM‐1, SKM‐1/AzaR, MOLM‐13, and MOLM‐13/AzaR are shown in Table 1 and Figure 1A–J. In the SKM‐1/AzaR and MOLM‐13/AzaR cells, the growth‐inhibitory effects of azacitidine were much lower than those in their parental cells. The two sublines acquired cross‐resistance to decitabine and cytarabine, gemcitabine, 5‐FU, FF‐10501, and mycophenolate mofetil, except for FF‐10501 in SKM‐1/AzaR. The shifts in IC50 values for decitabine and cytarabine in SKM‐1/AzaR and MOLM‐13/AzaR were more than 10‐fold, while the shifts were much less for gemcitabine in SKM‐1/AzaR and MOLM‐13/AzaR and for 5‐FU in MOLM‐13/AzaR (Table 1). Both IMPDH inhibitors showed concentration‐dependent inhibition of cell growth, both in parent and resistant cells. The effects of the two compounds were not affected in SKM‐1/AzaR. The maximum efficacies of FF‐10501 and mycophenolate mofetil were not affected in the resistant cells (Fig. 1G–J).
Table 1.
Effects of azacitidine, decitabine, cytarabine, gemcitabine, 5‐FU, FF‐10501, and mycophenolate mofetil on the growth of human leukemia cell lines, SKM‐1 and MOLM‐13, and the azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR
| Compound | IC50 (μmol/L) | |||||
|---|---|---|---|---|---|---|
| SKM‐1 | SKM‐1/AzaR | Fold change | MOLM‐13 | MOLM‐13/AzaR | Fold change | |
| Azacitidine | 1.85 ± 0.09 | 36.27 ± 0.17** | 20 | 0.30 ± 0.01 | 27.42 ± 0.80## | 91 |
| Decitabine | 0.13 ± 0.01 | >700 | >5400 | 0.01 ± 0.00 | 0.17 ± 0.01## | 17 |
| Cytarabine | 0.20 ± 0.00 | 2.08 ± 1.16 | 10 | 0.03 ± 0.01 | 0.38 ± 0.06## | 13 |
| Gemcitabine | 2.45 ± 0.31 | 8.54 ± 3.13 | 3 | 2.49 ± 0.39 | 9.39 ± 1.14** | 4 |
| 5‐FU | 2.93 ± 0.54 | 30.40 ± 5.42# | 10 | 1.45 ± 0.07 | 3.14 ± 0.62# | 2 |
| FF‐10501 | 23.47 ± 2.32 | 9.49 ± 0.42## | 0.4 | 7.11 ± 0.05 | 38.46 ± 0.34## | 5 |
| Mycophenolate mofetil | 0.70 ± 0.14 | 0.70 ± 0.18 | 1 | 0.34 ± 0.06 | 0.95 ± 0.11** | 3 |
The cells were cultured with or without test compound for 72 h, and the growth was analyzed using intracellular ATP as an index of viable cells. The data represent IC50 (μmol/L) values of test compounds against SKM‐1, SKM‐1/AzaR, MOLM‐13, and MOLM‐13/AzaR. The data are the means of three independent measurements ± SD. *P < 0.05, **P < 0.01 by Student's t‐test, # P < 0.05, ## P < 0.01 by Welch's t‐test compared with parent cells.
Figure 1.
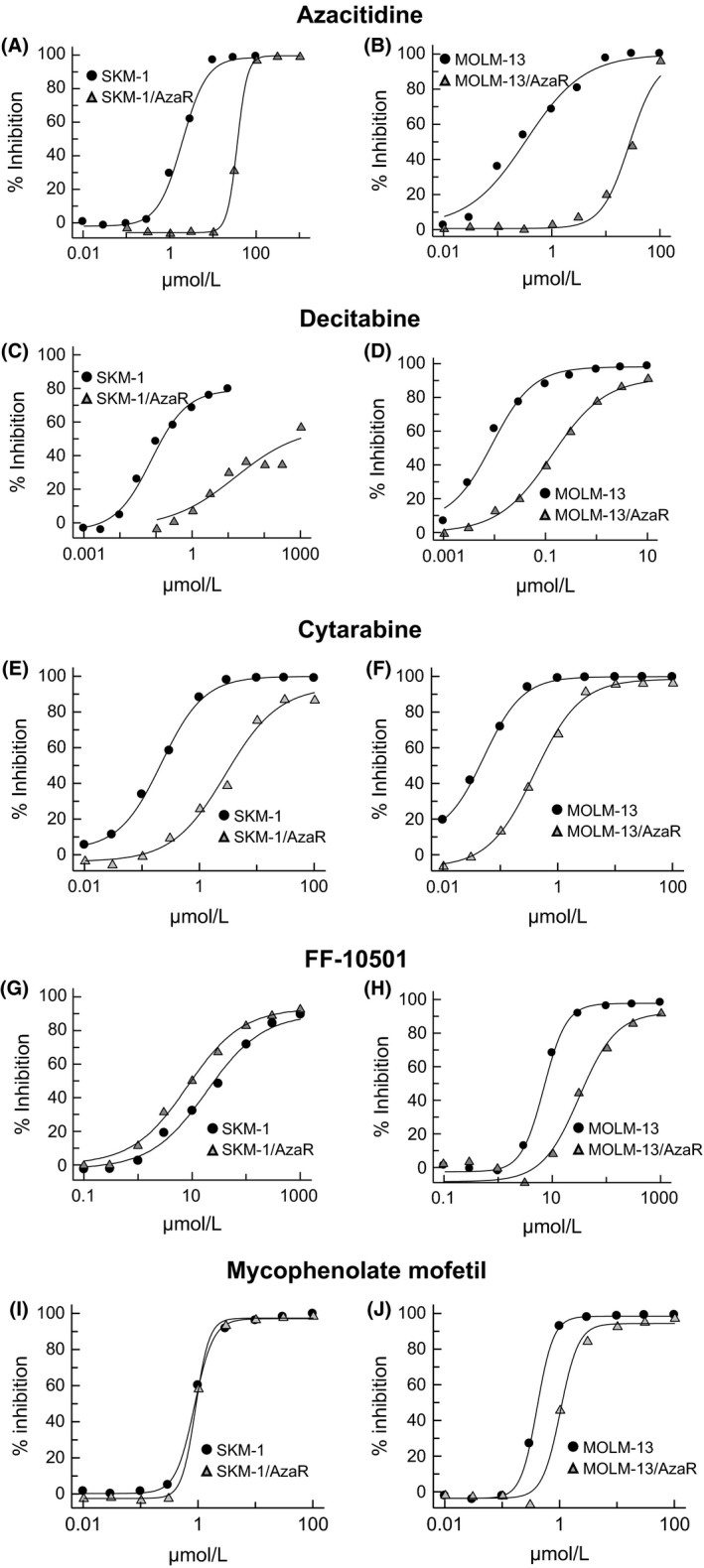
Concentration‐dependent inhibition of human myeloid leukemia cell lines, SKM‐1 and MOLM‐13, and the azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR, by azacitidine (A, B), decitabine (C, D), cytarabine (E, F), FF‐10501 (G, H), and mycophenolate mofetil (I, J). Azacitidine‐resistant cells were generated using SKM‐1 and MOLM‐13 leukemia cells that are able to grow in the presence of azacitidine, by gradually increasing concentration of azacitidine, and designated as SKM‐1/AzaR and MOLM‐13/AzaR. The azacitidine‐resistant cells and their parent cells were treated with the compounds for 72 h and analyzed using intracellular ATP as an index of viable cells. The figures indicate the % inhibition of cell growth of azacitidine‐resistant cells and their parent cells expressed as the average of three independent experiments.
These results indicate that the established sublines, SKM‐1/AzaR and MOLM‐13/AzaR, were azacitidine‐resistant and the efficacies of decitabine, cytarabine, gemcitabine, 5‐FU, FF‐10501, and mycophenolate mofetil were reduced in the sublines except for FF‐10501 and mycophenolate mofetil in SKM‐1/AzaR cells.
Protein expression relating to azacitidine and decitabine activation in azacitidine‐resistant cells
For the drugs for which conversion is necessary to exhibit their pharmacological activities, the efficacies are often limited by the expression levels of metabolic enzymes that catalyze them to active forms. In fact, decreased UCK activity and UCK2 gene mutation were previously reported in azacitidine‐resistant cells (Fukui et al. 1982; Sripayap et al. 2014) and overall survival of patients expressing lower levels of UCK1 was shorter than that of patients with median levels of UCK1 (Valencia et al. 2014), and loss of DCK function was also reported in decitabine‐resistant cells (Qin et al. 2009). To characterize the mechanisms of azacitidine resistance, protein expression of UCK2, one of the enzymes to activate azacitidine, and DCK, an enzyme to activate decitabine, was measured in four cell lines. UCK2 expression levels in the azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR, were decreased compared to their parent cells, SKM‐1 and MOLM‐13. In contrast, DCK expression levels were not changed based on comparison of β‐actin expressions (Fig. 2).
Figure 2.
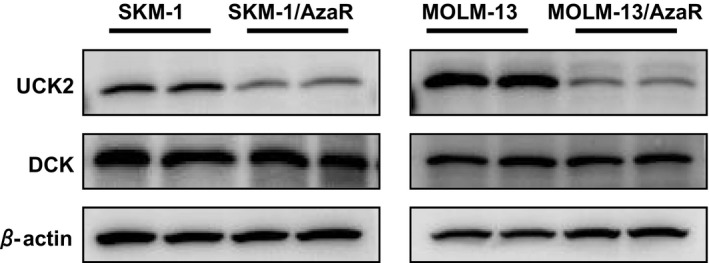
Expressions of UCK2 and DCK in azacitidine‐resistant cells, SKM‐1/AzaR, MOLM‐13/AzaR, and the parent cells, SKM‐1 and MOLM‐13. The cells were cultured in culture medium without any growth inhibitors. The growing cells were harvested and protein lysates were prepared. The lysates were subjected to western blot analyses and UCK2 and DCK antibodies were used to detect UCK2 and DCK. Each sample was loaded in duplicate to exclude possibility of inappropriate loading. β‐actin was also analyzed, as a loading control.
Intracellular levels of azacitidine and aza‐CTP in azacitidine‐resistant cells
To confirm the influence of decreased UCK expression on azacitidine activation in azacitidine‐resistant cells, intracellular azacitidine and its active metabolite aza‐CTP converted by UCK (Quintás‐Cardama et al. 2010) were measured using the LC‐MS/MS system. After treatment of the cells with azacitidine (10 and 100 μmol/L) for 6 h, increase in intracellular levels of azacitidine and aza‐CTP were observed in MOLM‐13 cells in a concentration‐dependent manner (Fig. 3A and B). More than 6 h incubation with 10 and 100 μmol/L of azacitidine caused significant cell death, which made collection of the cells difficult (data not shown). The increase in intracellular level of azacitidine was also observed in MOLM‐13/AzaR cells in a concentration‐dependent manner (Fig. 3C), whereas the intracellular level of aza‐CTP was drastically decreased in MOLM‐13/AzaR cells compared with MOLM‐13 cells (Fig. 3D). The same approach to SKM‐1 and SKM‐1/AzaR was not successful (data not shown) due to the appearance of interfering peaks.
Figure 3.
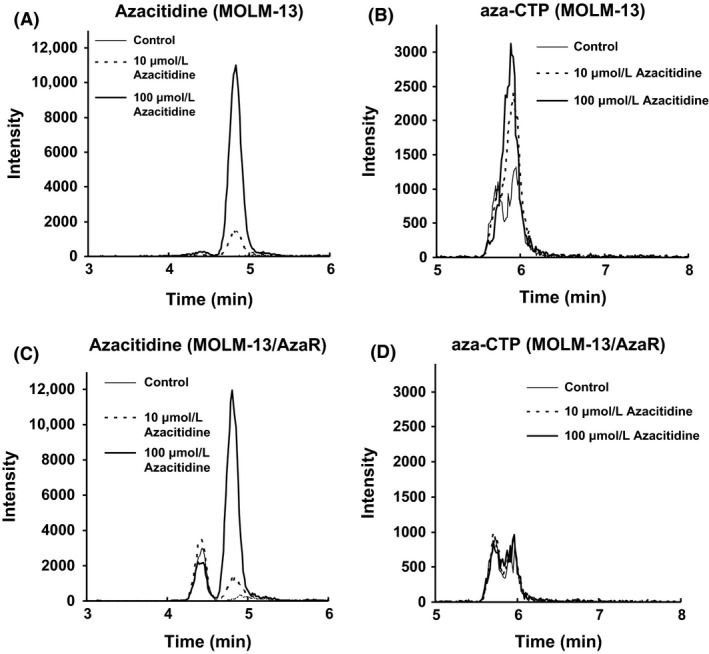
Extracted ion chromatograms of azacitidine and aza‐CTP in human myeloid leukemia cell line, MOLM‐13, and the azacitidine‐resistant cells, MOLM‐13/AzaR; azacitidine (A) and aza‐CTP (B) in parent cells, MOLM‐13, and azacitidine (C) and aza‐CTP (D) in azacitidine‐resistant cells, MOLM‐13/AzaR. Intracellular levels of azacitidine and aza‐CTP in MOLM‐13/AzaR and MOLM‐13 were analyzed using HPLC‐MS/MS. After 6 h treatment of 10 or 100 μmol/L of azacitidine to azacitidine‐resistant cells and the parent cells, the treated cells were harvested and azacitidine and aza‐CTP were extracted for subjection to HPLC‐MS/MS analyses. Because minor peaks appeared at the same retention time in control cells, specificity in measurement of azacitidine and aza‐CTP was insufficient for their quantification, and representative data are shown here.
IMPDH enzyme inhibition by IMPDH inhibitors, FF‐10501 and mycophenolic acid
Enzyme assay was performed using recombinant human IMPDH1 and IMPDH2 proteins to confirm that FF‐10501 is an inhibitor of both isozymes. Mycophenolic acid was used as an active control. Both FF‐10501RMP, an active form of FF‐10501, and mycophenolic acid inhibited the enzyme activities in a concentration‐dependent manner with equivalent potencies (Table 2A). In contrast, FF‐10501 itself did not inhibit enzyme activity (Table 2B). These data confirm that FF‐10501RMP is an active form of FF‐10501 for IMPDH inhibition and inhibits both IMPDH1 and IMPDH2.
Table 2.
Inhibitory effects of FF‐10501RMP, FF‐10501, and mycophenolic acid on the enzyme activities of IMPDH1 and IMPDH2
| Compound | IMPDH1 | IMPDH2 |
|---|---|---|
| (A) | IC50 (nmol/L) | |
| FF‐10501RMP | 29.0 ± 10.3 | 31.8 ± 5.6 |
| Mycophenolic acid | 26.4 ± 1.5 | 27.8 ± 8.3 |
| (B) | Enzyme activity inhibition (%) | |
| FF‐10501, 1000 nmol/L | 12.4 ± 11.5 | −6.2 ± 6.4 |
| FF‐10501, 10,000 nmol/L | 5.6 ± 16.6 | −11.1 ± 9.8 |
(A) IC50 values of FF‐10501RMP and mycophenolic acid against IMPDH1 and IMPDH2 were analyzed. (B) Enzyme activity inhibition (%) of FF‐10501 against IMPDH1 and IMPDH2 were measured. The data are the means of three independent measurements ± SD.
Protein expression of APRT and IMPDH
Western blotting analyses were performed to examine whether the azacitidine resistance affects expressions of APRT, which is necessary for FF‐10501 to be converted to its active form, FF‐10501RMP, and IMPDH, the target enzyme for IMPDH inhibitors. The results revealed that the expression levels of APRT, IMPDH, or hypoxanthine phosphoribosyltransferase (HPRT), an enzyme for salvage pathway, in the resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR, were comparable with those in the parent cells, SKM‐1 and MOLM‐13 (Fig. 4).
Figure 4.
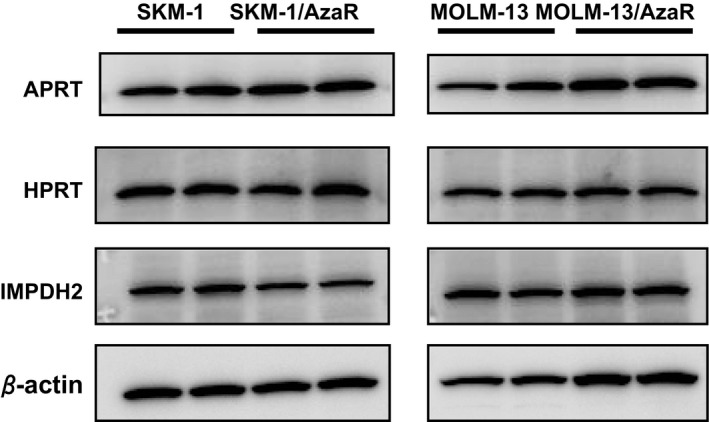
Expressions of APRT, HPRT, IMPDH2, and β‐actin in human myeloid leukemia cell lines, SKM‐1 and MOLM‐13, and azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR. The cells were cultured in the normal medium without any growth inhibitors. The growing cells were harvested, and protein lysates were prepared. The lysates were subjected to western blot analyses to detect APRT, HPRT, and IMPDH2 using APRT, HPRT, and IMPDH2 antibodies. Each sample was loaded in duplicate to exclude possibility of inappropriate loading. β‐actin was also analyzed, as a loading control.
Intracellular levels of FF‐10501RMP and GTP in azacitidine‐resistant cells
We next measured intracellular concentrations of FF‐10501RMP and GTP to confirm whether the FF‐10501 activation system and the target inhibition were intact in the azacitidine‐resistant cells. The cells were treated with 10 and 100 μmol/L of FF‐10501 for either 2 or 24 h, harvested, extracted, and FF‐10501RMP and GTP were measured using HPLC‐MS/MS. The concentrations of FF‐10501RMP at 2 h after treatment with FF‐10501 at 10 μmol/L were equivalent in SKM‐1 and SKM‐1/AzaR cells (Fig. 5A). There were significant decreases in average level of FF‐10501RMP at 100 μmol/L of FF‐10501 in azacitidine‐resistant cells compared with parent cells. The FF‐10501RMP levels after treatment with FF‐10501 at 10 and 100 μmol/L were lower in MOLM‐13/AzaR compared to MOLM‐13, however, they were not statistically significant (Fig. 5B). The intracellular GTP levels at 24 h after FF‐10501 treatment were identically reduced in a concentration‐dependent manner in both SKM‐1 and SKM‐1/AzaR cells, reaching approximately to less than 40% of control value at 100 μmol/L of FF‐10501 (Fig. 6A), while the decrease was significantly lower at 100 μmol/L of FF‐10501 in MOLM‐13/AzaR compared to MOLM‐13 cells (Fig. 6B).
Figure 5.
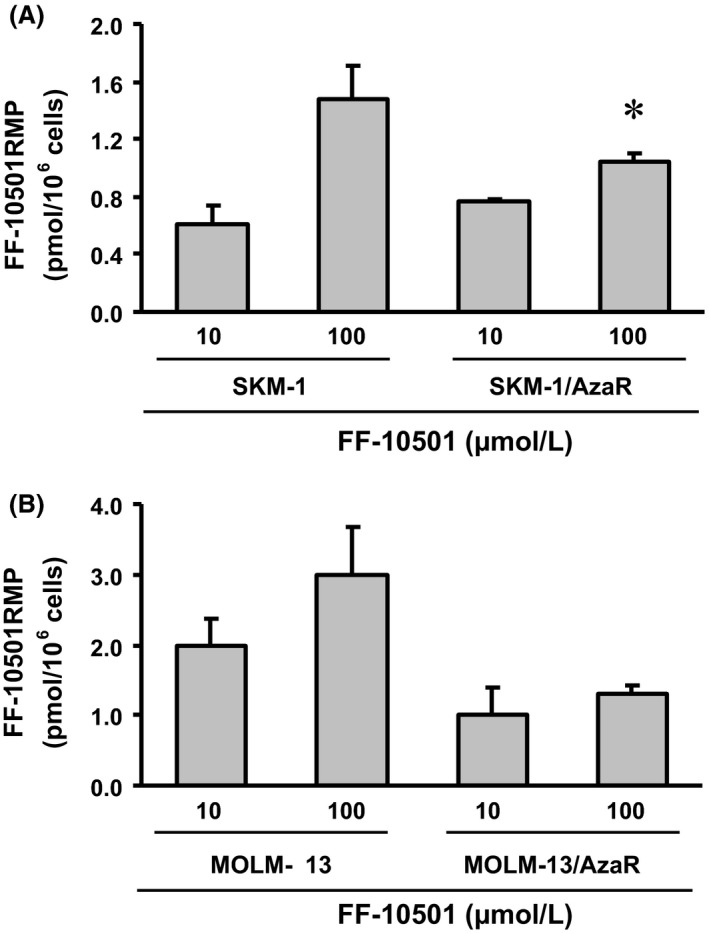
Intracellular concentrations of FF‐10501RMP in human myeloid leukemia cell lines, SKM‐1 and MOLM‐13, and the azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR. The figures represent intracellular concentrations of FF‐10501RMP in SKM‐1 and SKM‐1/AzaR (A), and in MOLM‐13 and MOLM‐13/AzaR (B). After 2 h treatment of 10 or 100 μmol/L FF‐10501 to azacitidine‐resistant cells and their parent cells, FF‐10501RMP concentration was analyzed by HPLC‐MS/MS. The data are the means of three independent measurements ± SD. *P < 0.05 compared with parent cells by Student's t‐test.
Figure 6.
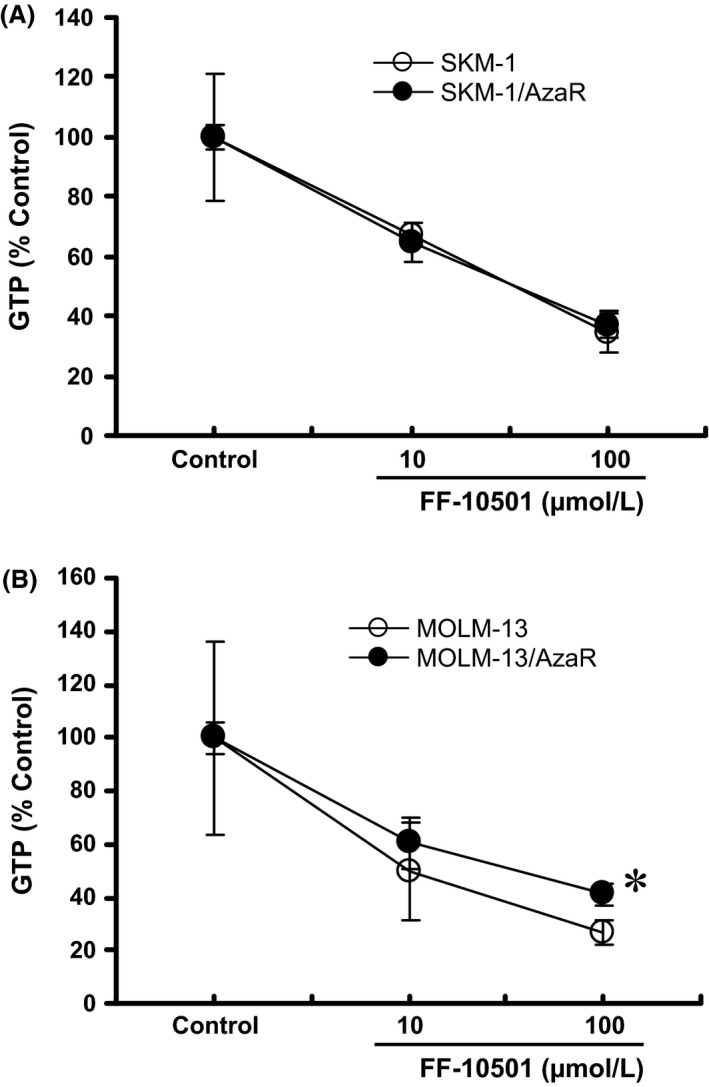
Decrease in intracellular concentrations of GTP after treatment with FF‐10501 in human myeloid leukemia cell lines, SKM‐1 and MOLM‐13, and the azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR. The figures represent the intracellular GTP in SKM‐1 and SKM‐1/AzaR (A), and MOLM‐13 and MOLM‐13/AzaR (B). Azacitidine‐resistant cells and the parent cells were treated with 10 or 100 μmol/L of FF‐10501, or PBS (control) for 24 h, and intracellular GTP concentrations were measured by HPLC‐MS/MS. The data represent % change in intracellular GTP from each control value. The data are the means of three independent measurements ± SD. *P < 0.05 compared with parent cells by Student's t‐test.
Discussion
In this study, we analyzed the mechanisms of azacitidine resistance using cells derived from leukemia cell lines, and examined the changes in metabolism of azacitidine. We then investigated the impact of the metabolic changes on the efficacies of other pyrimidine analogs, and explored robustness of IMPDH inhibitors as an alternative treatment against cells exhibiting azacitidine resistance, due to these metabolic changes.
Some cell lines such as SKM‐1, THP‐1, and HL60 were used to explore the mechanism of azacitidine resistance including cell proliferation and protein expression relating to azacitidine activation (Cluzeau et al. 2011; Sripayap et al. 2014). SKM‐1 cell line was established from a patient with AML following MDS (Kawaguchi et al. 1992; Nakagawa et al. 1993), and it has been used for elucidating the mechanism of azacitidine resistance (Cluzeau et al. 2014). MOLM‐13 was also established from a patient at relapse of AML following MDS (Matsuo et al. 1997). Therefore, these two cell lines were considered to be useful tools for investigating acquired azacitidine resistance in MDS/AML patients. Current results demonstrated that the sensitivities to azacitidine in the resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR, were greatly lost compared with the parent cells, and the cells acquired cross‐resistance to decitabine and cytarabine, and to a lesser extent to gemcitabine and 5‐FU.
Azacitidine requires phosphorylation by UCK1 or UCK2 in cells to enable its active form, aza‐CTP, to exert its action (Quintás‐Cardama et al. 2010). A previous report suggests that resistance to azacitidine results from downregulation of UCK activity (Grant et al. 1984). We therefore examined the changes in the expression of the metabolic enzymes for the activation of azacitidine. As expected, the expression levels of UCK2 in our azacitidine‐resistant cells, SKM‐1/AzaR and MOLM‐13/AzaR, were lower than those in their parent cells, and thus the levels of aza‐CTP would be decreased in the azacitidine‐resistant cells. In fact, the levels of aza‐CTP in MOLM‐13/AzaR cells were much lower than those in the parent cells. This is the first report demonstrating decreased aza‐CTP levels in azacitidine‐resistant cells. Although measurements of the aza‐CTP in SKM‐1 and SKM‐1/AzaR were not successful because of the high amount of endogenous UTP and CTP as reported previously (Derissen et al. 2014), the result of decreased levels of aza‐CTP in MOLM‐13/AzaR suggested that the activation pathway of azacitidine is impaired in the resistant cells, which leads to decreased sensitivity to azacitidine. Another study has also suggested that UCK2 gene mutation is related to azacitidine resistance (Sripayap et al. 2014). In clinical situations, furthermore, decreased expression of UCK1 was reported in azacitidine‐resistant MDS patients (Valencia et al. 2014).
Several various alterations in drug metabolism could contribute to the observed pattern of cross‐resistance. Loss of DCK function is reported to be one of the mechanisms of decitabine resistance (Qin et al. 2009, 2011). Unexpectedly, the expression levels of DCK in the azacitidine‐resistant cells in the present study were not changed, although those azacitidine‐resistant cells showed strong cross‐resistance to decitabine. It is possible that DCK dysfunction, rather than decreased expression, is the reason for the loss of sensitivity.
Resistance may also be acquired by other mechanisms, such as a decrease in equilibrative nucleoside transporters 1 (ENT1) (Hubeek et al. 2005; Hummel‐Eisenbeiss et al. 2013). ENT1 is involved in cellular uptake of azacitidine and decitabine, and is known to influence the inhibitory activities of azacitidine and decitabine. This is unlikely, however, in MOLM‐13 and MOLM‐13/AzaR, as our data showed that the levels of azacitidine at 6 h after the treatment were almost the same in both cell types.
Another possible factor in developing azacitidine resistance is increased activity of CDA, which metabolizes cytidine analogs. CDA was reported to be upregulated in azacitidine‐resistant cell line (Imanishi et al. 2014). Therefore, we further examined the effects of other cytidine analogs, cytarabine and gemcitabine, both of which are known to be activated by DCK and inactivated by CDA (Heinemann et al. 1988; Eliopoulos et al. 1998), and 5‐FU, which is a uridine analog and is not a substrate for DCK or CDA. It was found that the efficacy of cytarabine was the most affected in both of the resistant cell types; however, the shifts in IC50 for the cytidine and uridine analogs were not consistent (Table 1). These results suggest that the resistant mechanism of SKM‐1/AzaR and MOLM‐13/AzaR were based on multiple factors related to pyrimidine metabolism besides simple enzymatic activation or inactivation. There are various other factors affecting drug activities such as changes in gene expression of transporters (de Wolf et al. 2008; Fukuda and Schuetz 2012) or epigenetic modulation (Hauswald et al. 2009). Although it has yet to be determined what mechanism(s) confers resistance to our azacitidine‐resistant cell lines, neither decitabine nor cytarabine are good alternative compounds for growth inhibition against the azacitidine‐resistant cells. This indicates that the treatment with decitabine or cytarabine for leukemia patients after acquiring azacitidine resistance has a possible risk of cross‐resistance.
IMPDH is a key enzyme involved in de novo synthesis of GMP and GTP (Hedstrom 2009); therefore, IMPDH inhibitors have been tested against malignant neoplasms in expectation of the antiproliferative effects. Three IMPDH inhibitors, tiazofurin, AVN944, and mycophenolate mofetil, have been used as treatment in patients with leukemia, including MDS and AML, and positive responses have been reported (Tricot et al. 1987, 1989; Jayaram et al. 1992, 1999; Lin et al. 2002; Klisovic et al. 2007; Zuck et al. 2008; Remacha et al. 2010). FF‐10501 is another IMPDH inhibitor that was also tested against hematologic malignancies (Uzuka and Saito 1988; Kimura et al. 1989). We were intrigued by these results, and wondered whether IMPDH inhibitors would become an alternative treatment of MDS and AML, especially for the patients with acquired HMA resistance.
FF‐10501, formerly known as SM‐108, is an inhibitor of IMPDH (Fukui et al. 1982). However, whether FF‐10501 actually inhibits IMPDH2, which is overexpressed in cancer cells, has not been evaluated. If FF‐10501 were to be used against azacitidine‐resistant leukemia cells, it would need to be demonstrated whether FF‐10501 is an IMPDH2 inhibitor. The results of the present study demonstrated that FF‐10501RMP, active form of FF‐10501, significantly inhibited not only IMPDH1, but also IMPDH2 as expected, whereas FF‐10501 did not. This is consistent with the concept that FF‐10501 is a precursor compound of an IMPDH inhibitor (Fukui et al. 1982).
It was shown that the growth‐inhibitory activities of FF‐10501 remained in SKM‐1/AzaR and slightly reduced in MOLM‐13/AzaR. The growth inhibition by FF‐10501 is dependent on the expression/activity of APRT and IMPDH, because these are required for converting FF‐10501 to FF‐10501RMP and exerting efficacy through reduction of GMP and GTP, respectively (Jayaram et al. 1999). Western blot analyses showed that levels of APRT or IMPDH in azacitidine‐resistant cells were comparable with those in the parent cells. Intracellular FF‐10501RMP and GTP concentrations were increased and decreased, respectively, dependent on the concentration of FF‐10501. The effects of another IMPDH inhibitor, mycophenolate mofetil, were also tested and revealed that the activities remained in SKM‐1/AzaR and slightly reduced in MOLM‐13/AzaR. These results suggest that APRT and IMPDH were maintained in the azacitidine‐resistant cells.
Although IMPDH is a key enzyme for de novo GMP and GTP synthesis, there is another pathway to generate GMP and GTP, a salvage pathway in which GMP and GTP are synthesized from intermediates in the metabolic degradation to guanosine, and HPRT is involved in the pathway (Zoref‐Shani and Sperling 1980). To confirm whether this salvage pathway is affected in azacitidine‐resistant cells, the expression of HPRT was also measured. The result that the expression of HPRT was comparable in parent and azacitidine‐resistant cells and that GTP levels were almost equally decreased by FF‐10501 in SKM‐1/AzaR cells to the same levels as in the parent cells suggest that the contribution of HPRT to the growth inhibition by IMPDH inhibitors was low in the azacitidine‐resistant cells. The transporting mechanism of FF‐10501 and mycophenolate mofetil into cells or degradation remains unclear, and further investigation into these molecules may explain the differences observed in MOLM‐13/AzaR.
Taken together, these data suggest that the azacitidine resistance generated in MOLM‐13 and SKM‐1 was mainly caused by decreased expression of UCK2, an azacitidine‐activating enzyme. Development of large cross‐resistance to decitabine and cytarabine was speculated to be due to other metabolic changes, such as dysfunction of DCK and upregulation of CDA. In contrast, remaining efficacies of IMPDH inhibitors, FF‐10501 and mycophenolate mofetil, in the azacitidine‐resistant cells suggested unchanged functions of the target enzyme, IMPDH, and of APRT, for FF‐10501 to be transformed to its active form, FF‐10501RMP.
In conclusion, this study demonstrated that in spite of the potential induction of various drug resistance mechanisms, and the observed cross‐resistance to various drugs, there was not a large cross‐resistance to FF‐10501 with different behavior in growth inhibition of azacitidine‐resistant leukemia cells from that of azacitidine, decitabine, and cytarabine. It suggests that an IMPDH inhibitor like FF‐10501 is beneficial for leukemia patients with azacitidine failure. Clinical trials of FF‐10501 are currently being reconducted for the treatment of MDS and AML patients who are refractory and relapse after HMA treatment.
Conflict of Interest
None declared.
Acknowledgements
We thank Yoshikazu Yanagi for his critical advice, and Dr. Shigeki Kuwayama for his excellent performance of experiments.
Murase M., Iwamura H., Komatsu K., Saito M., Maekawa T., Nakamura T., Yokokawa T., Shimada Y., Lack of cross‐resistance to FF‐10501, an inhibitor of inosine‐5′‐monophosphate dehydrogenase, in azacitidine‐resistant cell lines selected from SKM‐1 and MOLM‐13 leukemia cell lines, 2015, Pharma Res Per, 4(1), 2016, e00206, doi: 10.1002/prp2.206
References
- Chen L, Pankiewicz KW (2007). Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr Opin Drug Discov Dev 10: 403–412. [PubMed] [Google Scholar]
- Cluzeau T, Robert G, Puissant A, Jean‐Michel K, Cassuto JP, Raynaud S, et al. (2011). Azacitidine‐resistant SKM1 myeloid cells are defective for AZA‐induced mitochondrial apoptosis and autophagy. Cell Cycle 10: 2339–2343. [DOI] [PubMed] [Google Scholar]
- Cluzeau T, Dubois A, Jacquel A, Luciano F, Renneville A, Preudhomme C, et al. (2014). Phenotypic and genotypic characterization of azacitidine‐sensitive and resistant SKM1 myeloid cell lines. Oncotarget 5: 4384–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colic M, Stojic‐Vukanic Z, Pavlovic B, Jandric D, Stefanoska I (2003). Mycophenolate mofetil inhibits differentiation, maturation and allostimulatory function of human monocyte‐derived dendritic cells. Clin Exp Immunol 134: 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart FR, Chubb CB, Mirkin BL, Huberman E (1992). Increased inosine‐5′‐phosphate dehydrogenase gene expression in solid tumor tissues and tumor cell lines. Cancer Res 52: 5826–5828. [DOI] [PubMed] [Google Scholar]
- Derissen EJB, Hillebrand MJX, Rosing H, Otten HMMB, Laille E, Schellens JHM, et al. (2014). Quantitative determination of azacitidine triphosphate in peripheral blood mononuclear cells using liquid chromatography coupled with high‐resolution mass spectrometry. J Pharm Biomed Anal 90: 7–14. [DOI] [PubMed] [Google Scholar]
- Eliopoulos N, Cournoyer D, Momparler RL (1998). Drug resistance to 5‐aza‐2′‐deoxycytidine, 2′,2′‐difluorodeoxycytidine, and cytosine arabinoside conferred by retroviral‐mediated transfer of human cytidine deaminase cDNA into murine cells. Cancer Chemother Pharmacol 42: 373–378. [DOI] [PubMed] [Google Scholar]
- Fenaux P, Mufti GJ, Hellstrom‐Lindberg E, Santini V, Finelli C, Giagounidis A, et al. (2009). Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher‐risk myelodysplastic syndromes: a randomised, open‐label, phase III study. Lancet Oncol 10: 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenaux P, Mufti GJ, Hellström‐Lindberg E, Santini V, Gattermann N, Germing U, et al. (2010). Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 28: 562–569. [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Schuetz JD (2012). ABC transporters and their role in nucleoside and nucleotide drug resistance. Biochem Pharmacol 83: 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui M, Inaba M, Tsukagoshi S, Sakurai Y (1982). New antitumor imidazole derivative, 5‐carbamoyl‐1H‐imidazol‐4‐yl piperonylate, as an inhibitor of purine synthesis and its activation by adenine phosphoribosyltransferase. Cancer Res 42: 1098–1102. [PubMed] [Google Scholar]
- Grant S, Bhalla K, Gleyzer M (1984). Effect of uridine on response of 5‐azacytidine‐resistant human leukemic cells to inhibitors of de novo pyrimidine synthesis. Cancer Res 44: 5505–5510. [PubMed] [Google Scholar]
- Hauswald S, Duque‐Afonso J, Wagner MM, Schertl FM, Lübbert M, Peschel C, et al. (2009). Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin Cancer Res 15: 3705–3715. [DOI] [PubMed] [Google Scholar]
- Hedstrom L (2009). IMP dehydrogenase: structure, mechanism, and inhibition. Chem Rev 109: 2903–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann V, Hertel LW, Grindey GB, Plunkett W (1988). Comparison of the cellular pharmacokinetics and toxicity of 2′,2′‐difluorodeoxycytidine and 1‐beta‐d‐arabinofuranosylcytosine. Cancer Res 48: 4024–4031. [PubMed] [Google Scholar]
- Hubeek I, Stam RW, Peters GJ, Broekhuizen R, Meijerink JPP, van Wering ER, et al. (2005). The human equilibrative nucleoside transporter 1 mediates in vitro cytarabine sensitivity in childhood acute myeloid leukaemia. Br J Cancer 93: 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel‐Eisenbeiss J, Hascher A, Hals P‐A, Sandvold ML, Müller‐Tidow C, Lyko F, et al. (2013). The role of human equilibrative nucleoside transporter 1 on the cellular transport of the DNA methyltransferase inhibitors 5‐azacytidine and CP‐4200 in human leukemia cells. Mol Pharmacol 84: 438–450. [DOI] [PubMed] [Google Scholar]
- Imanishi S, Umezu T, Ohtsuki K, Kobayashi C, Ohyashiki K, Ohyashiki JH (2014). Constitutive activation of the ATM/BRCA1 pathway prevents DNA damage‐induced apoptosis in 5‐azacytidine‐resistant cell lines. Biochem Pharmacol 89: 361–369. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Garcia‐Manero G, Batty N, Shan J, O'Brien S, Cortes J, et al. (2010). Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer 116: 3830–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram HN, Lapis E, Tricot G, Kneebone P, Paulik E, Zhen W, et al. (1992). Clinical pharmacokinetic study of tiazofurin administered as a 1‐hour infusion. Int J Cancer 51: 182–188. [DOI] [PubMed] [Google Scholar]
- Jayaram HN, Grusch M, Cooney DA, Krupitza G (1999). Consequences of IMP dehydrogenase inhibition, and its relationship to cancer and apoptosis. Curr Med Chem 6: 561–574. [PubMed] [Google Scholar]
- Kadia TM, Jabbour E, Kantarjian H (2011). Failure of hypomethylating agent‐based therapy in myelodysplastic syndromes. Semin Oncol 38: 682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. (2012). Multicenter, randomized, open‐label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low‐dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 30: 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R, Hosokawa Y, Komine A, Tsutsumi M, Hikiji K, Ishida‐Okawara A, et al. (1992). The monocytic cell line SKM‐1 strongly expresses the myeloperoxidase gene. Leukemia 6: 1296–1301. [PubMed] [Google Scholar]
- Kimura K, Yamada K, Uzuka Y, Masaoka T, Hirano M, Ohno R, et al. (1989). Phase II study of SM‐108 (4‐carbamoylimidazolium‐5‐olate) in hematological malignancies. Gan To Kagaku Ryoho 16: 123–130. [PubMed] [Google Scholar]
- Klisovic RB, Coutre S, Kovacsovics TJ, Kantarjian HM, Tricot GJ, Strovel JW, et al. (2007) Clinical‐biomarker correlations in adult AML patients in a phase I trial of AVN944 support observations of clinical effect and provide hypotheses for patient selection criteria for further clinical trials. Blood (ASH Annual Meeting Abstracts) 110 Abstract 896.
- Konno Y, Natsumeda Y, Nagai M, Yamaji Y, Ohno S, Suzuki K, et al. (1991). Expression of human IMP dehydrogenase types I and II in Escherichia coli and distribution in human normal lymphocytes and leukemic cell lines. J Biol Chem 266: 506–509. [PubMed] [Google Scholar]
- Lin JT, Wang WS, Yen CC, Chiou TJ, Liu JH, Hsiao LT, et al. (2002). Myelodysplastic syndrome complicated by autoimmune hemolytic anemia: remission of refractory anemia following mycophenolate mofetil. Ann Hematol 81: 723–726. [DOI] [PubMed] [Google Scholar]
- Lübbert M, Suciu S, Baila L, Rüter BH, Platzbecker U, Giagounidis A, et al. (2011). Low‐dose decitabine versus best supportive care in elderly patients with intermediate‐ or high‐risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol 29: 1987–1996. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, MacLeod RA, Uphoff CC, Drexler HG, Nishizaki C, Katayama Y, et al. (1997). Two acute monocytic leukemia (AML‐M5a) cell lines (MOLM‐13 and MOLM‐14) with interclonal phenotypic heterogeneity showing MLL‐AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 11: 1469–1477. [DOI] [PubMed] [Google Scholar]
- Nagai M, Natsumeda Y, Konno Y, Hoffman R, Irino S, Weber G (1991). Selective up‐regulation of type II inosine 5′‐monophosphate dehydrogenase messenger RNA expression in human leukemias. Cancer Res 51: 3886–3890. [PubMed] [Google Scholar]
- Nagai M, Natsumeda Y, Weber G (1992). Proliferation‐linked regulation of type II IMP dehydrogenase gene in human normal lymphocytes and HL‐60 leukemic cells. Cancer Res 52: 258–261. [PubMed] [Google Scholar]
- Nakagawa T, Matozaki S, Murayama T, Nishimura R, Tsutsumi M, Kawaguchi R, et al. (1993). Establishment of a leukaemic cell line from a patient with acquisition of chromosomal abnormalities during disease progression in myelodysplastic syndrome. Br J Haematol 85: 469–476. [DOI] [PubMed] [Google Scholar]
- Natsumeda Y, Ohno S, Kawasaki H, Konno Y, Weber G, Suzuki K (1990). Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem 265: 5292–5295. [PubMed] [Google Scholar]
- Prébet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. (2011). Outcome of high‐risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol 29: 3322–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prébet T, Gore SD, Thépot S, Esterni B, Quesnel B, Beyne Rauzy O, et al. (2012). Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br J Haematol 157: 764–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T, Jelinek J, Si J, Shu J, Issa J‐PJ (2009). Mechanisms of resistance to 5‐aza‐2′‐deoxycytidine in human cancer cell lines. Blood 113: 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin T, Castoro R, El Ahdab S, Jelinek J, Wang X, Si J, et al. (2011). Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS One 6: e23372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás‐Cardama A, Santos FPS, Garcia‐Manero G (2010). Therapy with azanucleosides for myelodysplastic syndromes. Nat Rev Clin Oncol 7: 433–444. [DOI] [PubMed] [Google Scholar]
- Remacha AF, Arrizabalaga B, Bueno J, Muñoz J, Bargay J, Pedro C (2010). Treatment with mycophenolate mofetil followed by recombinant human erythropoietin in patients with low‐risk myelodysplastic syndromes resistant to erythropoietin treatment. Haematologica 95: 339–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar‐Reissig R, et al. (2002). Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20: 2429–2440. [DOI] [PubMed] [Google Scholar]
- Sripayap P, Nagai T, Uesawa M, Kobayashi H, Tsukahara T, Ohmine K, et al. (2014). Mechanisms of resistance to azacitidine in human leukemia cell lines. Exp Hematol 42: 294.e2–306.e2. [DOI] [PubMed] [Google Scholar]
- Tefferi A (2010). Myelodysplastic syndromes‐many new drugs, little therapeutic progress. Mayo Clin Proc 85: 1042–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Vardiman JW (2009). Myelodysplastic syndromes. N Engl J Med 361: 1872–1885. [DOI] [PubMed] [Google Scholar]
- Tricot GJ, Jayaram HN, Nichols CR, Pennington K, Lapis E, Weber G, et al. (1987). Hematological and biochemical action of tiazofurin (NSC 286193) in a case of refractory acute myeloid leukemia. Cancer Res 47: 4988–4991. [PubMed] [Google Scholar]
- Tricot GJ, Jayaram HN, Lapis E, Natsumeda Y, Nichols CR, Kneebone P, et al. (1989). Biochemically directed therapy of leukemia with tiazofurin, a selective blocker of inosine 5′‐phosphate dehydrogenase activity. Cancer Res 49: 3696–3701. [PubMed] [Google Scholar]
- Uzuka Y, Saito Y (1988). Treatment of myelodysplastic syndrome using SM‐108 in relation to stem cell kinetics. Gan To Kagaku Ryoho 15: 1215–1222. [PubMed] [Google Scholar]
- Valencia A, Masala E, Rossi A, Martino A, Sanna A, Buchi F, et al. (2014). Expression of nucleoside‐metabolizing enzymes in myelodysplastic syndromes and modulation of response to azacitidine. Leukemia 28: 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wolf C, Jansen R, Yamaguchi H, de Haas M, van de Wetering K, Wijnholds J, et al. (2008). Contribution of the drug transporter ABCG2 (breast cancer resistance protein) to resistance against anticancer nucleosides. Mol Cancer Ther 7: 3092–3102. [DOI] [PubMed] [Google Scholar]
- Yoshida N, Nakamura M, Fukui M, Morisada S, Ogino S, Inaba M, et al. (1983). Optimal treatment schedule and antitumor spectrum of 4‐carbamoylimidazolium 5‐olate (SM‐108) in murine tumors. Cancer Res 43: 5851–5856. [PubMed] [Google Scholar]
- Zoref‐Shani E, Sperling O (1980). Characterization of purine nucleotide metabolism in cultured fibroblasts with deficiency of hypoxanthine‐guanine phosphoribosyltransferase and with superactivity of phosphoribosylpyrophosphate synthetase. Enzyme 25: 413–418. [DOI] [PubMed] [Google Scholar]
- Zuck K, Choe M J, Strand K J, Strovel JW, Hamilton J, Bol DK (2008) GTP as a biomarker of inosine monophosphate dehydrogenase (IMPDH) inhibition, in patients with advanced hematological malignancies treated with AVN944 in a phase I trial. J Clin Oncol, ASCO Annual Meeting Proceedings (Post‐Meeting Edition) 26(No. 15S): 14515. [Google Scholar]