

Abstract
Drug‐induced toxicity is a key issue for public health because some side effects can be severe and life‐threatening. These adverse effects can also be a major concern for the pharmaceutical companies since significant toxicity can lead to the interruption of clinical trials, or the withdrawal of the incriminated drugs from the market. Recent studies suggested that endoplasmic reticulum (ER) stress could be an important event involved in drug liability, in addition to other key mechanisms such as mitochondrial dysfunction and oxidative stress. Indeed, drug‐induced ER stress could lead to several deleterious effects within cells and tissues including accumulation of lipids, cell death, cytolysis, and inflammation. After recalling important information regarding drug‐induced adverse reactions and ER stress in diverse pathophysiological situations, this review summarizes the main data pertaining to drug‐induced ER stress and its potential involvement in different adverse effects. Drugs presented in this review are for instance acetaminophen (APAP), arsenic trioxide and other anticancer drugs, diclofenac, and different antiretroviral compounds. We also included data on tunicamycin (an antibiotic not used in human medicine because of its toxicity) and thapsigargin (a toxic compound of the Mediterranean plant Thapsia garganica) since both molecules are commonly used as prototypical toxins to induce ER stress in cellular and animal models.
Keywords: Adverse effects, drug, endoplasmic reticulum, ER stress, liver, toxicity
Abbreviations
- APAP
acetaminophen
- CHOP
C/EBP homologous protein
- COX
cyclooxygenase
- CYP
cytochrome P450
- ER
endoplasmic reticulum
- GI
gastrointestinal
- GRP
glucose‐related protein 78
- GSH
glutathione
- NAPQI
N‐acetyl‐p‐benzoquinone imine
- NRTI
nucleoside reverse transcriptase inhibitor
- NSAID
nonsteroidal anti‐inflammatory drug
- 4‐PBA
4‐phenylbutyrate
- PERK
PKR‐like ER kinase
- PI
protease inhibitor
- PPAR
peroxisome proliferator‐activated receptor
- ROS
reactive oxygen species
- SREBP
sterol regulatory element‐binding protein
- UPR
unfolded protein response
Introduction
Drug‐induced toxicity is an important issue for public health and the well‐being of patients. Indeed, some side effects can be severe and require a hospitalization, or even can cause the death of some patients. These adverse effects can also be a major concern for the pharmaceutical companies because significant toxicity can lead to the interruption of clinical trials, or the withdrawal of the incriminated drugs from the market (Labbe et al. 2008; Elangbam 2010). In the recent years, the endoplasmic reticulum (ER) stress emerged as a potential important event involved in drug liability, in addition to other key mechanisms such as mitochondrial dysfunction and oxidative stress. Thus, the main objective of the present review was to collect the available information regarding drug‐induced ER stress and its potential role in the occurrence of different adverse reactions. For this purpose, we performed a PubMed search of literature published in the English language using the following queries (by alphabetical order): adverse effect, adverse event, adverse reaction, drug, drug‐induced, ER stress, and toxicity. This data mining was also completed by using Google Scholar®. Besides the different drugs presented below, we also included data on tunicamycin and thapsigargin since both molecules are commonly used as prototypical toxins to induce ER stress in cellular and animal models.
Drug‐Induced Adverse Effects and Main Mechanisms of Toxicity
Many drugs of our modern pharmacopeia are able to induce adverse effects that can involve different tissues such as the liver, heart, kidney, lung, skeletal muscles, adipose tissue, and peripheral nerves (Marrer and Dieterle 2010; Begriche et al. 2011; Hohenegger 2012; Tocchetti et al. 2013; Miltenburg and Boogerd 2014). Notably, a single drug can damage several tissues in the same patient and induce several types of lesions in a given tissue. For instance, drug‐induced liver injury includes hepatic cytolysis, autoimmune‐like hepatitis, cholestasis, steatosis, steatohepatitis, cirrhosis, and hepatocellular adenoma (Wang et al. 2013a; Leise et al. 2014). This variety of liver lesions actually reflects the occurrence of different mechanisms of toxicity and the potential involvement of several types of hepatic cells such as hepatocytes, cholangiocytes, stellate cells, activated lymphocytes, and Kupffer cells (Stirnimann et al. 2010; Czaja 2011; Padda et al. 2011).
It is currently acknowledged that mitochondrial dysfunction and oxidative stress are two major mechanisms whereby drugs can induce injuries in different tissues such as the liver, heart, and kidney (Sardao et al. 2008; John and Herzenberg 2009; Baillie and Rettie 2011; Begriche et al. 2011; Deavall et al. 2012; Tocchetti et al. 2013; Miltenburg and Boogerd 2014). Actually, drugs can induce oxidative stress via several mechanisms including by increasing reactive oxygen species (ROS) production by the dysfunctional mitochondria and higher NADPH oxidase activity and by reducing cellular antioxidant defenses (Labbe et al. 2008; Baillie and Rettie 2011; Begriche et al. 2011; Leung et al. 2012).
Drugs can be harmful either directly or indirectly after their biotransformation into one or several toxic reactive metabolites by cellular enzymes such as cytochromes P450 (CYPs) (Baillie and Rettie 2011; Begriche et al. 2011; Leung et al. 2012). Importantly, CYPs and other xenobiotic metabolizing enzymes (XMEs) are expressed mainly in the liver but also in other tissues including the gastrointestinal (GI) tract, kidney, lung, brain, heart, and white adipose tissue (Dutheil et al. 2009; Thelen and Dressman 2009; Ellero et al. 2010; Knights et al. 2013; Ravindranath and Strobel 2013). Hence, the generation of toxic reactive metabolites can occur in liver and extra‐hepatic tissues (Gu et al. 2005; Ding and Kaminsky 2003). Finally, besides mitochondrial dysfunction and oxidative stress, there is increasing evidence that ER stress can be another important mechanism in drug‐induced adverse effects, as underlined in this review.
Definition and Cellular Consequences of ER Stress
The ER plays a crucial role in the synthesis of all the proteins that are secreted from cells, or inserted into organelle membranes. Efficient protein folding in the ER requires a tight coupling between the arrival of new proteins in the ER lumen and the ER folding capacity. Efficient folding requires ER‐resident proteins such as chaperones and foldases that are calcium‐binding/buffering proteins (Coe and Michalak 2009; Halperin et al. 2014). These proteins include for instance calreticulin, glucose‐regulated protein 78 (GRP78, also known as immunoglobulin‐binding protein or BiP), GRP94, and protein disulfide isomerase (PDI). When the demand for protein folding increases (e.g., enhanced protein synthesis, accumulation of mutated, or abnormal proteins, etc) and exceeds protein folding capacity, misfolded/unfolded proteins accumulate in the ER lumen and trigger an ER stress. Many physiological or pathological situations can interfere with protein folding and can thus impact ER homeostasis such as alterations of ER luminal calcium stores, energy depletion, redox disturbances, glucose starvation, lipid accumulation, viruses, ethanol intoxication, and xenobiotics (Malhi and Kaufman 2011; Cnop et al. 2012; Cheng et al. 2013; Chen et al. 2014). Notably, ER stress is leading to the activation of the so‐called unfolded protein response (UPR), the role of which is to maintain protein homeostasis by decreasing the load of unfolded proteins and increasing the protein folding capacity (Fig. 1). In addition, ER stress is also able to activate autophagy‐ and proteasome‐dependent proteolysis when misfolded proteins are in excess (Hoyer‐Hansen and Jäättelä 2007; Digaleh et al. 2013).
Figure 1.
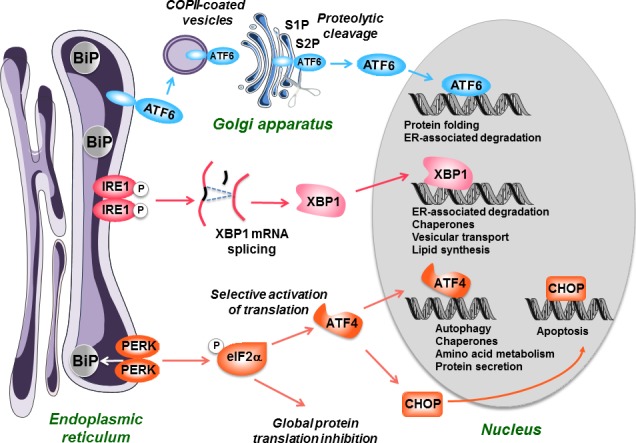
The unfolded protein response (UPR). When an endoplasmic reticulum (ER) stress occurs, the cell initiates an adaptive response called the UPR. It starts with the activation of three effectors, PKR‐like ER kinase (PERK), IRE1, and ATF6, following the removal of the chaperone BiP (GRP78) that maintains them in an inactivated state. PERK is a kinase which phosphorylates and inactivates the elongation initiation factor eIF2α, leading to a general decrease in protein translation. However, eIF2α selectively stimulates the translation of ATF4, a transcription factor which possesses a specific structure (uORF) on its mRNA. ATF4 then activates the synthesis of chaperones and proteins involved in autophagy, protein secretion, and amino acid metabolism. IRE1 possesses a kinase activity leading to its autophosphorylation and activation of a RNAse activity. This leads to the splicing of XBP1 mRNA, which is then translated into an active transcription factor. The transcription factor ATF6, which is bound to the ER membranes as an inactive precursor is transferred via COPII‐coated vesicles to the Golgi apparatus, where it is cleaved by the S1P and S2P proteases into an active form. XBP1 and ATF6 will then activate in the nucleus the transcription of a set of factors allowing to restore ER homeostasis including chaperones, foldases, and proteins involved in the degradation of unfolded polypeptides (ER‐associated degradation). If these mechanisms are not efficient to restore ER and cell homeostasis, the UPR will eventually activate mechanisms leading to cell apoptosis, in particular via the transcription factor C/EBP homologous protein (CHOP).
UPR activation involves three different effectors (also referred to as the 3 arms of the UPR): inositol requiring 1α (IRE1α), PKR‐like ER kinase (PERK), and activating transcription factor‐6α (ATF6α) (Cawley et al. 2011; Dara et al. 2011; Malhi and Kaufman 2011). One of the main consequences of UPR activation is the inhibition of translation, in order to curb the synthesis of new proteins. This response is mediated by the kinase PERK that phosphorylates the alpha subunit of eukaryotic translation‐initiation factor 2 (eIF2α) leading to a rapid reduction in the initiation of mRNA translation and thus reducing the load of new client proteins in the ER (Fig. 1).
Despite protein synthesis attenuation during ER stress, there is a small subset of genes that are specifically transcribed and translated via the three branches of the UPR (Fig. 1). For instance, these genes encode ER‐associated degradation (ERAD) proteins, ER chaperones (BiP/GRP78 and GRP94), and enzymes able to expand protein folding capacity including PDI and foldases (Kaufman 1999; Dara et al. 2011). This physiological response is also called the adaptive UPR. In contrast, if the UPR is strong enough and/or sustained, some deleterious consequences can be observed such as apoptosis, which can be dependent or not of mitochondria (Hom et al. 2007; Deniaud et al. 2008; Malhi and Kaufman 2011; Sano and Reed 2013). Importantly, the mitochondrial‐independent pathway of apoptosis triggered by ER stress is the consequence of the activation of different specific effectors such as the transcription factor C/EBP homologous protein (CHOP, also referred to as GADD153) and caspase 12 (Malhi and Kaufman 2011; Sano and Reed 2013). Depending of the stressors, caspase 12 activation can require the translocation of caspase 7 from the cytosol to the ER membranes, or a calcium‐mediated recruitment of calpain to the ER surface (Nakagawa and Yuan 2000; Rao et al. 2001; Xie et al. 2002).
Another consequence of ER stress in some cells can be the stimulation of lipid synthesis by activation of different transcription factors such as sterol regulatory element‐binding proteins (SREBPs), CAAT/enhancer‐binding proteins (C/EBP) and peroxisome proliferator‐activated receptor‐γ (PPARγ) (Werstuck et al. 2001; Colgan et al. 2007; Malhi and Kaufman 2011; Lee et al. 2012). It is also important to point out that ER stress and impaired protein folding can lead to the production of a significant amount of ROS (Malhotra and Kaufman 2007; Malhotra et al. 2008). Interestingly, it is estimated that 25% of the ROS generated in a cell results from the formation of disulfide bonds in the ER during normal protein synthesis (Tu and Weissman 2004; Sevier and Kaiser 2008). Conversely, ROS accumulation can induce the oxidation of resident ER proteins, including proteins of the polypeptide folding machinery such as PDI and BiP (van der Vlies et al. 2002, 2003), thus leading to an ER stress and creating a vicious cycle (Malhotra and Kaufman 2007). To alleviate the deleterious effects of ROS, the PERK branch of the UPR can activate the transcription of several key antioxidant enzymes via the phosphorylation of nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2) (Cullinan and Diehl 2006; Flamment et al. 2012).
Drug‐Induced ER Stress and Adverse Effects
Acetaminophen (APAP)
APAP is a popular drug for the management of pain and hyperthermia (Table 1). Although APAP is usually deemed as a safe drug, APAP intoxication after an overdose can lead to massive hepatocellular necrosis and acute liver failure (Craig et al. 2011). Moreover, there is evidence that the maximum recommended dose (i.e., 4 g/day) can induce mild to moderate hepatic cytolysis, even in healthy individuals (Watkins et al. 2006; Winnike et al. 2010). APAP can also induce acute kidney injury in some patients, either after poisoning or at therapeutic doses (Blakely and McDonald 1995; Kato et al. 2014). APAP‐induced acute nephrotoxicity manifests as acute tubular necrosis with oliguric renal failure, which can occur alone or in association with liver injury (Blakely and McDonald 1995; Jones and Prescott 1997). APAP overdose has also been reported to induce cardiotoxicity, pancreatitis, and ototoxicity (Jones and Prescott 1997; Yorgason et al. 2011).
Table 1.
Drugs for which adverse events have been linked to ER stress
| Drug(s) | Pharmacological class | Main side effects | Mechanism of ER stress | Additional information |
|---|---|---|---|---|
| Acetaminophen (APAP) | Antalgic and antipyretic | Hepatotoxicity and nephrotoxicity, commonly after an overdose | Currently unknown. Possible involvement of the APAP‐derived reactive metabolite NAPQI, which binds to several key microsomal proteins including PDI and calreticulin | ER stress could be a late event after APAP intoxication, compared to other deleterious events such as mitochondrial dysfunction and oxidative stress |
| Amiodarone | Antiarrhythmic and antianginal | Hypotension, cutaneous reactions, thyroid toxicity, liver injury, and pulmonary toxicity | Currently unknown | ER stress could be involved in some amiodarone‐induced adverse effects (e.g., thyroid and lung toxicity) in addition to mitochondrial dysfunction |
| Arsenic trioxide (As2O3) | Anticancer agent used to treat acute promyelocytic leukemia and other hematologic malignancies | GI disorders, rash, hematologic toxicity, infections, cardiac toxicity renal toxicity, myopathy, neuropathy, and hepatotoxicity | Possible involvement of oxidative stress and impairment of protein folding | In addition to ER stress, mitochondrial dysfunction is also likely to be involved in the pathogenesis of some adverse effects induced by arsenic trioxide |
| Bleomycin | Anticancer drug used in different malignancies such as lymphomas, head and neck cancers as well as ovarian and testicular cancers | GI disorders, cutaneous reactions, myelosuppression, and life‐threatening pulmonary toxicity | Currently unknown. Possible mechanisms could involve oxidative stress | ER stress could be involved in bleomycin‐induced pulmonary toxicity |
| Bortezomib (PS‐341) | Anticancer drug used to treat multiple myeloma and mantle cell lymphoma | GI symptoms, fatigue, peripheral neuropathy, and thrombocytopenia | Proteasome inhibition | ER stress is one mechanism whereby bortezomib is able to induced apoptosis in cancer cells. ER stress could be involved in bortezomib‐induced peripheral neuropathy |
| Cisplatin | Anticancer drug used in various malignancies such as testicular, gastric, lung, breast, and ovarian cancers | GI disorders, kidney injury, neurotoxicity, hepatotoxicity, and cardiotoxicity | Currently unknown. Possible mechanisms could involve oxidative stress and/or the covalent binding of cisplatin to key microsomal proteins | ER stress could be involved in cisplatin‐induced kidney injury |
| Clozapine and olanzapine | Antipsychotics | GI disorders, drowsiness, extrapyramidal symptoms, elevation in liver enzymes, and obesity (with related metabolic disorders such as insulin resistance and fatty liver) | Currently unknown. Possible involvement of increased cytosolic calcium | Although fatty liver induced by clozapine and olanzapine could be secondary to obesity, hepatocyte ER stress directly induced by these drugs might also be involved |
| Cyclosporin | Immunosuppressant | Infections, hypertension, neurotoxicity, nephrotoxicity, and hepatotoxicity (including cholestasis and cytolysis) | Currently unknown. In hepatocytes, a possible mechanism could be the covalent binding of cyclosporin metabolites to pivotal microsomal proteins | Possible involvement of ER stress in cyclosporin‐induced cholestasis. Cyclosporin‐induced ER stress in kidney could be indirect consequence of vascular dysfunction |
| Diclofenac | NSAID | GI complications (including gastric injury and intestinal damage), hypersensitivity reactions, hepatotoxicity, and kidney injury | Currently unknown. Possible involvement of increased intracellular calcium in gastric mucosal cells. Because diclofenac metabolism generates two p‐benzoquinone imines similar to APAP‐derived NAPQI, binding to key ER proteins could be involved (in liver and other tissues expressing CYPs) | Possible involvement of ER stress in diclofenac‐induced GI complications and liver toxicity |
| Efavirenz | Antiretroviral (non‐nucleoside reverse transcriptase inhibitor) | Rash, neuropsychological symptoms, lipodystrophy, and hepatotoxicity (in particular cytolysis and cholestasis) | Possible secondary consequence of mitochondrial dysfunction and release of mitochondrial calcium into the cytosol | Possible involvement of ER stress in efavirenz‐induced hepatotoxicity |
| Erlotinib | Anticancer drug used in different malignancies including non‐small cell lung cancer and pancreatic cancer | Rash and GI manifestations (in particular severe diarrhea) | Currently unknown | Possible involvement of ER stress in erlotinib‐induced small intestinal injury and diarrhea |
| Furosemide | Diuretic used to treat hypertension and edema | Dehydration, hypotension, hyponatremia, and hypokalemia | Currently unknown | Hepatic ER stress has been detected in mice treated by furosemide. Extrapolation to humans is doubtful since this drug induces virtually no hepatotoxicity in patients |
| Indomethacin | NSAID | GI complications (including gastric injury and intestinal damage), hypersensitivity reactions, hepatotoxicity, and kidney injury | Currently unknown. Possible involvement of increased intracellular calcium (in gastric mucosal cells) | Possible involvement of ER stress in indomethacin‐induced GI complications and liver toxicity |
| Paclitaxel (Taxol) | Anticancer agent used in different malignancies including ovarian, breast, and lung cancers | GI disorders, cardiac and skeletal muscle toxicity, myelosuppression, neurotoxicity, and acute liver injury (mostly hepatic cytolysis) | Currently unknown | Possible involvement of ER stress in paclitaxel‐induced neurotoxicity |
| Protease inhibitors (e.g., indinavir and ritonavir) | Antiretroviral | GI toxicity, rash, kidney injury, hepatotoxicity (including cytolysis, cholestasis, and steatosis), dyslipidemia, lipodystrophy, insulin resistance, and type 2 diabetes | Possible involvement of proteasome inhibition and oxidative stress | Significant ER stress has been showed with atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir, but not with amprenavir, darunavir, and tipranavir |
| Sertraline | Antidepressant (selective serotonin reuptake inhibitor) | Somnolence, GI disorders, tremor, sexual dysfunction, weight gain, and liver injury | Currently unknown. Possible role of mitogen‐activated protein kinase (MAPK) pathway activation | Possible involvement of ER stress in sertraline‐induced hepatotoxicity |
| Thapsigargin | Sesquiterpene lactone isolated from the plant Thapsia garganica, which has long been used in traditional Arabian medicine | Severe skin irritation, salivary hypersecretion, gastroenteritis, nervous disorders, and death | Inhibition of SERCA, thus leading to severe calcium depletion in the ER | Prototypical inducer of ER stress. In addition to ER stress, increase in free cytosolic calcium is inducing apoptosis in different types of cells treated with thapsigargin |
| Troglitazone | Antidiabetic (via activation of PPARγ) | Severe and fatal liver injury leading to the withdrawal of troglitazone from the market | Currently unknown. Possible role of mitogen‐activated protein kinase (MAPK) pathway activation | Possible involvement in troglitazone‐induced liver injury in addition with mitochondrial dysfunction and oxidative stress |
| Tunicamycin | Antibiotic active against different bacteria, fungi, and viruses | Major neurotoxicity and death in animals. Kidney and liver lesions are also observed in the treated animals | Impairment of glycosylation of newly synthesized proteins in the ER leading to the disruption of their folding | Prototypical inducer of ER stress. Tunicamycin has never been used in human medicine due to its toxicity |
| Zidovudine (AZT) | Antiretroviral (nucleoside reverse transcriptase inhibitor) | Lactic acidosis, myopathy, and hepatotoxicity (including cytolysis and steatosis) | Currently unknown. Possible impairment of proteasome activity | Although ER stress could be involved in steatosis, the current knowledge points to a major role of mitochondrial dysfunction in fat accretion induced by zidovudine |
ER, endoplasmic reticulum; CYPs, cytochromes P450; GI, gastrointestinal; PDI, protein disulfide isomerase; NAPQI, N‐acetyl‐p‐benzoquinone imine; NSAID, nonsteroidal anti‐inflammatory drug; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase; PPARγ, peroxisome proliferator‐activated receptor‐γ.
APAP is mainly metabolized in the liver into the nontoxic glucuronide and sulfate conjugates. However, a small amount of APAP is oxidized to the reactive metabolite N‐acetyl‐p‐benzoquinone imine (NAPQI) by CYPs 2E1 (CYP2E1) and 3A4 (Gonzalez 2007; Aubert et al. 2012). After its generation, NAPQI is normally detoxified by glutathione (GSH) when APAP is taken at the recommended dosage. After APAP overdose, or in the presence of predisposing factors, high levels of NAPQI can induce cytotoxicity. Indeed, once GSH is deeply depleted and no longer available for NAPQI detoxication, this reactive metabolite binds to different proteins, in particular at the mitochondrial level (Michaut et al. 2014). This is followed by profound mitochondrial dysfunction and ATP depletion, overproduction of ROS, c‐jun N‐terminal kinase (JNK) activation, and massive hepatocellular necrosis (Jaeschke et al. 2012; Michaut et al. 2014). APAP‐induced acute nephrotoxicity could also involve CYP2E1‐mediated generation of NAPQI, depletion of GSH, and secondary oxidative stress (Hart et al. 1994; Das et al. 2010).
In vivo and in vitro investigations reported that APAP was also able to induce an ER stress and that such deleterious effect could play an significant role in APAP‐induced cell death in liver, kidney, or inner ear (Lorz et al. 2004; Nagy et al. 2007, 2010; Uzi et al. 2013; Kalinec et al. 2014). In one of these studies, mortality induced by a lethal dose of APAP (1 g/kg) was completely prevented in CHOP knockout mice but data regarding liver injury induced by a lower dose of this painkiller (500 mg/kg) showed either a protection or no effect depending of the route of APAP administration (Uzi et al. 2013). In addition, other studies dealing with APAP hepatotoxicity did not find markers of ER stress (Van Summeren et al. 2011; Hur et al. 2012; van Summeren et al. 2013). Actually, some data in mice indicated that ER stress was a relatively late event after APAP intoxication (500 mg/kg), being significant only 12 hours following APAP administration (Hur et al. 2012; Uzi et al. 2013). In contrast, mitochondrial alterations, ATP depletion, JNK activation, oxidative stress, and increased cytosolic calcium occurred much earlier in mouse liver after the same dose of APAP (Burcham and Harman 1988; Jaeschke 1990; Ruepp et al. 2002; Aubert et al. 2012; Hur et al. 2012). Investigations in the human hepatoma HuH7 cell line also suggested that ER stress induced by APAP occurred well after mitochondrial alterations (Macanas‐Pirard et al. 2005). Thus, further studies are required to determine whether ER stress is a major pathway involved in APAP toxicity and cell death.
The mechanism whereby APAP induces ER stress is poorly understood. A first hypothesis could be the occurrence of microsomal alterations secondary to NAPQI generation. Indeed, it has been reported that APAP induced severe GSH depletion, lipid peroxidation, and an oxidative shift of the ER oxidoreductases ERp72 and PDI in liver microsomes (Nagy et al. 2007; Letelier et al. 2011). Furthermore, NAPQI can covalently bind to several microsomal proteins such as GSH‐S‐transferase, PDI, and calreticulin (Pumford et al. 1990; Weis et al. 1992; Zhou et al. 1996; Shin et al. 2007). Because PDI and calreticulin play a major role in protein folding and calcium sequestration within the ER (Coe and Michalak 2009), covalent binding of NAPQI to these proteins could induce an ER stress. Interestingly, it has been shown that other reactive benzoquinones induced an ER stress (Wang et al. 2006). Second, ER stress might also be a secondary consequence of mitochondrial dysfunction, as discussed later on with other drugs such as arsenic trioxide and efavirenz.
Amiodarone
This broad‐spectrum antiarrhythmic drug also presents an antianginal effect (Table 1). The main adverse effects of amiodarone include hypotension, thyroid toxicity (hyper‐ or hypothyroidism), pulmonary toxicity including bronchiolitis and pulmonary fibrosis, and hepatic lesions such as steatosis, steatohepatitis, and cirrhosis (Dusman et al. 1990; Fromenty and Pessayre 1995; Santangeli et al. 2012). Numerous studies have shown that mitochondrial dysfunction is a major mechanism of amiodarone‐induced toxicity in liver and other tissues (Fromenty and Pessayre 1995; Di Matola et al. 2000; Nicolescu et al. 2008; Begriche et al. 2011). Recently, amiodarone was shown to induce ER stress in thyrocytes and lung epithelial cells (Mahavadi et al. 2014; Lombardi et al. 2015), but the involved mechanism was not determined in these studies. In contrast, no ER stress was detected in hepatocytes treated with amiodarone, although it was observed with cyclosporin A in the same investigations (Van Summeren et al. 2011; van Summeren et al. 2013).
Arsenic Trioxide
Arsenic trioxide (As2O3) is used as an effective anticancer drug for the treatment of acute promyelocytic leukemia (APL) (Table 1). Interestingly, arsenic trioxide can be effective in APL patients refractory to all‐trans retinoic acid and other conventional anticancer drugs such as anthracyclines (Shen et al. 1997; Breccia and Lo‐Coco 2012). This compound could also be useful in other hematologic malignancies such as multiple myeloma and myelodysplastic syndromes, although higher doses are needed in these diseases (Douer and Tallman 2005; Xu et al. 2014). As2O3 exerts its powerful therapeutic effect in APL by promoting the degradation of a specific oncogenic protein, the so‐called PML‐RARα fusion protein (Emadi and Gore 2010; Zhang et al. 2010). In contrast, regarding the other hematologic malignancies, this drug could be effective by activating several pathways leading to cancer cell apoptosis including mitochondrial dysfunction, oxidative stress, and DNA damage (Pelicano et al. 2003; Emadi and Gore 2010; Kumar et al. 2014; Xu et al. 2014). Notably, some of these effects could be the consequence of a direct interaction of arsenic trioxide with GSH and protein thiols (Scott et al. 1993; Hughes 2002; Lu et al. 2007; Zhang et al. 2010). Some studies also suggested that ER stress could be involved in the anticancer action of arsenic trioxide (Du et al. 2006; Chen et al. 2012; Chiu et al. 2015).
As2O3 can induce different adverse effects including GI disorders, rash, hematologic toxicity (e.g., thrombocytopenia and neutropenia), infections, cardiac and renal toxicity, myopathy, neuropathy, and hepatotoxicity (Douer and Tallman 2005; Schiller et al. 2006; Echaniz‐Laguna et al. 2012). Some of these side effects can be serious and even fatal in a few cases (Westervelt et al. 2001; Schiller et al. 2006). It is also noteworthy that chronic arsenic poisoning via contaminated water and food is also associated with a wide array of deleterious effects, which overlap with arsenic trioxide toxicity (Yoshida et al. 2004; Emadi and Gore 2010). Unsurprisingly, investigations in noncancerous cells and in rodents suggested that some of these side effects could be secondary to oxidative stress and mitochondrial dysfunction (Aposhian and Aposhian 2006; Jomova et al. 2011; Mathews et al. 2013; Garcia‐Sevillano et al. 2014; Vineetha et al. 2015). Several studies also showed that arsenic trioxide triggered ER stress in different types of nonmalignant cells such as myoblasts, vascular endothelial cells, pancreatic β‐cells, neutrophils, and macrophages (Binet et al. 2010; Lu et al. 2011; Yen et al. 2012; Srivastava et al. 2013; Weng et al. 2014; King et al. 2015). In several of these studies, ER stress was associated with other deleterious events including loss of the mitochondrial membrane potential, increased intracellular free calcium, ROS overproduction, and apoptosis (Lu et al. 2011; Yen et al. 2012; Srivastava et al. 2013; Weng et al. 2014; King et al. 2015). Interestingly, arsenic trioxide‐induced ER stress could be prevented by the GSH precursor N‐acetylcysteine (Lu et al. 2011; Yen et al. 2012; Srivastava et al. 2013; King et al. 2015), or by the mitochondrial‐targeted antioxidant tiron (Weng et al. 2014). Since arsenic trioxide has been shown to increase mitochondrial ROS production possibly via an impairment of the respiratory chain (Paul et al. 2008; Vineetha et al. 2015), ROS released from mitochondria could thus play a significant role in arsenic trioxide‐induced ER stress. However, arsenic trioxide might also directly induce ER stress because some authors showed with in vitro assays that this compound was able to impair protein folding (Ramadan et al. 2009; Jacobson et al. 2012).
Bleomycin
This anticancer drug is used to treat several types of malignancies such as lymphomas, head, and neck cancers as well as ovarian and testicular cancers (Table 1). Bleomycin induces oxidative DNA damage and subsequent cell death after formation of a complex with iron (or copper) and oxygen, which generates free radicals able to create DNA single‐ and double‐strand breaks (Hecht 2000; Chen and Stubbe 2005). Notably, this drug is also able to oxidatively damage other cellular targets such as RNA, proteins, and lipids (Chen and Stubbe 2005). Bleomycin‐induced adverse effects include GI disorders, myelosuppression, cutaneous reactions, and pulmonary toxicity, in particular severe and life‐threatening lung fibrosis (Froudarakis et al. 2013; Della Latta et al. 2015). The pathophysiology of bleomycin lung toxicity seems complex, but could involve oxidative stress, inflammation, and overproduction of profibrotic cytokine transforming growth factor‐β (TGFβ) (Chen and Stubbe 2005; Kikuchi et al. 2011; Froudarakis et al. 2013; Della Latta et al. 2015). Recent investigations suggested that ER stress could also be involved (Zhao et al. 2014a,b; Tanaka et al. 2015). In one of these studies, bleomycin‐induced lung fibrosis was significantly reduced in CHOP−/− mice (Tanaka et al. 2015). Although the mechanism of bleomycin‐induced ER stress was not determined in these investigations, it is conceivable that oxidative damage of key ER components could be involved. In this regard, previous studies reported that N‐acetylcysteine was able to alleviate bleomycin‐induced lung injury (Hagiwara et al. 2000; Serrano‐Mollar et al. 2003; Kikuchi et al. 2011).
Bortezomib
This anticancer drug is approved for the treatment of several types of cancers including multiple myeloma and mantle cell lymphoma (Table 1). The mechanism of action of bortezomib (also known as PS‐341) involves the specific inhibition of proteasome, which is responsible for cytotoxicity and apoptosis in cancer cells (Holkova and Grant 2012; Dou and Zonder 2014). Indeed, proteasome inhibition secondarily leads to ER stress, ROS production, and activation of JNK and other signaling pathways inducing cell death (Lee et al. 2003; Fribley et al. 2004; Holkova and Grant 2012). The most frequent adverse events with this drug are GI symptoms, peripheral neuropathy, fatigue, and thrombocytopenia (Holkova and Grant 2012; Argyriou et al. 2014). One study suggested that bortezomib‐induced ER stress could be involved in peripheral neuropathy, possibly by impairing myelin synthesis (Shin et al. 2010).
Cisplatin
Cisplatin, also known as cisplatinum or cis‐diamminedichloroplatinum (II) (CDDP), is an antitumor agent used to treat various malignancies including testicular, gastric, lung, breast, and ovarian cancers (Table 1). Although the formation of cisplatin‐DNA adducts is deemed to be a key mechanism leading to cancer cell apoptosis, other mechanisms independent of DNA damage could also be involved such as ROS overproduction, increased plasma membrane fluidity, and ER stress (Mandic et al. 2003; Maccio and Madeddu 2013; Dasari and Tchounwou 2014).
Cisplatin treatment can induce several types of adverse effects including GI disorders, kidney injury, neurotoxicity, hepatotoxicity, and cardiotoxicity (Kitamura 2008; Pabla and Dong 2008; Florea and Büsselberg 2011). Many investigations have been performed to decipher the mechanisms of cisplatin‐induced acute kidney injury because this frequent adverse effect limits the use of cisplatin in cancer therapy (Kitamura 2008; Pabla and Dong 2008). The emerging picture to explain cisplatin‐induced tubular cell injury and death is a combination of different pathophysiological events including mitochondrial dysfunction, ROS overproduction, increased tumor necrosis factor‐α (TNFα) generation, and ER stress (Kruidering et al. 1997; Kitamura 2008; Pabla and Dong 2008; Servais et al. 2008; Mukhopadhyay et al. 2012). Indeed, markers of ER stress such as increased BiP/GRP78 and CHOP expression and caspase 12 activation have been found in different investigations performed in renal cells and kidneys of rodents treated with cisplatin (Liu and Baliga 2005; Peyrou et al. 2007; Khan et al. 2013; Kong et al. 2013; Gao et al. 2014; Wang et al. 2014; Chen et al. 2015). Interestingly, kidneys of CHOP−/− mice showed less evidence of cell death when treated with cisplatin (Zinszner et al. 1998). Despite these investigations, the precise mechanism(s) whereby cisplatin is able to induce ER stress is still unknown. Possible hypotheses could be cisplatin‐induced oxidative stress and/or irreversible binding of this drug to key ER components. In this regard, cisplatin has been shown to covalently bind to different proteins, including in the microsomal compartment (Pattanaik et al. 1992; Litterst and Schweitzer 1988; Huliciak et al. 2012).
Clozapine and olanzapine
These antipsychotic drugs are structurally similar to the sedative and anxiolytic benzodiazepines (Table 1). The antipsychotic action of clozapine and olanzapine is deemed to be due to dopamine D2 receptor blockade, although interactions with other neurotransmitter receptors have been reported (Reynolds and Kirk 2010; Brosda et al. 2014). The most common adverse effects of these compounds include GI manifestations, drowsiness, extrapyramidal symptoms, cardiac effects, elevation of liver enzymes, and obesity, which can be associated with insulin resistance and fatty liver (Melkersson and Dahl 2003; Begriche et al. 2011; De Fazio et al. 2015; Rojo et al. 2015). Although clozapine‐ and olanzapine‐induced fatty liver could be secondary to obesity, investigations in human hepatocytes also suggested a role of ER stress, SREBP1c activation and increased hepatic lipogenesis, possibly via a calcium‐dependent pathway (Lauressergues et al. 2012). Another study showed that olanzapine induced ER stress and reduced insulin secretion in hamster pancreatic β cells (Ozasa et al. 2013). However, the pathophysiological significance of this effect remains unclear since this drug classically induces hyperinsulinemia, even in the absence of weight gain (Melkersson and Dahl 2003; Teff et al. 2013).
Cyclosporin
Cyclosporin (also known as cyclosporin A) is a potent immunosuppressant isolated from the fungus Tolypocladium inflatum (Table 1). This drug significantly improves the short‐term transplant survival by reducing the incidence of acute allograft rejection. Like tacrolimus (FK506), cyclosporin exerts its immunosuppressive effect by inhibiting calcineurin, thus impairing the activation of the nuclear factor of activated T cells (NFAT) signaling pathway that plays a major role in T‐cell‐mediated adaptive immune response (Graham 1994; Jorgensen et al. 2003). Unfortunately, cyclosporin treatment can induce numerous side effects, which are sometimes severe and require the discontinuation of the treatment. Besides infections (that are the direct consequence of immunosuppression), cyclosporin can also induce hypertension, neurotoxicity (e.g., paresthesia and tremor), nephrotoxicity, and hepatotoxicity (Krupp and Monka 1990; Graham 1994; Servais et al. 2008). Liver toxicity not only includes cholestasis but also hepatic cytolysis and steatosis (Biour et al. 2004; Wang et al. 2013a).
Cyclosporin is extensively metabolized in liver and kidney by several CYPs, which generate at least 30 different metabolites (Kelly et al. 1999; Zheng et al. 2013). Some of these metabolites are able to form stable adducts with cellular proteins indicating that they are highly reactive in nature (Nagelkerke et al. 1987; Sadrieh and Thomas 1994; Christians and Sewing 1995). Notably, oxidative stress seems to be an important mechanism whereby cyclosporin can lead to cytolysis in different cell types (Wolf et al. 1997; Nishida et al. 2003; Navarro‐Antolin et al. 2007) and to kidney or liver damage in treated rodents (Tariq et al. 1999; Kaya et al. 2008; Haleagrahara et al. 2009). Cyclosporin‐induced oxidative stress could be the consequence of mitochondrial dysfunction and reduced antioxidant defenses (Kaya et al. 2008; Palomero et al. 2001; Redondo‐Horcajo et al. 2010; Xiao et al. 2013).
Several studies carried out in mouse hepatocytes and the human hepatoma cell lines HepG2 and HepaRG showed that cyclosporin was able to induce an ER stress (Van Summeren et al. 2011; van Summeren et al. 2013; Szalowska et al. 2013; van den Hof et al. 2014; Sharanek et al. 2014). Interestingly, the investigations performed in HepaRG cells showed that reduced canalicular efflux of taurocholate induced by high concentrations (50 μmol/L) of cyclosporin were alleviated by 4‐phenylbutyrate (4‐PBA), a chemical chaperone commonly used to protect against ER stress (Sharanek et al. 2014). However, lower concentrations of cyclosporin (10 μM) impaired taurocholate canalicular efflux without inducing ER stress (Sharanek et al. 2014). Notably, several studies showed that cyclosporin is a potent inhibitor of the bile salt export pump (BSEP) (Dawson et al. 2012; Pedersen et al. 2013). Thus, these investigations suggest that cyclosporin‐induced impairment of bile acid efflux and cholestasis could be secondary to ER stress only for high concentrations of this immunosuppressant. Whether ER stress plays a role in cyclosporin‐induced hepatic cytolysis and steatosis has not been addressed in the aforementioned studies. Moreover, it is still unknown how cyclosporin can induce ER stress in liver. A first mechanism could involve CYP‐mediated generation of reactive metabolites able to alter key ER proteins since some of these metabolites can bind covalently to microsomal proteins (Nagelkerke et al. 1987; Sadrieh and Thomas 1994). Another mechanism might be the occurrence of oxidative stress and lipid peroxidation within the ER (Barth et al. 1991; Serino et al. 1993).
Different in vitro and in vivo investigations also attempted to determine whether ER stress could be an important mechanism involved in cyclosporin‐induced nephrotoxicity (Han et al. 2008; Pallet et al. 2008; Lhotak et al. 2012; Sarro et al. 2012; Liu et al. 2015). These studies performed in cultured renal proximal tubule cells and kidneys of treated rats clearly showed that this immunosuppressant agent was able to induce an ER stress (e.g., activation of CHOP, BiP/GRP78, and GRP94). In addition, markers of ER stress have been found in renal biopsies of patients with cyclosporin‐induced acute or chronic nephrotoxicity (Lhotak et al. 2012; Hama et al. 2013). However, despite these data, a direct causal relationship between cyclosporin‐induced ER stress and nephrotoxicity is doubtful. Indeed, numerous experimental and clinical studies provided strong evidence that cyclosporin‐induced nephrotoxicity is mainly the consequence of reduced renal blood flow due to afferent and efferent arteriolar vasoconstriction (English et al. 1987; Kon et al. 1990; Smith et al. 1992; Grieve et al. 1993; Pannu and Nadim 2008). Since hypoxia can induce an ER stress in particular in the kidney (Inagi et al. 2014; Suh et al. 2014), renal ER stress observed during cyclosporin nephrotoxicity could be a mere consequence of vascular dysfunction induced by this drug. Finally, it is noteworthy that cyclosporin is not able to bind covalently to microsomal proteins in kidney, contrary to what happens in liver microsomes (Nagelkerke et al. 1987; Sadrieh and Thomas 1994).
Diclofenac and indomethacin
The nonsteroidal anti‐inflammatory drugs (NSAIDs) diclofenac and indomethacin are discussed together because of their similar pharmacological and toxicological profiles (Table 1). Indeed, these drugs are potent nonselective inhibitors of the cyclooxygenases 1 and 2 (COX1 and COX2) used for the treatment of chronic inflammatory diseases such as osteoarthritis and rheumatoid arthritis. As many other nonselective COX inhibitors, the most significant side effects induced by diclofenac and indomethacin are GI complications such as bleeding and ulceration, hypersensitivity reactions (e.g., anaphylaxis, skin eruptions, and bronchospasms), and kidney injury (Rossi et al. 1985; Richy et al. 2004; Gonzalez et al. 2009; Castellsague et al. 2012; Wallace 2012). These drugs can also induce hepatotoxicity, mostly cytolytic hepatitis and cholestasis (Banks et al. 1995; Biour et al. 2004; Wang et al. 2013a).
The most common NSAID‐induced GI toxicity seems to be gastric injury, albeit intestinal damage could also be a frequent side effect observed in treated patients (Wallace 2012; Boelsterli et al. 2013). Numerous studies have shown that a major mechanism of NSAID‐induced gastropathy is COX inhibition and the secondary decreased production of mucosal prostaglandins, which have a major cytoprotective action on the gastric mucosa (Rainsford and Willis 1982; Wallace 2012; Seminerio et al. 2014). Nevertheless, some investigations suggested that ER stress could also be involved for some NSAIDs including diclofenac and indomethacin (Tsutsumi et al. 2004; Ohyama et al. 2012). In one of these studies, indomethacin‐induced apoptosis in guinea‐pig gastric mucosal cells was partially but significantly prevented by expression of a dominant‐negative form of CHOP (Tsutsumi et al. 2004). One mechanism whereby some NSAIDs are able to induce ER stress and apoptosis in gastric mucosal cells could be an increase in intracellular calcium levels secondary to membrane permeabilization (Tanaka et al. 2005).
Contrary to NSAID‐induced gastropathy, COX inhibition does not seem to play a primary role in NSAID‐induced enteropathy, although decreased prostaglandin synthesis could render the intestine more susceptible to injury. Indeed, oxidative stress, mitochondrial dysfunction, and TNFα release could be important mechanisms in NSAID enteropathy (Wallace 2012; Boelsterli et al. 2013). Recent investigations carried out in rat intestinal slices suggested that diclofenac could be toxic via an ER stress in addition to mitochondrial injury and oxidative stress (Niu et al. 2014). ER stress was also observed in cultured rat enterocytes treated with indomethacin (Narabayashi et al. 2015).
An important mechanism of diclofenac‐induced hepatocellular injury is the CYP‐mediated generation of two p‐benzoquinone imines and possibly other reactive metabolites triggering oxidative stress, mitochondrial dysfunction, and cell death (Bort et al. 1999; Boelsterli 2003; Park et al. 2005). In addition, diclofenac is able to induce direct mitochondrial dysfunction (Begriche et al. 2011; Porceddu et al. 2012; Massart et al. 2013). Although ER stress with CHOP induction has been observed in hepatic cells treated with diclofenac (Franceschelli et al. 2011; Nadanaciva et al. 2013; Fredriksson et al. 2014), it is still unclear whether this event plays a primary role in diclofenac‐induced cell demise, compared to mitochondrial dysfunction. Moreover, the mechanism whereby diclofenac induces ER stress in hepatocytes is still unknown. Although diclofenac was shown to bind irreversibly to hepatic microsomal proteins in several studies (Kretz‐Rommel and Boelsterli 1994; Hargus et al. 1995; Obach et al. 2008), the targeted polypeptides are still unidentified. It will be interesting to determine whether diclofenac‐derived p‐benzoquinone imines can bind covalently to the microsomal PDI and calreticulin, as does APAP‐derived NAPQI (Zhou et al. 1996). Finally, it is noteworthy that indomethacin was also found to induce ER stress in hepatic cells (Franceschelli et al. 2011). However, similar to diclofenac, it is still unclear whether ER stress is the primary mechanism of liver injury induced by indomethacin since this drug is also able to directly impair mitochondrial function (Jacob et al. 2001; Porceddu et al. 2012).
Efavirenz
Efavirenz is a non‐nucleoside reverse transcriptase inhibitors (NNRTIs) used to treat the human immunodeficiency virus (HIV) infection (Table 1). Efavirenz can induce several types of side effects including neuropsychological manifestations (e.g., dizziness, headache, and depression), rash, lipodystrophy and hepatotoxicity, which can occur as hepatic cytolysis and cholestasis (Sulkowski et al. 2002; Biour et al. 2004; Maggiolo 2009; Margolis et al. 2014). Several studies also suggest that efavirenz could trigger endothelial dysfunction and increase the cardiovascular risk in HIV‐infected patients (Maggi et al. 2011; Gupta et al. 2012). The exact mechanism whereby efavirenz can be toxic is still unclear. Several studies showed that efavirenz can induce mitochondrial dysfunction in different experimental models including human hepatic cells (Apostolova et al. 2010; Blas‐García et al. 2010), primary rat neurons (Purnell and Fox 2014) and different brain regions of treated mice (Streck et al. 2011). A recent study reported that efavirenz was able to induce an ER stress in human hepatic Hep3B cells, with an upregulation of CHOP, BiP/GRP78, and phospho‐eIF2α (Apostolova et al. 2013). However, investigations demonstrated that this deleterious effect was actually secondary to mitochondrial dysfunction and the release of mitochondrial calcium into the cytosol (Apostolova et al. 2013). Two recent studies also showed that efavirenz‐induced ER stress in human endothelial cells (Bertrand and Toborek 2015; Weiß et al. 2015), but the involved mechanism was not determined in these investigations.
Protease inhibitors
Protease inhibitors (PIs) are antiretroviral drugs frequently used for the treatment of HIV‐infected patients (Table 1). This important pharmacological class includes numerous compounds such as amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and tipranavir. Taken as a whole, these drugs can induce GI toxicity such as nausea and diarrhea, rash, liver and kidney injury, dyslipidemia, lipodystrophy, insulin resistance, and type 2 diabetes (Gougeon et al. 2004; Pannu and Nadim 2008; Izzedine et al. 2009; Caron‐Debarle et al. 2010; Margolis et al. 2014). As regards hepatotoxicity, different types of lesions have been described in treated patients such as cytolysis, cholestasis, steatosis, and cirrhosis (Biour et al. 2004; Wang et al. 2013a). Some PIs such as indinavir, lopinavir, and ritonavir are suspected to increase the risk of cardiovascular diseases including myocardial infarction (Bavinger et al. 2013; Margolis et al. 2014).
PIs are toxic via different pathways, in particular by inducing mitochondrial dysfunction and oxidative stress and by promoting inflammation (Gougeon et al. 2004; Caron et al. 2007; Caron‐Debarle et al. 2010; Mencarelli et al. 2012; Bociaga‐Jasik et al. 2013; Cassol et al. 2013; Reyskens et al. 2013). Several studies also reported that some PIs are able to induce ER stress in different types of cells (e.g., macrophages, adipocytes, hepatocytes) and tissues (e.g., liver, intestine) (Zhou et al. 2005; Gupta et al. 2007; Cao et al. 2010; Touzet and Philips 2010; Wu et al. 2010; Zha et al. 2010, 2013; Brüning 2011; Zhou 2011; Brüning et al. 2012; Apostolova et al. 2013; Taura et al. 2013; Wang et al. 2013b). In these investigations, ER stress was consistently observed with atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir. In contrast, amprenavir, darunavir, and tipranavir induced only a weak or no ER stress in the different tested experimental models (Wu et al. 2010; Zhou 2011; Taura et al. 2013; Zha et al. 2013). Notably, some of these studies brought evidence that PI‐induced ER stress was involved in lipid metabolism alterations, inflammation, cell apoptosis, and tissue damage, in particular by using CHOP−/− mice or cells (Zhou et al. 2005; Cao et al. 2010; Wu et al. 2010; Zha et al. 2010, 2013; Zhou 2011; Wang et al. 2013b). According to these investigations, ER stress could thus be an important mechanism whereby some PIs are able to induce metabolic disturbances such as lipodystrophy, hepatic steatosis and cytolysis, dyslipidemia, and some cardiovascular complications. Lastly, it is noteworthy that PI‐induced ER stress and hepatocyte damage have been shown to be potentiated by ethanol exposure (Kao et al. 2012; Hu et al. 2015). Interestingly, one of these studies reported that the combination of PIs and ethanol significantly induced ER stress and cell death in mouse hepatocytes but not in mouse Kupffer and stellate cells (Hu et al. 2015). These experimental data could be clinically relevant because previous investigations reported a greater risk of PI‐induced severe hepatic injury in patients abusing alcohol (Nunez et al. 2001).
The precise mechanisms whereby some PIs can induce ER stress is still unclear, although two hypotheses have been proposed. The first mechanism could be PI‐induced inhibition of proteasome activity (Parker et al. 2005; Pyrko et al. 2007; Bono et al. 2012). Indeed, several studies carried out with other compounds (e.g., bortezomib) showed that specific impairment of proteasome function can potently trigger ER stress (Lee et al. 2003; Fribley et al. 2004; Bono et al. 2012). In this context, ER stress occurs because the proteasome cannot degrade the few misfolded proteins that are normally but constantly produced within the ER (Fribley and Wang 2006; Xu et al. 2005). Proteasome inhibition has been shown for atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir, although the potency of this inhibitory effect seemed variable between these drugs (Schmidtke et al. 1999; Pajonk et al. 2002; Piccinini et al. 2002; Parker et al. 2005; Bono et al. 2012). The second potential mechanism could involve PI‐induced oxidative stress, as suggested by some investigations (Touzet and Philips 2010; Taura et al. 2013). However, the precise origin of oxidative stress has not been addressed in these studies.
Thapsigargin
This sesquiterpene lactone of the Mediterranean plant Thapsia garganica is a prototypical and powerful inducer of ER stress (Zinszner et al. 1998; Rutkowski et al. 2008; Kammoun et al. 2009) (Table 1). More precisely, thapsigargin induces ER stress by inhibiting sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) (Fig. 2), thus leading to severe depletion of ER calcium (Kaufman 1999; Denmeade and Isaacs 2005; Schönthal 2012). In addition to ER stress‐induced activation of downstream effectors that can trigger cell death, the concomitant increase in free cytosolic calcium is also a potent proapoptotic signal in different types of cells (Jiang et al. 1994; Treiman et al. 1998; Kim et al. 2008). In mast cells, thapsigargin‐induced augmentation in cytosolic calcium is leading to the release of histamine, which could explain the potent skin irritating effects of Thapsia garganica (Jacobsen et al. 1987; Doan et al. 2015). Other toxic effects of this plant can include salivary hypersecretion, gastroenteritis, vomiting and diarrhea, nervous disorders, fever, and death in the most severe cases of intoxication (Bnouham et al. 2006; Hammiche et al. 2013). Despite its significant toxicity, Thapsia garganica has long been used in traditional Arabian medicine for the treatment of different illnesses including rheumatic pains, women infertility, and pulmonary diseases (Treiman et al. 1998; Bnouham et al. 2006; Hammiche et al. 2013). In the past few years, different thapsigargin analogs have been developed as potential drug candidates for the treatment of prostate cancers (Isaacs 2005; Dubois et al. 2013; Doan et al. 2015).
Figure 2.
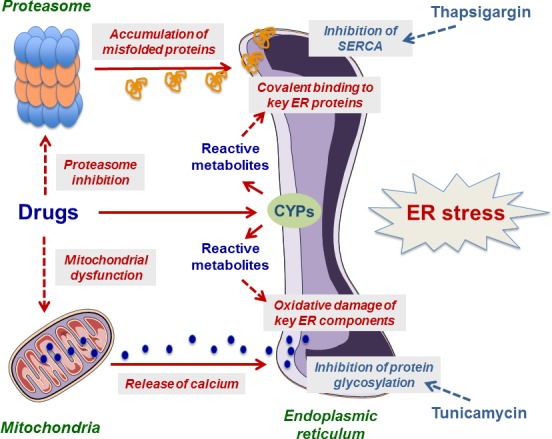
Potential mechanisms of drug‐induced endoplasmic reticulum (ER) stress. Drugs are able to induce ER stress via different mechanisms including proteasome inhibition, mitochondrial dysfunction, and alteration of key ER components. The latter mechanism is suspected with drugs that are transformed into one or several reactive metabolites able to bind covalently to ER proteins and/or to induce oxidative damage of ER components secondary to oxidative stress. Cytochromes P450 (CYPs) are often involved in the generation of reactive metabolites. The figure also indicates the respective targets of thapsigargin and tunicamycin, which are two prototypical inducers of ER stress. Further information is provided in the text and Table 1.
Tunicamycin
This glucosamine‐containing antibiotic produced by Streptomyces lysosuperificus is active against different types of microorganisms including gram‐positive bacteria, fungi, yeast, and viruses (Table 1). Early investigations have demonstrated that this antibiotic was able to inhibit the synthesis of N‐glycoproteins in yeast and bacteria (Kuo and Lampen 1974; Bettinger and Young 1975) as well as in animals and plants (Fig. 2) (Takatsuki and Tamura 1971; Tkacz and Lampen 1975; Ericson et al. 1977). More specifically, tunicamycin impairs the transfer of N‐glucosamine to the polyisoprenoid lipid dolichol phosphate, the first step in the synthesis of the lipid‐linked oligosaccharide that is used as a precursor for the N‐glycoproteins (Ericson et al. 1977; Bieberich 2014). Notably, this biosynthetic step takes place within the ER in eukaryotic cells (Bieberich 2014). Consequently, impairment of glycosylation of newly synthesized proteins is leading to the disruption of their folding in the ER (Kaufman 1999; Schönthal 2012). In addition, this alteration of peptide conformation can secondarily prevent the cellular secretion of some proteins such as different immunoglobulins (Hickman et al. 1977; Elbein 1981).
Tunicamycin has never been used in human medicine because of its major toxicity. In particular, acute tunicamycin administration in rodent, cattle and sheep is leading to severe neurotoxicity with disturbed consciousness, convulsions, and paralysis, which can occasionally lead to the death of the treated animals (Leaver et al. 1988; Bourke and Carrigan 1993). The lesions in the brain are mainly vascular and it is deemed that blood vessel damage could secondary lead to brain ischemia and hypoxic neuronal damage (Finnie and O'Shea 1988; Leaver et al. 1988). Indeed, tunicamycin damages the endothelial cells of small blood vessels with marked dilatation of the rough ER (i.e., typical of the presence of ER stress), which appears to strongly distort their cytoplasm and cause stenosis of the blood vessel lumen (Finnie and O'Shea 1988; Finnie and O'Shea 1990a). Tunicamycin‐induced direct endothelial toxicity has also been observed in vitro in vascular endothelial cells (Finnie and O'Shea 1990a; Galan et al. 2014; Suganya et al. 2014). Nevertheless, in vitro studies also showed that tunicamycin can induce neuronal cell death in different types of neurons (Chang and Korolev 1996; Kosuge et al. 2006; Galehdar et al. 2010). Some of these in vitro investigations demonstrated that tunicamycin‐induced endothelial and neuronal cell death was linked to ER stress (Kosuge et al. 2006; Galehdar et al. 2010; Suganya et al. 2014).
In addition to its neurotoxicity, tunicamycin is also able to induce acute liver and kidney injuries in the intoxicated animals (Zinszner et al. 1998; Finnie and O'Shea 1989; Finnie and O'Shea 1990b; Marciniak et al. 2004; Rutkowski et al. 2008; Carlisle et al. 2014). Notably, tunicamycin‐induced acute kidney injury is blunted in CHOP knockout mice, or in mice treated with the chemical chaperone 4‐PBA (Marciniak et al. 2004; Carlisle et al. 2014). In liver, the lesions observed in tunicamycin‐intoxicated animals include steatosis, bile ductular hyperplasia, swollen hepatocytes, and the presence of apoptotic bodies (Finnie and O'Shea 1989; Finnie and O'Shea 1990b; Finnie et al. 2004; Rutkowski et al. 2008). Interestingly, swollen hepatocytes are characterized by a marked dilatation of the rough ER (Finnie and O'Shea 1989; Finnie et al. 2004). The precise mechanism of tunicamycin‐induced hepatic steatosis is still elusive. Although some data suggested that tunicamycin could favor de novo lipogenesis in particular via a SREBP1c‐dependent pathway (Kammoun et al. 2009; Lee et al. 2012), other investigations did not support this mechanism (Rutkowski et al. 2008; Jo et al. 2013). Alternatively, tunicamycin could favor intrahepatic lipid accumulation by other mechanisms including reduced expression and activity of the transcription factor PPARα, a master regulator of fatty acid oxidation (Rutkowski et al. 2008; Yamamoto et al. 2010; Chikka et al. 2013), impairment of triglyceride excretion (Zhang et al. 2011), or increased lipid delivery to the liver (Jo et al. 2013; Bogdanovic et al. 2015).
Zidovudine
This nucleoside reverse transcriptase inhibitor (NRTI) is the first antiretroviral drug marketed for the treatment of HIV (Table 1). Other NRTIs include stavudine (d4T), lamivudine (3TC), didanosine (ddI), and tenofovir. Despite their pharmacological interest, these drugs can unfortunately induce a large array of side effects, which are sometimes fatal, such as hepatotoxicity, renal dysfunction, myopathy, pancreatitis, peripheral neuropathy, lipodystrophy, and lactic acidosis (Izzedine et al. 2009; Caron‐Debarle et al. 2010; Begriche et al. 2011; Margolis et al. 2014). NRTI‐induced liver injury includes hepatic cytolysis, microvesicular and/or macrovacuolar steatosis, steatohepatitis, and cirrhosis (Biour et al. 2004; Massart et al. 2013; Wang et al. 2013a). Numerous investigations have shown that the main mechanism involved in NRTI toxicity is mitochondrial dysfunction, as these drugs can reduce mitochondrial DNA (mtDNA) levels in various tissues via DNA polymerase γ inhibition (Gaou et al. 2001; Igoudjil et al. 2006; Schon and Fromenty 2016). A recent study showed that zidovudine‐induced hepatic steatosis in mice was associated with an ER stress, with a concomitant increase in SREBP1c expression and reduced PPARα expression (Banerjee et al. 2013). However, mtDNA levels and mitochondrial function were not measured in this study and thus it remains unclear whether zidovudine‐induced ER stress was the main mechanism of fat accumulation in mouse liver. In addition, the mechanism whereby zidovudine could induce an ER stress is still unknown, although a previous study showed that this drug was able to significantly inhibit the proteasome activity (Piccinini et al. 2002).
Other drugs
The antidepressant sertraline and the antidiabetic troglitazone can be toxic for the liver (Table 1) (Biour et al. 2004; Wang et al. 2013a). Importantly, troglitazone caused several cases of fatal liver failure and was thus withdrawn from the market (Labbe et al. 2008). The role and mechanisms of ER stress in sertraline and troglitazone‐induced hepatotoxicity have been discussed in a recent review (Chen et al. 2014). Hepatic ER stress has also been detected in a mouse model of liver toxicity induced by the diuretic drug furosemide (Table 1) (Qu et al. 2014). However, extrapolation of these experimental data to humans is doubtful because furosemide induces virtually no hepatotoxicity in treated patients (Biour et al. 2004; McGill et al. 2015).
Erlotinib is an anticancer drug that can induce severe diarrhea requiring an interruption of the treatment (Table 1) (Reck et al. 2011). A recent study provided some evidence that erlotinib‐induced ER stress in rat small intestine epithelial cells was involved in apoptosis and reduced expression of E‐cadherin (Fan et al. 2014). These effects might explain why erlotinib can impair the gut barrier integrity and induce diarrhea (Fan et al. 2014). ER stress could also be involved in the neuronal toxicity of the anticancer drug paclitaxel (Table 1) (Tanimukai et al. 2013), in addition to microtubule stabilization (Gornstein and Schwarz 2014).
Concluding Remarks and Remaining Issues
The present review underlines that ER stress could be involved in different adverse effects induced by drugs belonging to different pharmacological classes. However, it is still unclear whether ER stress is a common key pathophysiological signal involved in drug‐induced toxicity. This may be because ER stress has not been frequently assessed in the context of drug‐induced side effects, in contrast to other mechanisms of toxicity such as mitochondrial dysfunction and oxidative stress. It is also noteworthy that a great number of studies reported only a mere association between ER stress and cellular (or tissue) alterations. Hence, it is difficult to ascertain from these reports that ER stress is bona fine involved in drug‐induced toxicity. In contrast, some of the aforementioned studies brought causal relationship by using CHOP‐/‐ mice or cells (Zinszner et al. 1998; Marciniak et al. 2004; Wu et al. 2010; Zhou 2011; Uzi et al. 2013; Wang et al. 2013b; Zha et al. 2013; Tanaka et al. 2015). Thus, future investigations should use these experimental models, or similar ones with knockout of other major proteins involved in ER stress such as IRE1, PERK, or ATF6 (Fig. 1).
As previously mentioned, some investigators used 4‐PBA in order to determine whether this chemical chaperone could alleviate drug‐induced cellular (or tissue) damage. However, besides being protective against ER stress, 4‐PBA presents other biological effects. For instance, 4‐BPA was shown to be a histone deacetylase (HDAC) inhibitor and an autophagy inducer, to regulate Hsp70 expression and to present antioxidant and anti‐inflammatory properties (Iannitti and Palmieri 2011; Suaud et al. 2011; Roy et al. 2012; Rekha et al. 2015). Thus, protection afforded by 4‐BPA might not be fully related to ER stress defense.
In some studies, the presence of ER stress is based on the assessment of only a few UPR markers such as BiP/GRP78 and CHOP. However, accurate ER stress monitoring should include several markers activated by the three canonical arms of the UPR, as underscored in previous reviews (Samali et al. 2010; Cawley et al. 2011; Hiramatsu et al. 2011). Since the detection of cleaved ATF6, phospho‐PERK, and phospho‐IRE1 is rather difficult, different downstream targets of these three effectors can be studied (Samali et al. 2010). Hence, in addition to BiP/GRP78 and CHOP, other UPR markers should be investigated at the mRNA and/or protein level such as spliced X‐box binding protein 1 (XBP1), homocysteine‐inducible ER protein (HERP), ER degradation enhancer mannosidase alpha‐like 1 (EDEM1), phospho‐eIF2α, and ATF4 (Samali et al. 2010; Cawley et al. 2011; Hiramatsu et al. 2011). In addition, investigations on drug‐induced ER stress should systematically include a positive control such as tunicamycin or thapsigargin.
It must be also pointed out that the exact mechanism of ER stress was not addressed in most of the studies cited in this review. In contrast, investigations performed with a few drugs suggested that ER stress could be secondary to oxidative stress (i.e., arsenic trioxide), proteasome inhibition (i.e., bortezomib), or increased cytosolic calcium (i.e., efavirenz) (Fig. 2). The data reported with efavirenz are particularly interesting because ER stress and elevated cytosolic calcium were found to be secondary to mitochondrial dysfunction (Apostolova et al. 2013). Notably, mitochondrial dysfunction induced by other factors including toxins can secondarily cause an augmentation in cytosolic calcium (Luo et al. 1997; Sheehan et al. 1997; Biswas et al. 1999; Amuthan et al. 2002). Since numerous drugs are able to impair mitochondrial function (Labbe et al. 2008; Begriche et al. 2011; Porceddu et al. 2012; Jones et al. 2014), it will be important to determine whether some of these drugs are also able to induce an ER stress by increasing cytosolic calcium.
Another mechanism of drug‐induced ER stress that deserves to be carefully addressed is the covalent binding of reactive metabolites to ER proteins (Fig. 2). Indeed, drugs can be toxic because of their biotransformation by CYPs into one or several reactive metabolites, as previously discussed. Importantly, CYPs are mainly located in the ER membranes, although some CYPs could be also found in mitochondria (Avadhani et al. 2011; Knockaert et al. 2011). In addition, it is noteworthy that CYPs are expressed in various tissues, as previously mentioned. Thus, covalent binding of reactive metabolites to ER proteins could occur with some drugs in different tissues and might lead to ER stress and side effects.
Mitochondrial dysfunction can secondarily lead to ER stress, as previously mentioned. However, a reciprocal relationship has also been showed in some pathophysiological situations. For instance, ER stress induced by thapsigargin and tunicamycin can secondarily promote mitochondrial calcium accumulation and opening of the mitochondrial permeability transition (MPT) pore as well as other mitochondrial alterations leading to cell death (Hom et al. 2007; Deniaud et al. 2008). Actually, there is a close connection between ER and mitochondria and the physical association of the two organelles creates a structure known as the mitochondria‐associated ER membrane (MAM) (Csordas et al. 2006; Hayashi et al. 2009). In the physiological context, this connection allows a constant transfer of calcium between ER and mitochondria (Csordas et al. 2010; Giacomello et al. 2010). During ER stress, calcium can be massively transferred to the mitochondria through these ER‐mitochondria microdomains, or as a consequence of increased cytosolic calcium (Mandic et al. 2003; Benali‐Furet et al. 2005; Deniaud et al. 2008; Timmins et al. 2009). Hence, induction of ER stress can secondarily augment the mitochondrial overload of calcium and thus favor mitochondrial membrane permeabilization and the release of proapoptotic factors such as cytochrome c and apoptosis‐inducing factor (Deniaud et al. 2008; Pinton et al. 2008; Timmins et al. 2009; Fonseca et al. 2013). In the context of drug‐induced toxicity, it will thus be important to gain more information regarding the relationship between ER stress and mitochondrial dysfunction and to determine whether some drugs can specifically interact with MAM. This may help to find strategies to prevent, or alleviate, some adverse effects that can be severe and life‐threatening in some patients.
Disclosure
None declared.
Foufelle F., Fromenty B., Role of endoplasmic reticulum stress in drug‐induced toxicity, Pharma Res Per, 4(1), 2016, e00211, doi: 10.1002/prp2.211
References
- Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG (2002). Mitochondrial stress‐induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 21: 7839–7849. [DOI] [PubMed] [Google Scholar]
- Aposhian HV, Aposhian MM (2006). Arsenic toxicology: five questions. Chem Res Toxicol 19: 1–15. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez‐Sucerquia LJ, Moran A, Alvarez A, Blas‐Garcia A, Esplugues JV (2010). Enhanced oxidative stress and increased mitochondrial mass during efavirenz‐induced apoptosis in human hepatic cells. Br J Pharmacol 160: 2069–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, Gomez‐Sucerquia LJ, Alegre F, Funes HA, Victor VM, Barrachina MD, et al. (2013). ER stress in human hepatic cells treated with efavirenz: mitochondria again. J Hepatol 59: 780–789. [DOI] [PubMed] [Google Scholar]
- Argyriou AA, Cavaletti G, Bruna J, Kyritsis AP, Kalofonos HP (2014). Bortezomib‐induced peripheral neurotoxicity: an update. Arch Toxicol 88: 1669–1679. [DOI] [PubMed] [Google Scholar]
- Aubert J, Begriche K, Delannoy M, Morel I, Pajaud J, Ribault C, et al. (2012). Differences in early acetaminophen hepatotoxicity between obese ob/ob and db/db mice. J Pharmacol Exp Ther 342: 676–687. [DOI] [PubMed] [Google Scholar]
- Avadhani NG, Sangar MC, Bansal S, Bajpai P (2011). Bimodal targeting of cytochrome P450s to endoplasmic reticulum and mitochondria: the concept of chimeric signals. FEBS J 278: 4218–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie TA, Rettie AE (2011). Role of biotransformation in drug‐induced toxicity: influence of intra‐ and inter‐species differences in drug metabolism. Drug Metab Pharmacokinet 26: 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Abdelmegeed MA, Jang S, Song BJ (2013). Zidovudine (AZT) and hepatic lipid accumulation: implication of inflammation, oxidative and endoplasmic reticulum stress mediators. PLoS ONE 8: e76850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AT, Zimmerman HJ, Ishak KG, Harter JG (1995). Diclofenac‐associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 22: 820–827. [PubMed] [Google Scholar]
- Barth SA, Inselmann G, Engemann R, Heidemann HT (1991). Influences of Ginkgo biloba on cyclosporin a induced lipid peroxidation in human liver microsomes in comparison to vitamin E, glutathione and N‐acetylcysteine. Biochem Pharmacol 41: 1521–1526. [DOI] [PubMed] [Google Scholar]
- Bavinger C, Bendavid E, Niehaus K, Olshen RA, Olkin I, Sundaram V, et al. (2013). Risk of cardiovascular disease from antiretroviral therapy for HIV: a systematic review. PLoS ONE 8: e59551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begriche K, Massart J, Robin MA, Borgne‐Sanchez A, Fromenty B (2011). Drug‐induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol 54: 773–794. [DOI] [PubMed] [Google Scholar]
- Benali‐Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F, Lagorce D, et al. (2005). Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 24: 4921–4933. [DOI] [PubMed] [Google Scholar]
- Bertrand L, Toborek M (2015). Dysregulation of endoplasmic reticulum stress and autophagic responses by the antiretroviral drug efavirenz. Mol Pharmacol 88: 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettinger GE, Young FE (1975). Tunicamycin, an inhibitor of Bacillus peptidoglycan synthesis: a new site of inhibition. Biochem Biophys Res Commun 67: 16–21. [DOI] [PubMed] [Google Scholar]
- Bieberich E (2014). Synthesis, processing, and function of N‐glycans in N‐glycoproteins. Adv Neurobiol 9: 47–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet F, Chiasson S, Girard D (2010). Arsenic trioxide induces endoplasmic reticulum stress‐related events in neutrophils. Int Immunopharmacol 10: 508–512. [DOI] [PubMed] [Google Scholar]
- Biour M, Ben Salem C, Chazouillères O, Grangé JD, Serfaty L, Poupon R (2004). Drug‐induced liver injury; fourteenth updated edition of the bibliographic database of liver injuries and related drugs. Gastroenterol Clin Biol 28: 720–759. [DOI] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, et al. (1999). Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter‐organelle crosstalk. EMBO J 18: 522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely P, McDonald BR (1995). Acute renal failure due to acetaminophen ingestion: a case report and review of the literature. J Am Soc Nephrol 6: 48–53. [DOI] [PubMed] [Google Scholar]
- Blas‐García A, Apostolova N, Ballesteros D, Monleon D, Morales JM, Rocha M, et al. (2010). Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology 52: 115–125. [DOI] [PubMed] [Google Scholar]
- Bnouham M, Merhfour FZ, Elachoui M, Legssyer A, Mekhfi H, Lamnaouer D, et al. (2006). Toxic effects of some medicinal plants used in Moroccan traditional medicine. Moroccan J Biol 2–3: 21–30. [Google Scholar]
- Bociaga‐Jasik M, Polus A, Goralska J, Czech U, Gruca A, Sliwa A, et al. (2013). Metabolic effects of the HIV protease inhibitor–saquinavir in differentiating human preadipocytes. Pharmacol Rep 65: 937–950. [DOI] [PubMed] [Google Scholar]
- Boelsterli UA (2003). Diclofenac‐induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 192: 307–322. [DOI] [PubMed] [Google Scholar]
- Boelsterli UA, Redinbo MR, Saitta KS (2013). Multiple NSAID‐induced hits injure the small intestine: underlying mechanisms and novel strategies. Toxicol Sci 131: 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic E, Kraus N, Patsouris D, Diao L, Wang V, Abdullahi A, et al. (2015). Endoplasmic reticulum stress in adipose tissue augments lipolysis. J Cell Mol Med 19: 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bono C, Karlin L, Harel S, Mouly E, Labaume S, Galicier L, et al. (2012). The human immunodeficiency virus‐1 protease inhibitor nelfinavir impairs proteasome activity and inhibits the proliferation of multiple myeloma cells in vitro and in vivo. Haematologica 97: 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bort R, Ponsoda X, Jover R, Gómez‐Lechón MJ, Castell JV (1999). Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther 288: 65–72. [PubMed] [Google Scholar]
- Bourke CA, Carrigan MJ (1993). Experimental tunicamycin toxicity in cattle, sheep and pigs. Aust Vet J 70: 188–189. [DOI] [PubMed] [Google Scholar]
- Breccia M, Lo‐Coco F (2012). Arsenic trioxide for management of acute promyelocytic leukemia: current evidence on its role in front‐line therapy and recurrent disease. Expert Opin Pharmacother 13: 1031–1043. [DOI] [PubMed] [Google Scholar]
- Brosda J, Jantschak F, Pertz HH (2014). α2‐Adrenoceptors are targets for antipsychotic drugs. Psychopharmacology 231: 801–812. [DOI] [PubMed] [Google Scholar]
- Brüning A (2011). Analysis of nelfinavir‐induced endoplasmic reticulum stress. Methods Enzymol 491: 127–142. [DOI] [PubMed] [Google Scholar]
- Brüning A, Matsingou C, Brem GJ, Rahmeh M, Mylonas I (2012). Inhibin beta E is upregulated by drug‐induced endoplasmic reticulum stress as a transcriptional target gene of ATF4. Toxicol Appl Pharmacol 264: 300–304. [DOI] [PubMed] [Google Scholar]
- Burcham PC, Harman AW (1988). Effect of acetaminophen hepatotoxicity on hepatic mitochondrial and microsomal calcium contents in mice. Toxicol Lett 44: 91–99. [DOI] [PubMed] [Google Scholar]
- Cao R, Hu Y, Wang Y, Gurley EC, Studer EJ, Wang X, et al. (2010). Prevention of HIV protease inhibitor‐induced dysregulation of hepatic lipid metabolism by raltegravir via endoplasmic reticulum stress signaling pathways. J Pharmacol Exp Ther 334: 530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle RE, Brimble E, Werner KE, Cruz GL, Ask K, Ingram AJ, et al. (2014). 4‐Phenylbutyrate inhibits tunicamycin‐induced acute kidney injury via CHOP/GADD153 repression. PLoS ONE 9: e84663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron M, Auclair M, Donadille B, Béréziat V, Guerci B, Laville M, et al. (2007). Human lipodystrophies linked to mutations in A‐type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ 14: 1759–1767. [DOI] [PubMed] [Google Scholar]
- Caron‐Debarle M, Boccara F, Lagathu C, Antoine B, Cervera P, Bastard JP, et al. (2010). Adipose tissue as a target of HIV‐1 antiretroviral drugs. Potential consequences on metabolic regulations. Curr Pharm Des 16: 3352–3360. [DOI] [PubMed] [Google Scholar]
- Cassol E, Misra V, Holman A, Kamat A, Morgello S, Gabuzda D (2013). Plasma metabolomics identifies lipid abnormalities linked to markers of inflammation, microbial translocation, and hepatic function in HIV patients receiving protease inhibitors. BMC Infect Dis 13: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellsague J, Riera‐Guardia N, Calingaert B, Varas‐Lorenzo C, Fourrier‐Reglat A, Nicotra F, et al. (2012). Safety of Non‐Steroidal Anti‐Inflammatory Drugs (SOS) Project. Individual NSAIDs and upper gastrointestinal complications: a systematic review and meta‐analysis of observational studies (the SOS project). Drug Saf 35: 1127–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawley K, Deegan S, Samali A, Gupta S (2011). Assays for detecting the unfolded protein response. Methods Enzymol 490: 31–51. [DOI] [PubMed] [Google Scholar]
- Chang JY, Korolev VV (1996). Specific toxicity of tunicamycin in induction of programmed cell death of sympathetic neurons. Exp Neurol 137: 201–211. [DOI] [PubMed] [Google Scholar]
- Chen J, Stubbe J (2005). Bleomycins: towards better therapeutics. Nat Rev Cancer 5: 102–112. [DOI] [PubMed] [Google Scholar]
- Chen J, Wei H, Xie B, Wang B, Cheng J, Cheng J (2012). Endoplasmic reticulum stress contributes to arsenic trioxide‐induced apoptosis in drug‐sensitive and ‐resistant leukemia cells. Leuk Res 36: 1526–1535. [DOI] [PubMed] [Google Scholar]
- Chen S, Melchior WB Jr, Guo L (2014). Endoplasmic reticulum stress in drug‐ and environmental toxicant‐induced liver toxicity. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 32: 83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Liu G, Zou P, Li X, Hao Q, Jiang B, et al. (2015). Epigallocatechin‐3‐gallate protects against cisplatin‐induced nephrotoxicity by inhibiting endoplasmic reticulum stress‐induced apoptosis. Exp Biol Med 240: 1513–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Gong H, Xiao H, Petersen RB, Zheng L, Huang K (2013). Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta 1830: 4860–4871. [DOI] [PubMed] [Google Scholar]
- Chikka MR, McCabe DD, Tyra HM, Rutkowski DT (2013). C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J Biol Chem 288: 4405–4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HW, Tseng YC, Hsu YH, Lin YF, Foo NP, Guo HR, et al. (2015). Arsenic trioxide induces programmed cell death through stimulation of ER stress and inhibition of the ubiquitin‐proteasome system in human sarcoma cells. Cancer Lett 356: 762–772. [DOI] [PubMed] [Google Scholar]
- Christians U, Sewing KF (1995). Alternative cyclosporine metabolic pathways and toxicity. Clin Biochem 28: 547–559. [DOI] [PubMed] [Google Scholar]
- Cnop M, Foufelle F, Velloso LA (2012). Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med 18: 59–68. [DOI] [PubMed] [Google Scholar]
- Coe H, Michalak M (2009). Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys 28: F96–F103. [PubMed] [Google Scholar]
- Colgan SM, Tang D, Werstuck GH, Austin RC (2007). Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein‐2. Int J Biochem Cell Biol 39: 1843–1851. [DOI] [PubMed] [Google Scholar]
- Craig DG, Bates CM, Davidson JS, Martin KG, Hayes PC, Simpson KJ (2011). Overdose pattern and outcome in paracetamol‐induced acute severe hepatotoxicity. Br J Clin Pharmacol 71: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER–mitochondrial interface. Mol Cell 39: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA (2006). Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 38: 317–332. [DOI] [PubMed] [Google Scholar]
- Czaja AJ (2011). Drug‐induced autoimmune‐like hepatitis. Dig Dis Sci 56: 958–976. [DOI] [PubMed] [Google Scholar]
- Dara L, Ji C, Kaplowitz N (2011). The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53: 1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Ghosh J, Manna P, Sil PC (2010). Taurine protects acetaminophen‐induced oxidative damage in mice kidney through APAP urinary excretion and CYP2E1 inactivation. Toxicology 269: 24–34. [DOI] [PubMed] [Google Scholar]
- Dasari S, Tchounwou PB (2014). Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740: 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S, Stahl S, Paul N, Barber J, Kenna JG (2012). In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug‐induced liver injury in humans. Drug Metab Dispos 40: 130–138. [DOI] [PubMed] [Google Scholar]
- De Fazio P, Gaetano R, Caroleo M, Cerminara G, Maida F, Bruno A, et al. (2015). Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat 11: 1995–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deavall DG, Martin EA, Horner JM, Roberts R (2012). Drug‐induced oxidative stress and toxicity. J Toxicol 2012: 645460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Latta V, Cecchettini A, Del Ry S, Morales MA (2015). Bleomycin in the setting of lung fibrosis induction: From biological mechanisms to counteractions. Pharmacol Res 97: 122–130. [DOI] [PubMed] [Google Scholar]
- Deniaud A, el dein Sharaf O, Maillier E, Poncet D, Kroemer G, Lemaire C, et al. (2008) Endoplasmic reticulum stress induces calcium‐dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27:285–299. [DOI] [PubMed] [Google Scholar]
- Denmeade SR, Isaacs JT (2005). The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther 4: 14–22. [DOI] [PubMed] [Google Scholar]
- Di Matola T, D'Ascoli F, Fenzi G, Rossi G, Martino E, Bogazzi F, et al. (2000). Amiodarone induces cytochrome c release and apoptosis through an iodine‐independent mechanism. J Clin Endocrinol Metab 85: 4323–4330. [DOI] [PubMed] [Google Scholar]
- Digaleh H, Kiaei M, Khodagholi F (2013). Nrf2 and Nrf1 signaling and ER stress crosstalk: implication for proteasomal degradation and autophagy. Cell Mol Life Sci 70: 4681–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS (2003). Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue‐selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 43: 149–173. [DOI] [PubMed] [Google Scholar]
- Doan NT, Paulsen ES, Sehgal P, Moller JV, Nissen P, Denmeade SR, et al. (2015). Targeting thapsigargin towards tumors. Steroids 97: 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou QP, Zonder JA (2014). Overview of proteasome inhibitor‐based anti‐cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin‐proteasome system. Curr Cancer Drug Targets 14: 517–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douer D, Tallman MS (2005). Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol 23: 2396–2410. [DOI] [PubMed] [Google Scholar]
- Du Y, Wang K, Fang H, Li J, Xiao D, Zheng P, et al. (2006). Coordination of intrinsic, extrinsic, and endoplasmic reticulum‐mediated apoptosis by imatinib mesylate combined with arsenic trioxide in chronic myeloid leukemia. Blood 107: 1582–1590. [DOI] [PubMed] [Google Scholar]
- Dubois C, Vanden Abeele F, Sehgal P, Olesen C, Junker S, Christensen SB, et al. (2013). Differential effects of thapsigargin analogues on apoptosis of prostate cancer cells: complex regulation by intracellular calcium. FEBS J 280: 5430–5440. [DOI] [PubMed] [Google Scholar]
- Dusman RE, Stanton MS, Miles WM, Klein LS, Zipes DP, Fineberg NS, et al. (1990). Clinical features of amiodarone‐induced pulmonary toxicity. Circulation 82: 51–59. [DOI] [PubMed] [Google Scholar]
- Dutheil F, Dauchy S, Diry M, Sazdovitch V, Cloarec O, Mellottée L, et al. (2009). Xenobiotic‐metabolizing enzymes and transporters in the normal human brain: regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab Dispos 37: 1528–1538. [DOI] [PubMed] [Google Scholar]
- Echaniz‐Laguna A, Benoilid A, Vinzio S, Fornecker LM, Lannes B, Goullé JP, et al. (2012). Mitochondrial myopathy caused by arsenic trioxide therapy. Blood 119: 4272–4274. [DOI] [PubMed] [Google Scholar]
- Elangbam CS (2010). Drug‐induced valvulopathy: an update. Toxicol Pathol 38: 837–848. [DOI] [PubMed] [Google Scholar]
- Elbein AD (1981). The tunicamycins: useful tools for studies on glycoproteins. Trends Biochem Sci 6: 219–221. [Google Scholar]
- Ellero S, Chakhtoura G, Barreau C, Langouët S, Benelli C, Penicaud L, et al. (2010). Xenobiotic‐metabolizing cytochromes P450 in human white adipose tissue: expression and induction. Drug Metab Dispos 38: 679–686. [DOI] [PubMed] [Google Scholar]
- Emadi A, Gore SD (2010). Arsenic trioxide. An old drug rediscovered. Blood Rev 24: 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J, Evan A, Houghton DC, Bennett WM (1987). Cyclosporine‐induced acute renal dysfunction in the rat. Evidence of arteriolar vasoconstriction with preservation of tubular function. Transplantation 44: 135–141. [DOI] [PubMed] [Google Scholar]
- Ericson MC, Gafford JT, Elbein AD (1977). Tunicamycin inhibits GlcNAc‐lipid formation in plants. J Biol Chem 252: 7431–7433. [PubMed] [Google Scholar]
- Fan L, Hu L, Yang B, Fang X, Gao Z, Li W, et al. (2014). Erlotinib promotes endoplasmic reticulum stress‐mediated injury in the intestinal epithelium. Toxicol Appl Pharmacol 278: 45–52. [DOI] [PubMed] [Google Scholar]
- Finnie JW, O'Shea JD (1988). Pathological and pathogenetic changes in the central nervous system of guinea pigs given tunicamycin. Acta Neuropathol 75: 411–421. [DOI] [PubMed] [Google Scholar]
- Finnie JW, O'Shea JD (1989). Acute hepatotoxicity with resultant pulmonary and cerebral embolism in guinea pigs given tunicamycin. Pathology 21: 194–199. [DOI] [PubMed] [Google Scholar]
- Finnie JW, O'Shea JD (1990a). Effect of tunicamycin on the blood‐brain barrier and on endothelial cells in vitro. J Comp Pathol 102: 363–374. [DOI] [PubMed] [Google Scholar]
- Finnie JW, O'Shea JD (1990b). Unusual concretions in the hepatocyte rough endoplasmic reticulum of sheep given tunicamycin. J Comp Pathol 103: 121–123. [DOI] [PubMed] [Google Scholar]
- Finnie JW, Read SH, Swift JG (2004). Apoptosis in liver damage produced by tunicamycin. Aust Vet J 82: 87–90. [DOI] [PubMed] [Google Scholar]
- Flamment M, Hajduch E, Ferré P, Foufelle F (2012). New insights into ER stress‐induced insulin resistance. Trends Endocrinol Metab 23: 381–390. [DOI] [PubMed] [Google Scholar]
- Florea AM, Büsselberg D (2011). Cisplatin as an anti‐tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers 3: 1351–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF (2013). Activation of the endoplasmic reticulum stress response by the amyloid‐beta 1‐40 peptide in brain endothelial cells. Biochim Biophys Acta 1832: 2191–2203. [DOI] [PubMed] [Google Scholar]
- Franceschelli S, Moltedo O, Amodio G, Tajana G, Remondelli P (2011). In the Huh7 Hepatoma Cells Diclofenac and indomethacin activate differently the unfolded protein response and induce ER stress apoptosis. Open Biochem J 5: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson L, Wink S, Herpers B, Benedetti G, Hadi M, de Bont H, et al. (2014). Drug‐induced endoplasmic reticulum and oxidative stress responses independently sensitize toward TNFα‐mediated hepatotoxicity. Toxicol Sci 140: 144–159. [DOI] [PubMed] [Google Scholar]
- Fribley A, Wang CY (2006). Proteasome inhibitor induces apoptosis through induction of endoplasmic reticulum stress. Cancer Biol Ther 5: 745–748. [DOI] [PubMed] [Google Scholar]
- Fribley A, Zeng Q, Wang CY (2004). Proteasome inhibitor PS‐341 induces apoptosis through induction of endoplasmic reticulum stress‐reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 24: 9695–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromenty B, Pessayre D (1995). Inhibition of mitochondrial beta‐oxidation as a mechanism of hepatotoxicity. Pharmacol Ther 67: 101–154. [DOI] [PubMed] [Google Scholar]
- Froudarakis M, Hatzimichael E, Kyriazopoulou L, Lagos K, Pappas P, Tzakos AG, et al. (2013). Revisiting bleomycin from pathophysiology to safe clinical use. Crit Rev Oncol Hematol 87: 90–100. [DOI] [PubMed] [Google Scholar]
- Galan M, Kassan M, Kadowitz PJ, Trebak M, Belmadani S, Matrougui K (2014). Mechanism of endoplasmic reticulum stress‐induced vascular endothelial dysfunction. Biochim Biophys Acta 1843: 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP (2010). Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4‐CHOP‐mediated induction of the Bcl‐2 homology 3‐only member PUMA. J Neurosci 30: 16938–16948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Liu G, Hu Z, Li X, Yang X, Jiang B, et al. (2014). Grape seed proanthocyanidin extract protects from cisplatin‐induced nephrotoxicity by inhibiting endoplasmic reticulum stress‐induced apoptosis. Mol Med Rep 9: 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaou I, Malliti M, Guimont MC, Lettéron P, Demeilliers C, Peytavin G, et al. (2001). Effect of stavudine on mitochondrial genome and fatty acid oxidation in lean and obese mice. J Pharmacol Exp Ther 297: 516–523. [PubMed] [Google Scholar]
- Garcia‐Sevillano MA, Contreras‐Acuna M, García‐Barrera T, Navarro F, Gomez‐Ariza JL (2014). Metabolomic study in plasma, liver and kidney of mice exposed to inorganic arsenic based on mass spectrometry. Anal Bioanal Chem 406: 1455–1469. [DOI] [PubMed] [Google Scholar]
- Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, et al. (2010). Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store‐operated Ca2+ channels. Mol Cell 38: 280–290. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ (2007). The 2006 Bernard B. Brodie Award Lecture. Cyp2E1. Drug Metab Dispos 35: 1–8. [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Lopez‐Herce J, Moraleda C (2009). A child presenting with acute renal failure secondary to a high dose of indomethacin: a case report. J Med Case Rep 3: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornstein E, Schwarz TL (2014). The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology 76: 175–183. [DOI] [PubMed] [Google Scholar]
- Gougeon ML, Pénicaud L, Fromenty B, Leclercq P, Viard JP, Capeau J (2004). Adipocytes targets and actors in the pathogenesis of HIV‐associated lipodystrophy and metabolic alterations. Antivir Ther 9: 161–177. [PubMed] [Google Scholar]
- Graham RM (1994). Cyclosporine: mechanisms of action and toxicity. Cleve Clin J Med 61: 308–313. [DOI] [PubMed] [Google Scholar]
- Grieve EM, Hawksworth GM, Simpson JG, Whiting PH (1993). The reversal of experimental cyclosporin A nephrotoxicity by thromboxane synthetase inhibition. Biochem Pharmacol 45: 1351–1354. [DOI] [PubMed] [Google Scholar]
- Gu J, Cui H, Behr M, Zhang L, Zhang QY, Yang W, et al. (2005). In vivo mechanisms of tissue‐selective drug toxicity: effects of liver‐specific knockout of the NADPH‐cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol Pharmacol 67: 623–630. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Li B, Cerniglia GJ, Ahmed MS, Hahn SM, Maity A (2007). The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia 9: 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta SK, Shen C, Moe SM, Kamendulis LM, Goldman M, Dubé MP (2012). Worsening endothelial function with efavirenz compared to protease inhibitors: a 12‐month prospective study. PLoS ONE 7: e45716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara SI, Ishii Y, Kitamura S (2000). Aerosolized administration of N‐acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med 162: 225–231. [DOI] [PubMed] [Google Scholar]
- Haleagrahara N, Yee TM, Chakravarthi S, Lee N (2009). Protective effect of N‐acetylcysteine on cyclosporine A‐induced lipid peroxide levels and renal dysfunction in rats. Arch Med Sci 1: 16–22. [Google Scholar]
- Halperin L, Jung J, Michalak M (2014). The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 66: 318–326. [DOI] [PubMed] [Google Scholar]
- Hama T, Nakanishi K, Mukaiyama H, Shima Y, Togawa H, Sako M, et al. (2013). Endoplasmic reticulum stress with low‐dose cyclosporine in frequently relapsing nephrotic syndrome. Pediatr Nephrol 28: 903–909. [DOI] [PubMed] [Google Scholar]
- Hammiche V, Merad R, Azzouz M (2013). Thapsia monographies Pp. 285–289 in Hammiche V., Merad R. and Azzouz M., eds. Plantes toxiques à usage médicinal du pourtour méditerranéen. Springler‐Verlag France, Paris. [Google Scholar]
- Han SW, Li C, Ahn KO, Lim SW, Song HG, Jang YS, et al. (2008). Prolonged endoplasmic reticulum stress induces apoptotic cell death in an experimental model of chronic cyclosporine nephropathy. Am J Nephrol 28: 707–714. [DOI] [PubMed] [Google Scholar]
- Hargus SJ, Martin BM, George JW, Pohl LR (1995). Covalent modification of rat liver dipeptidyl peptidase IV (CD26) by the nonsteroidal anti‐inflammatory drug diclofenac. Chem Res Toxicol 8: 993–996. [DOI] [PubMed] [Google Scholar]
- Hart SG, Beierschmitt WP, Wyand DS, Khairallah EA, Cohen SD (1994). Acetaminophen nephrotoxicity in CD‐1 mice. I. Evidence of a role for in situ activation in selective covalent binding and toxicity. Toxicol Appl Pharmacol 126: 267–275. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009). MAM: more than just a housekeeper. Trends Cell Biol 19: 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SM (2000). Bleomycin: new perspectives on the mechanism of action. J Nat Prod 63: 158–168. [DOI] [PubMed] [Google Scholar]
- Hickman S, Kulczycki A Jr, Lynch RG, Kornfeld S (1977). Studies of the mechanism of tunicamycin inhibition of IgA and IgE secretion by plasma cells. J Biol Chem 252: 4402–4408. [PubMed] [Google Scholar]
- Hiramatsu N, Joseph VT, Lin JH (2011). Monitoring and manipulating mammalian unfolded protein response. Methods Enzymol 491: 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Hof WF, Van Summeren A, Lommen A, Coonen ML, Brauers K, van Herwijnen M, et al. (2014). Integrative cross‐omics analysis in primary mouse hepatocytes unravels mechanisms of cyclosporin A‐induced hepatotoxicity. Toxicology 324: 18–26. [DOI] [PubMed] [Google Scholar]
- Hohenegger M (2012). Drug induced rhabdomyolysis. Curr Opin Pharmacol 12: 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkova B, Grant S (2012). Proteasome inhibitors in mantle cell lymphoma. Best Pract Res Clin Haematol 25: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y (2007). Thapsigargin induces biphasic fragmentation of mitochondria through calcium‐mediated mitochondrial fission and apoptosis. J Cell Physiol 212: 498–508. [DOI] [PubMed] [Google Scholar]
- Hoyer‐Hansen M, Jäättelä M (2007). Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14: 1576–1582. [DOI] [PubMed] [Google Scholar]
- Hu J, Han H, Lau MY, Lee H, MacVeigh‐Aloni M, Ji C (2015). Effects of combined alcohol and anti‐HIV drugs on cellular stress responses in primary hepatocytes and hepatic stellate and Kupffer cells. Alcohol Clin Exp Res 39: 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MF (2002). Arsenic toxicity and potential mechanisms of action. Toxicol Lett 133: 1–16. [DOI] [PubMed] [Google Scholar]
- Huliciak M, Vacek J, Sebela M, Orolinová E, Znaleziona J, Havlikova M, et al. (2012). Covalent binding of cisplatin impairs the function of Na+/K+‐ATPase by binding to its cytoplasmic part. Biochem Pharmacol 83: 1507–1513. [DOI] [PubMed] [Google Scholar]
- Hur KY, So JS, Ruda V, Frank‐Kamenetsky M, Fitzgerald K, Koteliansky V, et al. (2012). IRE1α activation protects mice against acetaminophen‐induced hepatotoxicity. J Exp Med 209: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannitti T, Palmieri B (2011). Clinical and experimental applications of sodium phenylbutyrate. Drugs RD 11: 227–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoudjil A, Begriche K, Pessayre D, Fromenty B (2006). Mitochondrial, metabolic and genotoxic effects of antiretroviral nucleoside reverse‐transcriptase Inhibitors. AntiInfect Agents Med Chem 5: 273–292. [Google Scholar]
- Inagi R, Ishimoto Y, Nangaku M (2014). Proteostasis in endoplasmic reticulum‐ new mechanisms in kidney disease. Nat Rev Nephrol 10: 369–378. [DOI] [PubMed] [Google Scholar]
- Isaacs JT (2005). New strategies for the medical treatment of prostate cancer. BJU Int 96: 35–40. [DOI] [PubMed] [Google Scholar]
- Izzedine H, Harris M, Perazella MA (2009). The nephrotoxic effects of HAART. Nat Rev Nephrol 5: 563–573. [DOI] [PubMed] [Google Scholar]
- Jacob M, Bjarnason I, Rafi S, Wrigglesworth J, Simpson RJ (2001). A study of the effects of indometacin on liver mitochondria from rats, mice and humans. Aliment Pharmacol Ther 15: 1837–1842. [DOI] [PubMed] [Google Scholar]
- Jacobsen S, Hansen HS, Jensen B (1987). Synergism between thapsigargin and the phorbol ester 12‐O‐tetradecanoylphorbol 13‐acetate on the release of [14C]arachidonic acid and histamine from rat peritoneal mast cells. Biochem Pharmacol 36: 621–626. [DOI] [PubMed] [Google Scholar]
- Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, et al. (2012). Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J Cell Sci 125: 5073–5083. [DOI] [PubMed] [Google Scholar]
- Jaeschke H (1990). Glutathione disulfide formation and oxidant stress during acetaminophen‐induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther 255: 935–941. [PubMed] [Google Scholar]
- Jaeschke H, McGill MR, Ramachandran A (2012). Oxidant stress, mitochondria, and cell death mechanisms in drug‐induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44: 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Chow SC, Nicotera P, Orrenius S (1994). Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+‐ATPase inhibitor thapsigargin. Exp Cell Res 212: 84–92. [DOI] [PubMed] [Google Scholar]
- Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, et al. (2013). Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low‐density lipoprotein receptor. Hepatology 57: 1366–1377. [DOI] [PubMed] [Google Scholar]
- John R, Herzenberg AM (2009). Renal toxicity of therapeutic drugs. J Clin Pathol 62: 505–515. [DOI] [PubMed] [Google Scholar]
- Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, et al. (2011). Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol 31: 95–107. [DOI] [PubMed] [Google Scholar]
- Jones AL, Prescott LF (1997). Unusual complications of paracetamol poisoning. QJM 90: 161–168. [DOI] [PubMed] [Google Scholar]
- Jones JD, Kirsch HL, Wortmann RL, Pillinger MH (2014). The causes of drug‐induced muscle toxicity. Curr Opin Rheumatol 26: 697–703. [DOI] [PubMed] [Google Scholar]
- Jorgensen KA, Koefoed‐Nielsen PB, Karamperis N (2003). Calcineurin phosphatase activity and immunosuppression. A review on the role of calcineurin phosphatase activity and the immunosuppressive effect of cyclosporin A and tacrolimus. Scand J Immunol 57: 93–98. [DOI] [PubMed] [Google Scholar]
- Kalinec GM, Thein P, Parsa A, Yorgason J, Luxford W, Urrutia R, et al. (2014). Acetaminophen and NAPQI are toxic to auditory cells via oxidative and endoplasmic reticulum stress‐dependent pathways. Hear Res 313: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. (2009). GRP78 expression inhibits insulin and ER stress‐induced SREBP‐1c activation and reduces hepatic steatosis in mice. J Clin Invest 119: 1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao E, Shinohara M, Feng M, Lau MY, Ji C (2012). Human immunodeficiency virus protease inhibitors modulate Ca2+ homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology 56: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Fujigaki Y, Inoue R, Asakawa S, Shin S, Shima T, et al. (2014). Therapeutic dose of acetaminophen as a possible risk factor for acute kidney injury: learning from two healthy young adult cases. Intern Med 53: 1531–1534. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (1999). Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13: 1211–1233. [DOI] [PubMed] [Google Scholar]
- Kaya H, Koc A, Sogut S, Duru M, Yilmaz HR, Uz E, et al. (2008). The protective effect of N‐acetylcysteine against cyclosporine A‐induced hepatotoxicity in rats. J Appl Toxicol 28: 15–20. [DOI] [PubMed] [Google Scholar]
- Kelly PA, Wang H, Napoli KL, Kahan BD, Strobel HW (1999). Metabolism of cyclosporine by cytochromes P450 3A9 and 3A4. Eur J Drug Metab Pharmacokinet 24: 321–328. [DOI] [PubMed] [Google Scholar]
- Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD (2013). Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J 27: 2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Yamadori T, et al. (2011). Aggravation of bleomycin‐induced pulmonary inflammation and fibrosis in mice lacking peroxiredoxin I. Am J Respir Cell Mol Biol 45: 600–609. [DOI] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC (2008). Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7: 1013–1030. [DOI] [PubMed] [Google Scholar]
- King YA, Chiu YJ, Chen HP, Kuo DH, Lu CC, Yang JS (2015). Endoplasmic reticulum stress contributes to arsenic trioxide‐induced intrinsic apoptosis in human umbilical and bone marrow mesenchymal stem cells. Environ Toxicol. doi:10.1002/tox.22046. [DOI] [PubMed] [Google Scholar]
- Kitamura M (2008). Endoplasmic reticulum stress and unfolded protein response in renal pathophysiology: Janus faces. Am J Physiol Renal Physiol 295: F323–F334. [DOI] [PubMed] [Google Scholar]
- Knights KM, Rowland A, Miners JO (2013). Renal drug metabolism in humans: the potential for drug‐endobiotic interactions involving cytochrome P450 (CYP) and UDP‐glucuronosyltransferase (UGT). Br J Clin Pharmacol 76: 587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockaert L, Fromenty B, Robin MA (2011). Mechanisms of mitochondrial targeting of cytochrome P450 2E1: physiopathological role in liver injury and obesity. FEBS J 278: 4252–4260. [DOI] [PubMed] [Google Scholar]
- Kon V, Sugiura M, Inagami T, Harvie BR, Ichikawa I, Hoover RL (1990). Role of endothelin in cyclosporine‐induced glomerular dysfunction. Kidney Int 37: 1487–1491. [DOI] [PubMed] [Google Scholar]
- Kong D, Zhuo L, Gao C, Shi S, Wang N, Huang Z, et al. (2013). Erythropoietin protects against cisplatin‐induced nephrotoxicity by attenuating endoplasmic reticulum stress‐induced apoptosis. J Nephrol 26: 219–227. [DOI] [PubMed] [Google Scholar]
- Kosuge Y, Sakikubo T, Ishige K, Ito Y (2006). Comparative study of endoplasmic reticulum stress‐induced neuronal death in rat cultured hippocampal and cerebellar granule neurons. Neurochem Int 49: 285–293. [DOI] [PubMed] [Google Scholar]
- Kretz‐Rommel A, Boelsterli UA (1994). Mechanism of covalent adduct formation of diclofenac to rat hepatic microsomal proteins. Retention of the glucuronic acid moiety in the adduct. Drug Metab Dispos 22: 956–961. [PubMed] [Google Scholar]
- Kruidering M, Van de Water B, de Heer E, Mulder GJ, Nagelkerke JF (1997) Cisplatin‐induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther 280: 638–649. [PubMed] [Google Scholar]
- Krupp P, Monka C (1990). Side‐effect profile of cyclosporin A in patients treated for psoriasis. Br J Dermatol 122: 47–56. [DOI] [PubMed] [Google Scholar]
- Kumar S, Yedjou CG, Tchounwou PB (2014). Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL‐60) cells. J Exp Clin Cancer Res 33: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo SC, Lampen JO (1974). Tunicamycin: an inhibitor of yeast glycoprotein synthesis. Biochem Biophys Res Commun 58: 287–295. [DOI] [PubMed] [Google Scholar]
- Labbe G, Pessayre D, Fromenty B (2008). Drug‐induced liver injury through mitochondrial dysfunction: mechanisms and detection during preclinical safety studies. Fundam Clin Pharmacol 22: 335–353. [DOI] [PubMed] [Google Scholar]
- Lauressergues E, Bert E, Duriez P, Hum D, Majd Z, Staels B, et al. (2012). Does endoplasmic reticulum stress participate in APD‐induced hepatic metabolic dysregulation? Neuropharmacology 62: 784–796. [DOI] [PubMed] [Google Scholar]
- Leaver DD, Schneider KM, Rand MJ, Anderson RM, Gage PW, Malbon R (1988). The neurotoxicity of tunicamycin. Toxicology 49: 179–187. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH (2003). Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 100: 9946–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Zheng Z, Mendez R, Ha SW, Xie Y, Zhang K (2012). Pharmacologic ER stress induces non‐alcoholic steatohepatitis in an animal model. Toxicol Lett 211: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leise MD, Poterucha JJ, Talwalkar JA (2014). Drug‐induced liver injury. Mayo Clin Proc 89: 95–106. [DOI] [PubMed] [Google Scholar]
- Letelier ME, Lopez‐Valladares M, Peredo‐Silva L, Rojas‐Sepulveda D, Aracena P (2011). Microsomal oxidative damage promoted by acetaminophen metabolism. Toxicol In Vitro 25: 1310–1313. [DOI] [PubMed] [Google Scholar]
- Leung L, Kalgutkar AS, Obach RS (2012). Metabolic activation in drug‐induced liver injury. Drug Metab Rev 44: 18–33. [DOI] [PubMed] [Google Scholar]
- Lhotak S, Sood S, Brimble E, Carlisle RE, Colgan SM, Mazzetti A, et al. (2012). ER stress contributes to renal proximal tubule injury by increasing SREBP‐2‐mediated lipid accumulation and apoptotic cell death. Am J Physiol Renal Physiol 303: F266–F278. [DOI] [PubMed] [Google Scholar]
- Litterst CL, Schweitzer VG (1988). Covalent binding of platinum to renal protein from sensitive and resistant guinea pigs treated with cisplatin: possible role in nephrotoxicity. Res Commun Chem Pathol Pharmacol 61: 35–48. [PubMed] [Google Scholar]
- Liu H, Baliga R (2005). Endoplasmic reticulum stress‐associated caspase 12 mediates cisplatin‐induced LLC‐PK1 cell apoptosis. J Am Soc Nephrol 16: 1985–1992. [DOI] [PubMed] [Google Scholar]
- Liu QF, Deng ZY, Ye JM, He AL, Li SS (2015). Ginsenoside Rg1 protects chronic cyclosporin a nephropathy from tubular cell apoptosis by inhibiting endoplasmic reticulum stress in rats. Transplant Proc 47: 566–569. [DOI] [PubMed] [Google Scholar]
- Lombardi A, Inabnet WB, Owen R, Farenholtz KE, Tomer Y (2015). Endoplasmic reticulum stress as a novel mechanism in amiodarone‐induced destructive thyroiditis. J Clin Endocrinol Metab 100: E1–E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorz C, Justo P, Sanz A, Subira D, Egido J, Ortiz A (2004). Paracetamol‐induced renal tubular injury: a role for ER stress. J Am Soc Nephrol 15: 380–389. [DOI] [PubMed] [Google Scholar]
- Lu J, Chew EH, Holmgren A (2007). Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci USA 104: 12288–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TH, Su CC, Chen YW, Yang CY, Wu CC, Hung DZ, et al. (2011). Arsenic induces pancreatic β‐cell apoptosis via the oxidative stress‐regulated mitochondria‐dependent and endoplasmic reticulum stress‐triggered signaling pathways. Toxicol Lett 201: 15–26. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bond JD, Ingram VM (1997). Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci USA 94: 9705–9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macanas‐Pirard P, Yaacob NS, Lee PC, Holder JC, Hinton RH, Kass GE (2005). Glycogen synthase kinase‐3 mediates acetaminophen‐induced apoptosis in human hepatoma cells. J Pharmacol Exp Ther 313: 780–789. [DOI] [PubMed] [Google Scholar]
- Maccio A, Madeddu C (2013). Cisplatin: an old drug with a newfound efficacy ‐ from mechanisms of action to cytotoxicity. Expert Opin Pharmacother 14: 1839–1857. [DOI] [PubMed] [Google Scholar]
- Maggi P, Bellacosa C, Carito V, Perilli F, Lillo A, Volpe A, et al. (2011). Cardiovascular risk factors in patients on long‐term treatment with nevirapine‐ or efavirenz‐based regimens. J Antimicrob Chemother 66: 896–900. [DOI] [PubMed] [Google Scholar]
- Maggiolo F (2009). Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother 64: 910–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahavadi P, Henneke I, Ruppert C, Knudsen L, Venkatesan S, Liebisch G, et al. (2014). Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone‐induced lung fibrosis. Toxicol Sci 142: 285–297. [DOI] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ (2011). Endoplasmic reticulum stress in liver disease. J Hepatol 54: 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ (2007). Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double‐edged sword? Antioxid Redox Signal 9: 2277–2293. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, et al. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA 105: 18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic A, Hansson J, Linder S, Shoshan MC (2003). Cisplatin induces endoplasmic reticulum stress and nucleus‐independent apoptotic signaling. J Biol Chem 278: 9100–9106. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis AM, Heverling H, Pham PA, Stolbach A (2014). A review of the toxicity of HIV medications. J Med Toxicol 10: 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrer E, Dieterle F (2010). Impact of biomarker development on drug safety assessment. Toxicol Appl Pharmacol 243: 167–179. [DOI] [PubMed] [Google Scholar]
- Massart J, Begriche K, Buron N, Porceddu M, Borgne‐Sanchez A, Fromenty B (2013). Drug‐induced inhibition of mitochondrial fatty acid oxidation and steatosis. Curr Pathobiol Rep 1: 147–157. [Google Scholar]
- Mathews VV, Paul MV, Abhilash M, Manju A, Abhilash S, Nair RH (2013). Myocardial toxicity of acute promyelocytic leukaemia drug‐arsenic trioxide. Eur Rev Med Pharmacol Sci 17: 34–38. [PubMed] [Google Scholar]
- McGill MR, Du K, Xie Y, Bajt ML, Ding WX, Jaeschke H (2015). The role of the c‐Jun N‐terminal kinases 1/2 and receptor‐interacting protein kinase 3 in furosemide‐induced liver injury. Xenobiotica 45: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkersson KI, Dahl ML (2003). Relationship between levels of insulin or triglycerides and serum concentrations of the atypical antipsychotics clozapine and olanzapine in patients on treatment with therapeutic doses. Psychopharmacology 170: 157–166. [DOI] [PubMed] [Google Scholar]
- Mencarelli A, Francisci D, Renga B, D'Amore C, Cipriani S, Basile F, et al. (2012). Ritonavir‐induced lipoatrophy and dyslipidaemia is reversed by the anti‐inflammatory drug leflunomide in a PPAR‐γ‐dependent manner. Antivir Ther 17: 669–678. [DOI] [PubMed] [Google Scholar]
- Michaut A, Moreau C, Robin MA, Fromenty B (2014). Acetaminophen‐induced liver injury in obesity and nonalcoholic fatty liver disease. Liver Int 34: e171–e179. [DOI] [PubMed] [Google Scholar]
- Miltenburg NC, Boogerd W (2014). Chemotherapy‐induced neuropathy: A comprehensive survey. Cancer Treat Rev 40: 872–882. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horvath B, Zsengeller Z, Zielonka J, Tanchian G, Holovac E, et al. (2012). Mitochondrial‐targeted antioxidants represent a promising approach for prevention of cisplatin‐induced nephropathy. Free Radic Biol Med 52: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadanaciva S, Aleo MD, Strock CJ, Stedman DB, Wang H, Will Y (2013). Toxicity assessments of nonsteroidal anti‐inflammatory drugs in isolated mitochondria, rat hepatocytes, and zebrafish show good concordance across chemical classes. Toxicol Appl Pharmacol 272: 272–280. [DOI] [PubMed] [Google Scholar]
- Nagelkerke JF, Tijdens RB, Schwarz EP, Winters MF, Paul LC, Mulder GJ (1987). The covalent binding of cyclosporin A to rat liver macromolecules in vivo and in vitro: the role of cytochrome P‐450. Toxicology 47: 277–284. [DOI] [PubMed] [Google Scholar]
- Nagy G, Kardon T, Wunderlich L, Szarka A, Kiss A, Schaff Z, et al. (2007). Acetaminophen induces ER dependent signaling in mouse liver. Arch Biochem Biophys 459: 273–279. [DOI] [PubMed] [Google Scholar]
- Nagy G, Szarka A, Lotz G, Doczi J, Wunderlich L, Kiss A, et al. (2010). BGP‐15 inhibits caspase‐independent programmed cell death in acetaminophen‐induced liver injury. Toxicol Appl Pharmacol 243: 96–103. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J (2000). Cross‐talk between two cysteine protease families. Activation of caspase‐12 by calpain in apoptosis. J Cell Biol 150: 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narabayashi K, Ito Y, Eid N, Maemura K, Inoue T, Takeuchi T, et al. (2015). Indomethacin suppresses LAMP‐2 expression and induces lipophagy and lipoapoptosis in rat enterocytes via the ER stress pathway. J Gastroenterol 50: 541–554. [DOI] [PubMed] [Google Scholar]
- Navarro‐Antolin J, Redondo‐Horcajo M, Zaragoza C, Alvarez‐Barrientos A, Fernandez AP, Leon‐Gomez E, et al. (2007). Role of peroxynitrite in endothelial damage mediated by Cyclosporine A. Free Radic Biol Med 42: 394–403. [DOI] [PubMed] [Google Scholar]
- Nicolescu AC, Ji Y, Comeau JL, Hill BC, Takahashi T, Brien JF, et al. (2008). Direct mitochondrial dysfunction precedes reactive oxygen species production in amiodarone‐induced toxicity in human peripheral lung epithelial HPL1A cells. Toxicol Appl Pharmacol 227: 370–379. [DOI] [PubMed] [Google Scholar]
- Nishida M, Ogawa H, Tamai M, Ishiwari K, Hamaoka K (2003). Role of hydrogen peroxide in cyclosporine‐induced renal tubular cell (LLC‐PK1) injury. J Pharmacol Sci 91: 255–258. [DOI] [PubMed] [Google Scholar]
- Niu X, de Graaf IA, van der Bij HA, Groothuis GM (2014). Precision cut intestinal slices are an appropriate ex vivo model to study NSAID‐induced intestinal toxicity in rats. Toxicol In Vitro 28: 1296–1305. [DOI] [PubMed] [Google Scholar]
- Nunez M, Lana R, Mendoza JL, Martín‐Carbonero L, Soriano V (2001). Risk factors for severe hepatic injury after introduction of highly active antiretroviral therapy. J Acquir Immune Defic Syndr 27: 426–431. [DOI] [PubMed] [Google Scholar]
- Obach RS, Kalgutkar AS, Soglia JR, Zhao SX (2008). Can in vitro metabolism‐dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chem Res Toxicol 21: 1814–1822. [DOI] [PubMed] [Google Scholar]
- Ohyama K, Shiokawa A, Ito K, Masuyama R, Ichibangase T, Kishikawa N, et al. (2012). Toxicoproteomic analysis of a mouse model of nonsteroidal anti‐inflammatory drug‐induced gastric ulcers. Biochem Biophys Res Commun 420: 210–215. [DOI] [PubMed] [Google Scholar]
- Ozasa R, Okada T, Nadanaka S, Nagamine T, Zyryanova A, Harding H, et al. (2013). The antipsychotic olanzapine induces apoptosis in insulin‐secreting pancreatic β cells by blocking PERK‐mediated translational attenuation. Cell Struct Funct 38: 183–195. [DOI] [PubMed] [Google Scholar]
- Pabla N, Dong Z (2008). Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007. [DOI] [PubMed] [Google Scholar]
- Padda MS, Sanchez M, Akhtar AJ, Boyer JL (2011). Drug‐induced cholestasis. Hepatology 53: 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajonk F, Himmelsbach J, Riess K, Sommer A, McBride WH (2002). The human immunodeficiency virus (HIV)‐1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non‐HIV‐associated human cancer cells. Cancer Res 62: 5230–5235. [PubMed] [Google Scholar]
- Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, et al. (2008). Cyclosporine‐induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. Am J Transplant 8: 2283–2296. [DOI] [PubMed] [Google Scholar]
- Palomero J, Galan AI, Munoz ME, Tunon MJ, Gonzalez‐Gallego J, Jimenez R (2001). Effects of aging on the susceptibility to the toxic effects of cyclosporin A in rats. Changes in liver glutathione and antioxidant enzymes. Free Radic Biol Med 30: 836–845. [DOI] [PubMed] [Google Scholar]
- Pannu N, Nadim MK (2008). An overview of drug‐induced acute kidney injury. Crit Care Med 36: S216–S223. [DOI] [PubMed] [Google Scholar]
- Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP (2005). The role of metabolic activation in drug‐induced hepatotoxicity. Annu Rev Pharmacol Toxicol 45: 177–202. [DOI] [PubMed] [Google Scholar]
- Parker RA, Flint OP, Mulvey R, Elosua C, Wang F, Fenderson W, et al. (2005). Endoplasmic reticulum stress links dyslipidemia to inhibition of proteasome activity and glucose transport by HIV protease inhibitors. Mol Pharmacol 67: 1909–1919. [DOI] [PubMed] [Google Scholar]
- Pattanaik A, Bachowski G, Laib J, Lemkuil D, Shaw CF, Petering DH, et al. (1992). Properties of the reaction of cis‐dichlorodiammineplatinum(II) with metallothionein. J Biol Chem 267: 16121–16128. [PubMed] [Google Scholar]
- Paul MK, Kumar R, Mukhopadhyay AK (2008). Dithiothreitol abrogates the effect of arsenic trioxide on normal rat liver mitochondria and human hepatocellular carcinoma cells. Toxicol Appl Pharmacol 226: 140–152. [DOI] [PubMed] [Google Scholar]
- Pedersen JM, Matsson P, Bergström CA, Hoogstraate J, Noren A, LeCluyse EL, et al. (2013). Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11). Toxicol Sci 136: 328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, et al. (2003). Inhibition of mitochondrial respiration: a novel strategy to enhance drug‐induced apoptosis in human leukemia cells by a reactive oxygen species‐mediated mechanism. J Biol Chem 278: 37832–37839. [DOI] [PubMed] [Google Scholar]
- Peyrou M, Hanna PE, Cribb AE (2007). Cisplatin, gentamicin, and p‐aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci 99: 346–353. [DOI] [PubMed] [Google Scholar]
- Piccinini M, Rinaudo MT, Chiapello N, Ricotti E, Baldovino S, Mostert M, et al. (2002). The human 26S proteasome is a target of antiretroviral agents. AIDS 16: 693–700. [DOI] [PubMed] [Google Scholar]
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008). Calcium and apoptosis: ER‐mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27: 6407–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porceddu M, Buron N, Roussel C, Labbe G, Fromenty B, Borgne‐Sanchez A (2012). Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol Sci 129: 332–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumford NR, Roberts DW, Benson RW, Hinson JA (1990). Immunochemical quantitation of 3‐(cystein‐S‐yl)acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol 40: 573–579. [DOI] [PubMed] [Google Scholar]
- Purnell PR, Fox HS (2014). Efavirenz induces neuronal autophagy and mitochondrial alterations. J Pharmacol Exp Ther 351: 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC (2007). HIV‐1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res 67: 10920–10928. [DOI] [PubMed] [Google Scholar]
- Qu Q, Liu J, Zhou HH, Klaassen CD (2014). Nrf2 protects against furosemide‐induced hepatotoxicity. Toxicology 324: 35–42. [DOI] [PubMed] [Google Scholar]
- Rainsford KD, Willis C (1982). Relationship of gastric mucosal damage induced in pigs by antiinflammatory drugs to their effects on prostaglandin production. Dig Dis Sci 27: 624–635. [DOI] [PubMed] [Google Scholar]
- Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C (2009). Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry 48: 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Hermel E, Castro‐Obregon S, del Rio G, Ellerby LM, Ellerby HM, et al. (2001). Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276: 33869–33874. [DOI] [PubMed] [Google Scholar]
- Ravindranath V, Strobel HW (2013). Cytochrome P450‐mediated metabolism in brain: functional roles and their implications. Expert Opin Drug Metab Toxicol 9: 551–558. [DOI] [PubMed] [Google Scholar]
- Reck M, Mok T, Wolf J, Heigener D, Wu YL (2011). Reviewing the safety of erlotinib in non‐small cell lung cancer. Expert Opin Drug Saf 10: 147–157. [DOI] [PubMed] [Google Scholar]
- Redondo‐Horcajo M, Romero N, Martinez‐Acedo P, Martinez‐Ruiz A, Quijano C, Lourenço CF, et al. (2010). Cyclosporine A‐induced nitration of tyrosine 34 MnSOD in endothelial cells: role of mitochondrial superoxide. Cardiovasc Res 87: 356–365. [DOI] [PubMed] [Google Scholar]
- Rekha RS, Rao Muvva SJ, Wan M, Raqib R, Bergman P, Brighenti S, et al. (2015). Phenylbutyrate induces LL‐37‐dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy 11: 1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Kirk SL (2010). Metabolic side effects of antipsychotic drug treatment ‐ pharmacological mechanisms. Pharmacol Ther 125: 169–179. [DOI] [PubMed] [Google Scholar]
- Reyskens KM, Fisher TL, Schisler JC, O'Connor WG, Rogers AB, Willis MS, et al. (2013). Cardio‐metabolic effects of HIV protease inhibitors (lopinavir/ritonavir). PLoS ONE 8: e73347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richy F, Bruyere O, Ethgen O, Rabenda V, Bouvenot G, Audran M, et al. (2004). Time dependent risk of gastrointestinal complications induced by non‐steroidal anti‐inflammatory drug use: a consensus statement using a meta‐analytic approach. Ann Rheum Dis 63: 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo LE, Gaspar PA, Silva H, Risco L, Arena P, Cubillos‐Robles K, et al. (2015). Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol Res. doi:10.1016/j.phrs.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Rossi E, Ferraccioli GF, Cavalieri F, Menta R, Dall'Aglio PP, and Migone L (1985). Diclofenac‐associated acute renal failure. Report of 2 cases. Nephron 40: 491–493. [DOI] [PubMed] [Google Scholar]
- Roy A, Ghosh A, Jana A, Liu X, Brahmachari S, Gendelman HE, et al. (2012). Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson's disease. PLoS ONE 7: e38113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp SU, Tonge RP, Shaw J, Wallis N, Pognan F (2002). Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol Sci 65: 135–150. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, et al. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress‐mediated suppression of transcriptional master regulators. Dev Cell 15: 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrieh N, Thomas PE (1994). Characterization of rat cytochrome P450 isozymes involved in the covalent binding of cyclosporin A to microsomal proteins. Toxicol Appl Pharmacol 127: 222–232. [DOI] [PubMed] [Google Scholar]
- Samali A, Fitzgerald U, Deegan S, Gupta S (2010). Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int J Cell Biol 2010: 830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC (2013). ER stress‐induced cell death mechanisms. Biochim Biophys Acta 1833: 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangeli P, Di Biase L, Burkhardt JD, Bai R, Mohanty P, Pump A, et al. (2012). Examining the safety of amiodarone. Expert Opin Drug Saf 11: 191–214. [DOI] [PubMed] [Google Scholar]
- Sardao VA, Pereira SL, Oliveira PJ (2008). Drug‐induced mitochondrial dysfunction in cardiac and skeletal muscle injury. Expert Opin Drug Saf 7: 129–146. [DOI] [PubMed] [Google Scholar]
- Sarro E, Jacobs‐Cacha C, Itarte E, Meseguer A (2012). A pharmacologically‐based array to identify targets of cyclosporine A‐induced toxicity in cultured renal proximal tubule cells. Toxicol Appl Pharmacol 258: 275–287. [DOI] [PubMed] [Google Scholar]
- Schiller GJ, Slack J, Hainsworth JD, Mason J, Saleh M, Rizzieri D, et al. (2006). Phase II multicenter study of arsenic trioxide in patients with myelodysplastic syndromes. J Clin Oncol 24: 2456–2464. [DOI] [PubMed] [Google Scholar]
- Schmidtke G, Holzhütter HG, Bogyo M, Kairies N, Groll M, de Giuli R, et al. (1999). How an inhibitor of the HIV‐I protease modulates proteasome activity. J Biol Chem 274: 35734–35740. [DOI] [PubMed] [Google Scholar]
- Schon E, Fromenty B (2016). Alterations of mitochondrial DNA in liver diseases Pp. 283–313 in Kaplowitz N. and Han D., eds. Mitochondria in Liver Disease. Taylor & Francis, New‐York. [Google Scholar]
- Schönthal AH (2012). Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012: 857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott N, Hatlelid KM, MacKenzie NE, Carter DE (1993). Reactions of arsenic(III) and arsenic(V) species with glutathione. Chem Res Toxicol 6: 102–106. [DOI] [PubMed] [Google Scholar]
- Seminerio J, McGrath K, Arnold CA, Voltaggio L, Singhi AD (2014). Medication‐associated lesions of the GI tract. Gastrointest Endosc 79: 140–150. [DOI] [PubMed] [Google Scholar]
- Serino F, Grevel J, Napoli KL, Kahan BD, Strobel HW (1993). Generation of oxygen free radicals during the metabolism of cyclosporine A: a cause‐effect relationship with metabolism inhibition. Mol Cell Biochem 122: 101–112. [DOI] [PubMed] [Google Scholar]
- Serrano‐Mollar A, Closa D, Prats N, Blesa S, Martinez‐Losa M, Cortijo J, et al. (2003). In vivo antioxidant treatment protects against bleomycin‐induced lung damage in rats. Br J Pharmacol 138: 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais H, Ortiz A, Devuyst O, Denamur S, Tulkens PM, Mingeot‐Leclercq MP (2008). Renal cell apoptosis induced by nephrotoxic drugs: cellular and molecular mechanisms and potential approaches to modulation. Apoptosis 13: 11–32. [DOI] [PubMed] [Google Scholar]
- Sevier CS, Kaiser CA (2008). Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta 1783: 549–556. [DOI] [PubMed] [Google Scholar]
- Sharanek A, Azzi PB, Al‐Attrache H, Savary CC, Humbert L, Rainteau D, et al. (2014). Different dose‐dependent mechanisms are involved in early cyclosporine A‐induced cholestatic effects in HepaRG cells. Toxicol Sci 141: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, et al. (1997). Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci 17: 4612–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. (1997). Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89: 3354–3360. [PubMed] [Google Scholar]
- Shin NY, Liu Q, Stamer SL, Liebler DC (2007). Protein targets of reactive electrophiles in human liver microsomes. Chem Res Toxicol 20: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YK, Jang SY, Lee HK, Jung J, Suh DJ, Seo SY, et al. (2010). Pathological adaptive responses of Schwann cells to endoplasmic reticulum stress in bortezomib‐induced peripheral neuropathy. Glia 58: 1961–1976. [DOI] [PubMed] [Google Scholar]
- Smith SR, Creech EA, Schaffer AV, Martin LL, Rakhit A, Douglas FL, et al. (1992). Effects of thromboxane synthase inhibition with CGS 13080 in human cyclosporine nephrotoxicity. Kidney Int 41: 199–205. [DOI] [PubMed] [Google Scholar]
- Srivastava RK, Li C, Chaudhary SC, Ballestas ME, Elmets CA, Robbins DJ, et al. (2013). Unfolded protein response (UPR) signaling regulates arsenic trioxide‐mediated macrophage innate immune function disruption. Toxicol Appl Pharmacol 272: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnimann G, Kessebohm K, Lauterburg B (2010). Liver injury caused by drugs: an update. Swiss Med Wkly 140: w13080. [DOI] [PubMed] [Google Scholar]
- Streck EL, Ferreira GK, Scaini G, Rezin GT, Gonçalves CL, Jeremias IC, et al. (2011). Non‐nucleoside reverse transcriptase inhibitors efavirenz and nevirapine inhibit cytochrome c oxidase in mouse brain regions. Neurochem Res 36: 962–966. [DOI] [PubMed] [Google Scholar]
- Suaud L, Miller K, Panichelli AE, Randell RL, Marando CM, Rubenstein RC (2011). 4‐Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT‐3 in cystic fibrosis epithelial cells. J Biol Chem 286: 45083–45092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganya N, Bhakkiyalakshmi E, Suriyanarayanan S, Paulmurugan R, Ramkumar KM (2014). Quercetin ameliorates tunicamycin‐induced endoplasmic reticulum stress in endothelial cells. Cell Prolif 47: 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh HN, Lee YJ, Kim MO, Ryu JM, Han HJ (2014). Glucosamine‐induced Sp1 O‐GlcNAcylation ameliorates hypoxia‐induced SGLT dysfunction in primary cultured renal proximal tubule cells. J Cell Physiol 229: 1557–1568. [DOI] [PubMed] [Google Scholar]
- Sulkowski MS, Thomas DL, Mehta SH, Chaisson RE, Moore RD (2002). Hepatotoxicity associated with nevirapine or efavirenz‐containing antiretroviral therapy: role of hepatitis C and B infections. Hepatology 35: 182–189. [DOI] [PubMed] [Google Scholar]
- van Summeren A, Renes J, Lizarraga D, Bouwman FG, Noben JP, van Delft JH, et al. (2013). Screening for drug‐induced hepatotoxicity in primary mouse hepatocytes using acetaminophen, amiodarone, and cyclosporin A as model compounds: an omics‐guided approach. OMICS 17: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalowska E, Stoopen G, Groot MJ, Hendriksen PJ, Peijnenburg AA (2013). Treatment of mouse liver slices with cholestatic hepatotoxicants results in down‐regulation of Fxr and its target genes. BMC Med Genomics 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuki A, Tamura G (1971). Effect of tunicamycin on the synthesis of macromolecules in cultures of chick embryo fibroblasts infected with Newcastle disease virus. J Antibiot 24: 785–794. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Tomisato W, Hoshino T, Ishihara T, Namba T, Aburaya M, et al. (2005). Involvement of intracellular Ca2+ levels in nonsteroidal anti‐inflammatory drug‐induced apoptosis. J Biol Chem 280: 31059–31067. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Ishitsuka Y, Hayasaka M, Yamada Y, Miyata K, Endo M, et al. (2015). The exacerbating roles of CCAAT/enhancer‐binding protein homologous protein (CHOP) in the development of bleomycin‐induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol Res 99: 52–62. [DOI] [PubMed] [Google Scholar]
- Tanimukai H, Kanayama D, Omi T, Takeda M, Kudo T (2013). Paclitaxel induces neurotoxicity through endoplasmic reticulum stress. Biochem Biophys Res Commun 437: 151–155. [DOI] [PubMed] [Google Scholar]
- Tariq M, Morais C, Sobki S, Al Sulaiman M, Al Khader A (1999). N‐acetylcysteine attenuates cyclosporin‐induced nephrotoxicity in rats. Nephrol Dial Transplant 14: 923–929. [DOI] [PubMed] [Google Scholar]
- Taura M, Kariya R, Kudo E, Goto H, Iwawaki T, Amano M, et al. (2013). Comparative analysis of ER stress response into HIV protease inhibitors: lopinavir but not darunavir induces potent ER stress response via ROS/JNK pathway. Free Radic Biol Med 65: 778–788. [DOI] [PubMed] [Google Scholar]
- Teff KL, Rickels MR, Grudziak J, Fuller C, Nguyen HL, Rickels K (2013). Antipsychotic‐induced insulin resistance and postprandial hormonal dysregulation independent of weight gain or psychiatric disease. Diabetes 62: 3232–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen K, Dressman JB (2009). Cytochrome P450‐mediated metabolism in the human gut wall. J Pharm Pharmacol 61: 541–558. [DOI] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, et al. (2009). Calcium/calmodulin‐dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest 119: 2925–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkacz JS, Lampen O (1975). Tunicamycin inhibition of polyisoprenyl N‐acetylglucosaminyl pyrophosphate formation in calf‐liver microsomes. Biochem Biophys Res Commun 65: 248–257. [DOI] [PubMed] [Google Scholar]
- Tocchetti CG, Gallucci G, Coppola C, Piscopo G, Cipresso C, Maurea C, et al. (2013). The emerging issue of cardiac dysfunction induced by antineoplastic angiogenesis inhibitors. Eur J Heart Fail 15: 482–489. [DOI] [PubMed] [Google Scholar]
- Touzet O, Philips A (2010). Resveratrol protects against protease inhibitor‐induced reactive oxygen species production, reticulum stress and lipid raft perturbation. AIDS 24: 1437–1447. [DOI] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB (1998). A tool coming of age: thapsigargin as an inhibitor of sarco‐endoplasmic reticulum Ca2+‐ATPases. Trends Pharmacol Sci 19: 131–135. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Gotoh T, Tomisato W, Mima S, Hoshino T, Hwang HJ, et al. (2004). Endoplasmic reticulum stress response is involved in nonsteroidal anti‐inflammatory drug‐induced apoptosis. Cell Death Differ 11: 1009–1016. [DOI] [PubMed] [Google Scholar]
- Tu BP, Weissman JS (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164: 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez‐Rodriguez A, et al. (2013). CHOP is a critical regulator of acetaminophen‐induced hepatotoxicity. J Hepatol 59: 495–503. [DOI] [PubMed] [Google Scholar]
- Van Summeren A, Renes J, Bouwman FG, Noben JP, van Delft JH, Kleinjans JC, et al. (2011). Proteomics investigations of drug‐induced hepatotoxicity in HepG2 cells. Toxicol Sci 120: 109–122. [DOI] [PubMed] [Google Scholar]
- Vineetha VP, Soumya RS, Raghu KG (2015). Phloretin ameliorates arsenic trioxide induced mitochondrial dysfunction in H9c2 cardiomyoblasts mediated via alterations in membrane permeability and ETC. complexes. Eur J Pharmacol 754: 162–172. [DOI] [PubMed] [Google Scholar]
- van der Vlies D, Pap EH, Post JA, Celis JE, Wirtz KW (2002). Endoplasmic reticulum resident proteins of normal human dermal fibroblasts are the major targets for oxidative stress induced by hydrogen peroxide. Biochem J 366: 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vlies D, Makkinje M, Jansens A, Braakman I, Verkleij AJ, Wirtz KW, et al. (2003). Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal 5: 381–387. [DOI] [PubMed] [Google Scholar]
- Wallace JL (2012). NSAID gastropathy and enteropathy: distinct pathogenesis likely necessitates distinct prevention strategies. Br J Pharmacol 165: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Thomas B, Sachdeva R, Arterburn L, Frye L, Hatcher PG, et al. (2006). Mechanism of arylating quinone toxicity involving Michael adduct formation and induction of endoplasmic reticulum stress. Proc Natl Acad Sci USA 103: 3604–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lin Z, Liu Z, Harris S, Kelly R, Zhang J, et al. (2013a). A unifying ontology to integrate histological and clinical observations for drug‐induced liver injury. Am J Pathol 182: 1180–1187. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang L, Wu X, Gurley EC, Kennedy E, Hylemon PB, et al. (2013b). The role of CCAAT enhancer‐binding protein homologous protein in human immunodeficiency virus protease‐inhibitor‐induced hepatic lipotoxicity in mice. Hepatology 57: 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Chen W, Zhang Y, Liu C, Yuan L, Fu L, et al. (2014). Deletion of p18(INK4c) aggravates cisplatin‐induced acute kidney injury. Int J Mol Med 33: 1621–1626. [DOI] [PubMed] [Google Scholar]
- Watkins PB, Kaplowitz N, Slattery JT, Colonese CR, Colucci SV, Stewart PW, et al. (2006). Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 296: 87–93. [DOI] [PubMed] [Google Scholar]
- Weis M, Morgenstern R, Cotgreave IA, Nelson SD, Moldeus P (1992). N‐acetyl‐p‐benzoquinone imine‐induced protein thiol modification in isolated rat hepatocytes. Biochem Pharmacol 43: 1493–1505. [DOI] [PubMed] [Google Scholar]
- Weiß M, Kost B, Renner‐Müller I, Wolf E, Mylonas I, Brüning A (2015). Efavirenz causes oxidative stress, endoplasmic reticulum stress, and autophagy in endothelial cells. Cardiovasc Toxicol. doi:10.1007/s12012‐015‐9314‐2. [DOI] [PubMed] [Google Scholar]
- Weng CY, Chiou SY, Wang L, Kou MC, Wang YJ, Wu MJ (2014). Arsenic trioxide induces unfolded protein response in vascular endothelial cells. Arch Toxicol 88: 213–226. [DOI] [PubMed] [Google Scholar]
- Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, et al. (2001). Homocysteine‐induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 107: 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westervelt P, Brown RA, Adkins DR, Khoury H, Curtin P, Hurd D, et al. (2001). Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood 98: 266–271. [DOI] [PubMed] [Google Scholar]
- Winnike JH, Li Z, Wright FA, Macdonald JM, O'Connell TM, Watkins PB (2010). Use of pharmaco‐metabonomics for early prediction of acetaminophen‐induced hepatotoxicity in humans. Clin Pharmacol Ther 88: 45–51. [DOI] [PubMed] [Google Scholar]
- Wolf A, Trendelenburg CF, Diez‐Fernandez C, Prieto P, Houy S, Trommer WE, et al. (1997). Cyclosporine A‐induced oxidative stress in rat hepatocytes. J Pharmacol Exp Ther 280: 1328–1334. [PubMed] [Google Scholar]
- Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, et al. (2010). HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology 138: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Shan J, Li C, Luo L, Lu J, Li S, et al. (2013). Mechanisms of cyclosporine‐induced renal cell apoptosis: a systematic review. Am J Nephrol 37: 30–40. [DOI] [PubMed] [Google Scholar]
- Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, et al. (2002). Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress‐induced caspase‐12 activation. Hepatology 36: 592–601. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly‐Maitre B, Reed JC (2005). Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115: 2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wang Y, Tong H, Qian W, Jin J (2014). Downregulation of hTERT: an important As2O3 induced mechanism of apoptosis in myelodysplastic syndrome. PLoS ONE 9: e113199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, et al. (2010). Induction of liver steatosis and lipid droplet formation in ATF6α‐knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell 21: 2975–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH (2012). Arsenic induces apoptosis in myoblasts through a reactive oxygen species‐induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol 86: 923–933. [DOI] [PubMed] [Google Scholar]
- Yorgason JG, Luxford W, Kalinec F (2011). In vitro and in vivo models of drug ototoxicity: studying the mechanisms of a clinical problem. Expert Opin Drug Metab Toxicol 7: 1521–1534. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Yamauchi H, Fan Sun G (2004). Chronic health effects in people exposed to arsenic via the drinking water: dose‐response relationships in review. Toxicol Appl Pharmacol 198: 243–252. [DOI] [PubMed] [Google Scholar]
- Zha W, Liang G, Xiao J, Studer EJ, Hylemon PB, Pandak WM, et al. (2010). Berberine inhibits HIV protease inhibitor‐induced inflammatory response by modulating ER stress signaling pathways in murine macrophages. PLoS ONE 5: e9069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha BS, Wan X, Zhang X, Zha W, Zhou J, Wabitsch M, et al. (2013). HIV protease inhibitors disrupt lipid metabolism by activating endoplasmic reticulum stress and inhibiting autophagy activity in adipocytes. PLoS ONE 8: e59514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, et al. (2010). Arsenic trioxide controls the fate of the PML‐RARalpha oncoprotein by directly binding PML. Science 328: 240–243. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. (2011). The unfolded protein response transducer IRE1α prevents ER stress‐induced hepatic steatosis. EMBO J 30: 1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Wu QQ, Cao LF, Qing HY, Zhang C, Chen YH, et al. (2014a). Melatonin inhibits endoplasmic reticulum stress and epithelial‐mesenchymal transition during bleomycin‐induced pulmonary fibrosis in mice. PLoS ONE 9: e97266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Qin HY, Cao LF, Chen YH, Tan ZX, Zhang C, et al. (2014b). Phenylbutyric acid inhibits epithelial‐mesenchymal transition during bleomycin‐induced lung fibrosis. Toxicol Lett 232: 213–220. [DOI] [PubMed] [Google Scholar]
- Zheng S, Tasnif Y, Hebert MF, Davis CL, Shitara Y, Calamia JC, et al. (2013). CYP3A5 gene variation influences cyclosporine A metabolite formation and renal cyclosporine disposition. Transplantation 95: 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H (2011). HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Methods Enzymol 490: 107–119. [DOI] [PubMed] [Google Scholar]
- Zhou L, McKenzie BA, Eccleston ED Jr, Srivastava SP, Chen N, Erickson RR, et al. (1996). The covalent binding of [14C]acetaminophen to mouse hepatic microsomal proteins: the specific binding to calreticulin and the two forms of the thiol:protein disulfide oxidoreductases. Chem Res Toxicol 9: 1176–1182. [DOI] [PubMed] [Google Scholar]
- Zhou H, Pandak WM Jr, Lyall V, Natarajan R, Hylemon PB (2005). HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol 68: 690–700. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]