

Abstract
Liquid chromatography–mass spectrometry (LC-MS)-based proteomics is a powerful technique for the profiling of protein expression in cells in a high-throughput fashion. Herein we report a protocol using LC-MS/MS-based proteomics for the screening of enzymes involved in natural product biosynthesis, such as nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) from bacterial strains. Taking advantage of the large size of modular NRPSs and PKSs (often >200 kDa), size-based separation (SDS-PAGE) is employed prior to LC-MS/MS analysis. Based upon the protein identifications obtained through software search, we can accurately pinpoint the expressed NRPS and/or PKS gene clusters from a given strain and growth condition. The proteomics screening result can be used to guide the discovery of potentially new nonribosomal peptide and polyketide natural products.
Keywords: Proteomics, Liquid chromatography, Mass spectrometry, Nonribosomal peptide, Polyketide, Natural product
1 Introduction
Many natural products with antibiotic, anticancer, and antifungal activities are synthesized by nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs) [1]. Multimodular NRPS and PKS enzymes are often very large proteins (>200 kDa) and contain many functional domains for the assembly of simple building blocks into natural products [2]. Traditionally, natural product discovery has relied on a bioactivity-guided screening approach, which includes repeated fractionation to isolate the compounds of interest [3]. This approach often suffers from a high frequency of rediscovering known natural products [4]. With the advent of the genomic revolution and whole genome sequencing, researchers have realized that microorganisms possess a far greater genetic potential for natural product biosynthesis than what had been observed [5]. Through rational prediction followed by targeted detection of the natural products based on the genetic information, an approach known as “genome mining” has led to the discovery of a number of new natural products [6–8]. On the other hand, genome mining approaches also have faced roadblocks in discovery as some biosynthetic pathways are not actively expressed or expressed at extremely low levels in laboratory conditions, also known as “cryptic” pathways [9, 10].
To complement the genome mining and bioassay-based natural product discovery approaches, a proteomics approach was developed by the Kelleher group, termed Proteomic Investigation of Secondary Metabolism (PrISM) [11]. By initially identifying the actively expressed biosynthetic proteins, PrISM avoids pursuing “cryptic” pathways. Natural products are predicted according to the genetic information and discovered in a targeted way. The PrISM approach has led to the discovery of several new natural products including koranimine, flavopeptins, gobichelin A and B, as well as the identification of biosynthetic pathways for many known natural products [11–15]. Most recently, the “depth” at which PrISM could be applied (i.e., detecting expression of gene clusters at low levels) was evaluated using a single Actinomycete strain with its genome sequence; this was called Genome-enabled PrISM [16].
In this chapter, we present the detailed protocol for the PrISM screening of Actinobacteria strains for the expressed NRPS and PKS proteins. In this protocol, Actinobacteria strains are grown under a variety of culture conditions and harvested at several time points. Cells are lysed by mechanical disruption using bead beating, and the proteomes are fractionated by size using one dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The high molecular weight proteins (>150 kDa), often representing NRPSs or PKSs, are subjected to in-gel trypsin digestion followed by liquid chromatography coupled with mass spectrometric (LC-MS) analysis. The LC-MS data are automatically analyzed using proteomics software and the protein identifications reveal which NRPS and/or PKS pathways are actively expressed under the growth conditions—guiding the discovery of the corresponding natural products.
2 Materials
2.1 Culture Media
Malt Yeast Glucose: 10 g/L malt extract, 4 g/L yeast extract, 4 g/L glucose, pH to 7.
ATCC 172: 10 g/L glucose, 20 g/L soluble starch, 5 g/L yeast extract, 5 g/L N–Z amine type A, 1 g/L CaCO3.
ISP4: 10 g/L soluble starch, 1 g/L K2HPO4, 1 g/L MgSO4, 1 g/L NaCl, 2 g/L (NH4)2SO4, 2 g/L CaCO3, 0.001 g/L FeSO4•7H2O, 0.001 g/L ZnSO4•7H2O, 0.001 g/L MnCl2•4H2O, pH to 7.0.
Arginine Glycerol Salts: 0.85 g/L l-arginine, 12.5 g/L glycerol, 1 g/L K2HPO4, 1 g/L NaCl, 0.5 g/L MgSO4•7H2O, 0.01 g/L FeSO4•7H2O, 0.001 g/L CuSO4•5H2O, 0.001 g/L ZnSO4•7H2O, 0.001 g/L MnSO4•H2O, pH to 7.0.
Soy Flour Mannitol: 20 g/L d-mannitol, 20 g/L soy flour, pH to 7.0 (This medium needs to be autoclaved twice).
4× R2A: 2 g/L peptone, 2 g/L starch, 2 g/L glucose, 2 g/L yeast extract, 2 g/L casein hydrolysate, 1.2 g/L K2HPO4, 1.2 g/L sodium pyruvate, 0.1 g/L MgSO4.
- MSB medium: To make 1 L MSB medium, mix separately sterilized A (40 mL), B (10 mL), C (5 mL), and ddH2O. Supplement with 10 mM (final concentration) of sodium succinate and 0.05 % casamino acids.
- 1 M Na2HPO4 + KH2PO4, pH 7.3. Mix ~38.25 mL 1 M Na2HPO4 stock solution and ~11.5 mL KH2PO4 stock solutions and adjust pH to 7.3.
-
Dissolve 20 g nitrilotriacetic acid and 14 g KOH in 700 mL H2O. Then add the following chemicals individually in order: 28 g MgSO4, 6.67 g CaCl2•2H2O (then add KOH to raise pH till dissolved), 0.0185 g (NH4)6Mo7O24•4H2O, 0.198 g FeSO4•7H2O, 100 mL Hunter’s metals 44*. Adjust pH to 6.8 with KOH. Adjust volume to 1 L.*Hunter’s metals 44: 2.5 g EDTA (free acid) dissolved in 800 mL dH2O, add 10.95 g ZnSO4•7H2O, 5 g FeSO4•7H2O, 1.54 g MnSO4•H2O, 0.392 g CuSO4•5H2O, 0.250 g Co(NO3)2•6H2O, 0.177 g Na2B4O7•10H2O, add five drops of concentrated sulfuric acid, adjust volume to 1 L and store at 4 °C.
- 20 % (wt/vol) (NH4)2SO4.
2.2 Reagents
All solutions are prepared using ultrapure water (prepared by purifying deionized water to attain a resistivity of 18 MΩ • cm at 25 °C) and analytical grade reagents.
SDS lysis buffer (4×): 125 mM Tris (pH 6.8), 4 % (wt/vol) SDS, 40 % (vol/vol) glycerol, 10 % (vol/vol) β-mercaptoethanol, 0.01 % (wt/vol) bromophenol blue.
SDS-PAGE running buffer: 25 mM Tris, pH 8.3, 192 mM glycine, 0.1 % SDS.
Ammonium bicarbonate (NH4HCO3) solution: 100 mM in water, pH 7.8 and 50 mM in water, pH 7.8.
Tris-(2-carboxyethyl) phosphine (TCEP): 10 mM in 50 mM NH4HCO3, pH 7.
Iodoacetamide (IAA): 50 mM solution in water.
Trypsin: dissolve one vial of lyophilized trypsin in 40 μL trypsin resuspension buffer provided by manufacturer, and heat at 30 °C for 15 min, then dilute 40-fold using 50 mM NH4HCO3 to a final concentration of 12.5 μg/mL. This solution should be freshly prepared.
1:2 5 % formic acid–acetonitrile: 1 part 5 % v/v formic acid solution in water to 2 parts LC-MS grade acetonitrile.
LC-MS buffer A: LC-MS grade water with 0.1 % formic acid.
LC-MS buffer B: LC-MS grade acetonitrile with 0.1 % formic acid.
2.3 Equipment
LTQ-Orbitrap mass spectrometer (Thermo-Fisher Scientific, MA, USA) with a nanoelectrospray source.
Nano LC system with autosampler.
Nano LC trap column (2 cm length × 100 μm ID, 5 μm C18 particle).
Nano LC analytical column (10 cm length × 75 μm ID, 5 μm C18 particle).
Vortexer.
Vortexer adapter for 1.5 mL tubes, horizontal orientation (MOBIO, CA, USA).
Sonic dismembrator with 1/8 in. microtip.
Electrophoresis system and power supply.
SpeedVac concentrator.
Ultrasonic cleanser.
Shaker incubator.
18 × 150 mm glass tubes with caps (autoclave).
Centrifuge for 1.5 mL tube.
Protein LoBind Tubes (Eppendorf, Germany).
15 mL conical centrifuge tubes.
14 mm culture dish.
Razor blades.
Carbide beads (0.25 mm, MOBIO, CA, USA).
3 Methods
3.1 Bacteria Strain Growth
This protocol uses Actinobacteria strains as an example, but the PrISM methodology can be applied to other species of bacteria as well. We suggest to use at least three types of culture media and to collect samples at 4 time points (see Note 1).
Inoculate 5 mL of culture media in an 18 × 150 mm sterile glass tube with a few single colonies of the strain of interest to generate a starting culture.
The tubes are placed at 45° angle in an incubator shaker and are allowed to grow at 30 °C with rotation at 250 rpm.
After ~2–3 days, when the strain has reached log growth phase, transfer an aliquot (500 μL) of the starting culture to the screening media (5 mL each) which are also contained in 18 × 150 mm glass tubes.
For each screening medium, four tubes are prepared to be harvested at 4 time points (see Note 2). For a typical Actinobacteria strain, we recommend harvesting at 24 h, 48 h, 72 h, and 96 h after transferring to screening media.
3.2 Cell Lysis
At each time point for harvesting, the entire culture is transferred to a 15 mL conical centrifuge tube and subjected to centrifugation at > 4000 × g for 10 min (see Note 3). The culture supernatants are removed and the cell pellets can be stored at −80 °C until ready for lysis.
Resuspend the cell pellet in an equal volume of 4× SDS lysis buffer, and transfer it to a 1.5 mL centrifuge tube. Place the sample on a hotplate set at 95 °C for 10 min (see Note 4).
Add a small amount (the same volume as the cell pellet) of carbide beads (0.25 mm) to the sample, and place the tubes on a vortexer with a horizontal orientation. Vortex for 30 min at maximum speed (see Note 5).
Sonicate the sample for 1 min using a sonic dismembrator with a 1/8 in. microtip (see Note 6).
Heat the samples at 95 °C for 10 min.
Centrifuge the samples at 20,000 × g for 10 min. Extract the supernatant to a new 1.5 mL centrifuge tube for SDS-PAGE analysis. This is the proteomic sample for a given strain under a certain growth condition. The samples can be stored at −80 °C until ready for SDS-PAGE separation.
3.3 Separation of the Proteomic Samples Using SDS-PAGE
Assemble the 10 % T precast gels (15 well) onto the electrophoresis system following the manufacturer’s instructions. Fill the inner and outside chambers of the cassette with the SDS running buffer.
Load 5 μL of the prestained protein standard into the first lane. Load 15 μL of the proteomic samples from Subheading 3.3 into each other lane. To avoid cross-contamination, try to load samples from the same strain on the same gel. Perform the electrophoresis at a constant voltage of 180 V, until the bromophenol blue dye reaches the bottom of the gel.
After SDS-PAGE, pry the gel plates open with a spatula. Place the gel in a clean 14 mm culture dish. Wash the gel with 200 mL of water three times for 5 min each. Remove the water and add ~50 mL of Bio-Safe™ Coomassie stain to the dish and let shake for 1 h. After staining, remove the stain and rinse with water for 3 × 10 min. Take a picture of the Coomassie stained gel and check the efficiency of the cell lysis (see Note 7) (Fig. 1).
Fig. 1.
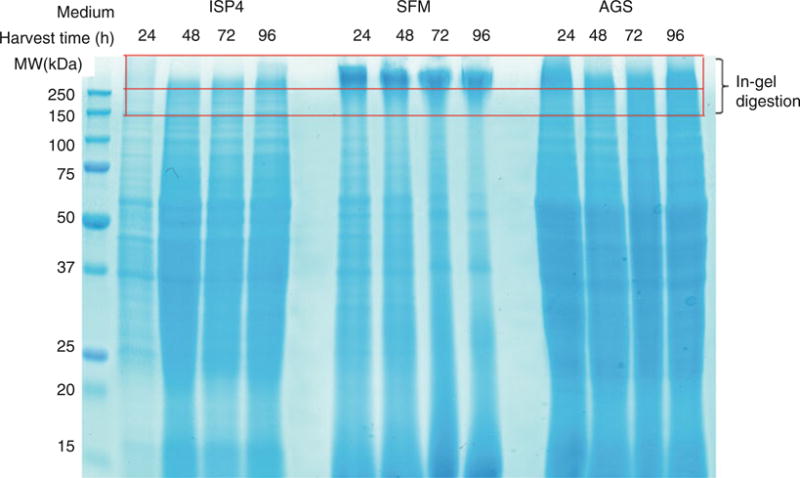
PrISM screening for an Actinobacteria strain grown in three different media (ISP4, SFM, and AGS) and harvested at 4 time points (24, 48, 72, and 96 h). The proteome samples are separated on an SDS-PAGE gel and stained by Coomassie. The high molecular weight regions (>150 kDa, shown within red box) are subjected to in-gel trypsin digestion
3.4 In-Gel Tryptic Digestion
Remove most of the water from the dish—leaving a small amount to prevent the gel from drying. For each lane, a razor blade is used to excise the region above 250 kDa as one slice and the 150–250 kDa region as another slice (see Note 8). Cut each gel slice into 1 × 1 mm gel pieces and transfer using the razor blade into a 1.5 ml LoBind Eppendorf tube (see Note 9). Always use a new razor blade and a new tube for each lane from a gel sample.
Add 300 μL H2O to each tube and vortex for 15 min.
Add 300 μL acetonitrile and wash for another 15 min.
Remove the supernatant carefully without picking up the gel pieces. Wash the gel pieces with 300 μL of 100 mM NH4HCO3 for 15 min. Discard the supernatant. Wash the gel pieces with 300 μL of 100 mM NH4HCO3–acetonitrile (50:50 vol/vol) for 15 min (see Note 10).
Add 100 μL acetonitrile to dehydrate the gel pieces for 5 min.
Discard the supernatant. Dry the gel pieces in a SpeedVac for 5 min. The samples can be stored at −80 °C until ready to move onto the next step.
Reduce the disulfide bonds of proteins by adding 50 μL of 10 mM TCEP in 50 mM NH4HCO3, incubate at room temperature for 30 min. Discard the supernatant.
Add 50 μL of 50 mM iodoacetamide in 50 mM NH4HCO3 (freshly prepared). Incubate at room temperature in the dark for 30 min. Discard the supernatant.
Wash the gel pieces with 300 μL of 100 mM NH4HCO3 for 15 min. Discard the supernatant.
Wash the gel pieces with 300 μL of 50 mM NH4HCO3–acetonitrile (50:50 vol/vol) for 15 min. Discard the supernatant.
Add 100 μL of acetonitrile to dehydrate the gel pieces for 5 min. Discard the supernatant.
Dry the gel pieces in a SpeedVac for 5 min. The samples can be stored at −80 °C until ready to move onto the next step.
Add 20 μL of the trypsin solution (12.5 μg/mL) to each tube. Allow the gel bands to rehydrate in trypsin solution for 30 min on ice.
If the digestion solution is fully absorbed after 30 min, add an additional 10 μL trypsin solution to cover the gel pieces. Let the gel sit on ice for an additional 1 h.
Remove the excessive trypsin solution and add 50 mM NH4HCO3 (sufficient volume to cover the gel pieces). Keep a record of the quantity of liquid added to the gel pieces.
Incubate at 30 °C overnight.
Cool the sample to room temperature and add 2 μL of 100 % formic acid to quench the digestion.
Add acetonitrile (an equal volume as added from 15) to the tube and shake vigorously on a vortexer for 30 min.
Transfer the supernatant to a new clean Eppendorf LoBind tube. This contains the extracted peptides.
Add 100 μL of 1:2 5 % formic acid–acetonitrile to the gel pieces and let shake for 20 min. Transfer the supernatant to the tube in step (19).
Repeat step (20).
Add 100 μL of acetonitrile to the gel pieces. Shake for 10 min. Transfer the supernatant to the tube in step (19).
Repeat step (22).
Dry the peptides in the tube at step (19) completely using the SpeedVac. Store the sample in −80 °C freezer until ready for LC-MS/MS analysis.
3.5 LC-MS/MS
Add 20 μL of LC-MS buffer A to each tube. To solubilize the peptides, place the tubes in an ultrasonic cleanser containing ice water and sonicate for 10 min.
Centrifuge the samples at 20,000 × g for 10 min to precipitate any insoluble materials. Transfer the supernatant to an LC sample vial.
The nanoLC method is configured as follows: the samples are initially loaded using 100 % LC-MS buffer A to the nanoLC trap column with a flow rate of 3 μL/min for 10 min. The eluent is diverted to the waste at this step.
-
Switch the valve so that the eluent is diverted to the nanoLC analytical column. The LC gradient is set as follows, with a flow rate at 300 nL/min:
Time (min) % B
0 0
55 45
63 80
67 0
90 0
- The eluted peptides are subjected to positive nanoelectrospray ionization (nanoESI) for mass spectrometric analysis using a Thermo LTQ-Orbitrap instrument (see Note 11). The instrument settings are configured as follows:
- FTMS full scan Automatic Gaining Control (AGC): 1e6.
- FTMS full scan maximum ion time: 2000 ms.
- Ion trap MSn AGC: 1e4.
- Ion trap MSn maximum ion time: 300 ms.
- Mass spectrometry method setup:
- FTMS full scan from m/z 400–2000 using a 30,000 resolving power. For each full scan, the top five most intense ions are selected for data dependent fragmentation using collision induced dissociation (CID) at 35 eV. The fragments are detected in the ion trap. Minimum signal required for MS2 is 500. Dynamic exclusion is set to 60 s, repeat count = 2. 1+ and unassigned charge states are rejected for MS2 events.
3.6 Data Analysis
To process the LC-MS data, extract the peak file lists from the .raw data files by using the DTA generator module from COMPASS software suite (http://www.chem.wisc.edu/~coon/software.php). Assume the precursor charge state to be 2+ through 4+.
Search the data using a proteomics software. In this protocol we use the Mascot software (Matrix Science, UK) for demonstration but other software can be used. If the target strain has a sequenced genome or draft genome, the target protein database should be used for the search. Otherwise, the publicly available NCBI nonredundant protein database (NCBInr) can be used, selecting bacteria as the taxonomy.
The following searching parameters are recommended: 10 ppm precursor mass error tolerance; 0.8 Da MS2 error tolerance; carbamidomethylation of cysteines as fixed modification; oxidation of methionines as variable modifications; maximal number of missed cleavages is 2; allowed peptide charge states are 2+, 3+, and 4+; data format is Mascot generic; and instrument type is ESI-TRAP.
After the Mascot search has finished, open the search results and perform filtering. We recommend the “ion score or expect cut-off” to be set to 30.
Browse the “protein family summary” and look for proteins that are associated with NRPS or PKS biosynthesis. For strains with sequenced genomes, the protein identification list will inform which high molecular weight NRPS and/or PKS proteins are expressed. The number of peptides identified from each proteomic sample is generally correlated with the expression level of the NRPS/PKS proteins under each growth conditions (Table 1).
If the genome sequence of the target strain is not available and the Mascot search is performed using NCBInr as the database, the protein identification list usually contains NRPS/PKS proteins from different organisms (Table 2). Depending on the sequence homology between the actual protein sequence in your sample and the sequence in the database, you may get one or two protein identifications that contain many peptide hits, or you may get many protein identifications each containing only one or two peptide hits. In the latter case and when the LC-MS/MS data are of high quality (as shown in Table 2), it is likely that the NRPS/PKS in your sample is a novel gene cluster that is not represented in the database.
Table 1.
An example of protein identification list (partial) from Mascot search for an Actinobacteria strain using its own sequenced genome as the database
| Protein description | Protein mass (kDa) | #Matches (sig) | #Sequence (sig) | %Coverage |
|---|---|---|---|---|
| NAD-glutamate dehydrogenase | 185 | 262 68 | 48.60 | |
| GAF sensor hybrid histidine kinase | 194 | 35 | 15 | 10.33 |
| Protein of unknown function DUF3686 | 175 | 30 | 16 | 12.54 |
| Hypothetical protein Sfla_5835 | 185 | 27 | 14 | 9.96 |
| Hypothetical protein Sfla_5465 | 169 | 18 | 15 | 11.81 |
| DNA-directed RNA polymerase, beta subunit | 144 | 18 | 12 | 11.39 |
| Amino acid adenylation domain protein | 265 | 15 | 8 | 3.80 |
| Bacterioferritin | 18 | 8 | 6 | 39.62 |
| Amino acid adenylation domain protein | 340 | 8 | 5 | 1.93 |
| Amino acid adenylation domain protein | 511 | 8 | 5 | 1.60 |
Three “amino acid adenylation proteins” (i.e., NRPSs, bolded), 511 kDa, 340 kDa, and 265 kDa each, are identified, indicating these three NRPS proteins are expressed under this growth condition
Table 2.
An example of Mascot search result (partial) using NCBInr as the database for an Actinobacteria strain which has no sequenced genome available
| Peptide sequence | Protein description | Accession # |
|---|---|---|
| DAEALVAYcDR | Nonribosomal peptide synthetase [Streptomyces kitasatoensis] | BAH68474 |
| FVADPFGEPGER | Nonribosomal peptide synthetase [Streptomyces griseolus] | BAH68409 |
| ADGAVEYIGR | Pyoverdine sidechain peptide synthetase I, epsilon Lys module [Pseudomonas syringae pv. tabaci ATCC 11528] | ZP_05637427 |
| GVGPESVVGVAVPR | Nonribosomal peptide synthetase [Rhodococcus opacus B4] | YP_002779301 |
| ADGNVDFLGR | Amino acid adenylation domain-containing protein [Opitutus terrae PB90-1] | YP_001818855 |
| FVADPYGAPGSR | Nonribosomal peptide synthetase [Streptomyces gibsonii] | BAH68663 |
| LGAGDDIPIGTPVAGR | Peptide synthetase 2 [Streptomyces roseosporus NRRL 11379] | AAX31558 |
| DVVFGTTVSGR | Amino acid adenylation domain protein [Acetivibrio cellulolyticus CD2] | ZP_07326042 |
| FTADPYGPAGSR | Putative NRPS [Streptomyces griseus subsp. griseus NBRC 13350] | YP_001824777 |
| GAGPETLVAVALPR | Mannopeptimycin peptide synthetase MppB [Streptomyces hygroscopicus] | AAU34203 |
| TVAALAALAR | Putative nonribosomal peptide synthetase [Nocardia farcinica IFM 10152] | YP_119006 |
| LGAGDDIPIGSPVAGR | Amino acid adenylation domain-containing protein [Frankia sp. EAN1pec] | YP_001510190 |
| VVAIALPR | Peptide synthetase ScpsA [Saccharothrix mutabilis subsp. capreolus] | AAM47272 |
| VAGPALTDFLADQR | Nonribosomal peptide synthetase [Streptomyces griseolus] | BAH68411 |
| LVLITDEAR | Nonribosomal peptide synthetase [Streptomyces griseolus] | BAH68411 |
| DIATAYAAR | Amino acid adenylation domain-containing protein [Streptomyces roseosporus NRRL 15998] | ZP_04696845 |
| IPLSYAQR | Nonribosomal peptide synthetase [Streptomyces fungicidicus] | ABD65957 |
All the peptides related to NRPS/PKS are extracted. These peptides match to NRPS proteins from different organisms, indicating the detected NRPS share a low sequence homology with any known NRPS gene clusters in the database and likely represent a new NRPS biosynthetic pathway
Acknowledgments
This work was supported by National Institute of General Medical Sciences of the National Institutes of Health under award number R01 GM067725. We thank the Agricultural Research Service, US Department of Agriculture for providing the bacterial strains.
Footnotes
For a general PrISM screening of Actinobacteria strains, the typical medium for starting culture is Malt Yeast Glucose (MYG) medium or ATCC 172 medium, in which most strains grow vigorously. For the screening media, the researchers can use whichever media they prefer. In our practice, we have used ISP4, Arginine Glycerol Salts (AGS), Soy Flour Mannitol (SFM), 4× R2A, and MSB media. All media are prepared using tap deionized water (do not use ultrapure water) and autoclaved.
This growth condition (30 °C, 3 day, 250 rpm) is suitable for most Actinobacteria strains. However, if the strain of interest has special growth needs (e.g., higher/lower temperature, special growth factors, light/dark environment, or faster/slower growth rate), the specific growth conditions should be adjusted accordingly.
For some strains, the cell pellet can be very loose. Be careful when pouring out the culture supernatant. Use a pipette when necessary. If the cells do not pellet down completely, repeat the centrifugation step at a higher speed then remove the medium completely.
Estimate the volume of the cell pellet. Then add an equal volume of the lysis buffer, e.g., 25–100 μL.
Bead beating has been demonstrated as the most powerful and universal method for lysis of cells and tissue. We have tested that carbide bead with 0.25 mm diameter is best for the lysis of bacteria samples. Placing the tubes in the horizontal orientation increases lysis efficiency. If the cells are not lysed using bead beating, other methods should be considered, sonication, freeze–thaw, French press, manual grinding, etc.
This short sonication step is to disrupt DNA in the cells. The undisrupted DNA molecules will cause streaking bands on the SDS-PAGE.
The Coomassie staining step is primarily to check the efficiency of cell lysis. A successfully lysed sample should show an abundant amount of proteins on the SDS-PAGE gel. If the cells are not lysed properly, little or no protein bands will show on the gel. In this case, there is no need to proceed to the following steps. Other cell lysis protocols should be attempted to extract the proteome. If the proteome sample is much diluted (>100 μL volume), and higher sensitivity of detection is desired, the sample can be separated on a preparative SDS-PAGE gel, which contains a single lane with 250 μL loading capacity.
We recommend performing in-gel trypsin digestion to all samples, whether they show visible protein bands at the high molecular weight region by Coomassie stain. Coomassie stain has a much lower sensitivity of detection than nanoLC-MS. We also suggest cutting the high molecular weight region into two separate gel slices, one contains proteins >250 kDa and one contains 150–250 kDa proteins. Additional size based fractionation will result in more protein identifications.
In-gel digestion procedure is very sensitive to the contamination of keratin. To minimize contamination, always wear gloves when handling gels and performing in-gel digestion. Always use LoBind tubes and use clean spatula when preparing reagents.
If the band pieces are still blue after this step, repeat the washing steps using 300 μL of 100 mM NH4HCO3 for 15 min and then 300 μL of 100 mM NH4HCO3–acetonitrile (50:50 vol/vol) for 15 min.
Other types of mass spectrometers that are capable of proteomic analysis can also be used, Q-Exactive, QqTOF, Ion trap, FT-ICR, etc. Users need to adjust the mass spectrometric method according to the type of instrument used.
References
- 1.Walsh CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Koehn FE. High impact technologies for natural products screening. Prog Drug Res. 2008;65:177–210. doi: 10.1007/978-3-7643-8117-2_5. [DOI] [PubMed] [Google Scholar]
- 4.Baltz RH. Marcel Faber Roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. 2006;33:507–513. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- 5.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lautru S, Deeth RJ, Bailey LM, et al. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol. 2005;1:265–269. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 7.Corre C, Challis GL. New natural product biosynthetic chemistry discovered by genome mining. Nat Prod Rep. 2009;26:977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 8.Gross H, Stockwell VO, Henkels MD, et al. The genomisotopic approach: a systematic method to isolate products of orphan biosynthetic gene clusters. Chem Biol. 2007;14:53–63. doi: 10.1016/j.chembiol.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Scherlach K, Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem. 2009;7:1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 10.van Wezel GP, McDowall KJ. The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat Prod Rep. 2011;28:1311–1333. doi: 10.1039/c1np00003a. [DOI] [PubMed] [Google Scholar]
- 11.Bumpus SB, Evans BS, Thomas PM, et al. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27:951–956. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans BS, Ntai I, Chen Y, et al. Proteomics-based discovery of koranimine, a cyclic imine natural product. J Am Chem Soc. 2011;133:7316–7319. doi: 10.1021/ja2015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, McClure RA, Zheng Y, et al. Proteomics guided discovery of flavopeptins: anti-proliferative aldehydes synthesized by a reductase domain containing non-ribosomal peptide synthetase. J Am Chem Soc. 2013;135:10449–10456. doi: 10.1021/ja4031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Ntai I, Ju KS, et al. A proteomic survey of nonribosomal peptide and polyketide biosynthesis in Actinobacteria. J Proteome Res. 2012;11:85–94. doi: 10.1021/pr2009115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Unger M, Ntai I, et al. Gobichelin A and B: mixed-ligand siderophores discovered using proteomics. Medchemcomm. 2013;4:233–238. doi: 10.1039/C2MD20232H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albright JC, Goering AW, Doroghazi JR, et al. Strain-specific proteogenomics accelerates the discovery of natural products via their biosynthetic pathways. J Ind Microbiol Biotechnol. 2014;41:451–459. doi: 10.1007/s10295-013-1373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]