

Abstract
Introduction
Sialidosis is a neurosomatic, lysosomal storage disease (LSD) caused by mutations in the NEU1 gene, encoding the lysosomal sialidase NEU1. Deficient enzyme activity results in impaired processing/degradation of sialo-glycoproteins, and accumulation of oversialylated metabolites. Sialidosis is considered an orphan disorder for which no therapy is currently available.
Areas covered
The review describes the clinical forms of sialidosis and the NEU1 mutations so far identified; NEU1 requirement to complex with the protective protein/cathepsin A for stability and activation; and the pathogenic effects of NEU1 deficiency. Studies of the molecular mechanisms of pathogenesis in animal models uncovered basic cellular pathways downstream of NEU1 and its substrates, which may be implicated in more common adult (neurodegenerative) diseases. The development of a Phase I/II clinical trial for patients with galactosialidosis may prove suitable for sialidosis patients with the attenuated form of the disease.
Expert opinion
Recently, there has been a renewed interest in the development of therapies for orphan LSDs, like sialidosis. Given the small number of potentially eligible patients, the way to treat sialidosis would be through the coordinated effort of clinical centers, which provide diagnosis and care for these patients, and the basic research labs that work towards understanding the disease pathogenesis.
Keywords: NEU1, sialidosis, mechanisms of pathogenesis, lysosomal storage disease, lysosomal exocytosis, chaperone-mediated therapy
1. Sialidosis: Clinical Phenotypes
Sialidosis is an autosomal recessive lysosomal storage disease, belonging to the glycoproteinosis subgroup. The underlying cause is the deficiency of the sialic acid-cleaving enzyme, sialidase 1 or neuraminidase 1 (NEU1) 1, 2. Historically, the name sialidosis was first used by Durand et al. (1977) 3 to designate the syndrome of two siblings of 22 and 13 years of age who developed visual impairment and mild neurological manifestations in their adolescence. Enzymatic assays in culture fibroblasts and leukocytes from these patients revealed an isolated deficiency of NEU1. Until then, deficiency of this enzyme was linked to classical mucolipidosis I 4, a severe and rapidly progressive lysosomal storage disease with onset at birth or shortly after birth. It became apparent at that point that patients with isolated deficiency of NEU1 developed a broad range of clinical manifestations associated with seemingly distinct diseases. The proper nosology of neuraminidase deficiency was given by Lowden and O’Brien (1979) 5, who classified patients with sialidosis into two main types: Type I (normomorphic) and Type II (dysmorphic).
1.1 Type I sialidosis
Type I sialidosis is the attenuated, non-neuropathic form of the disease, also known as cherry-red spot myoclonus syndrome. Generally, type I patients have no obvious physical defects, and their intelligence is normal or only slightly impaired. Symptoms appear in the second or third decade of life with gait abnormalities, decreased visual acuity, or both 6. The visual loss is progressive and is associated with bilateral macular cherry-red spots that may fade later in the course of the disease 6–8. The most prominent clinical aspect is myoclonus that may be precipitated by voluntary movements, the thought of movement, passive joint movements, or light touch or sound stimuli. Initially there are intention tremors and difficulty with fine motor movements, but eventually the myoclonus can become very debilitating, despite maintaining normal muscle strength. Leg tremors and generalized seizures may also occur. In the severe cases, as the disease progresses, the patients may be confined to a wheelchair. Despite becoming disabled, type 1 patients remain intellectually active. A few vacuolated lymphocytes and histiocytes may be present in peripheral blood and bone marrow smears, respectively. At the ultrastructural level swollen lysosome are visible in bone marrow cells and in Kupffer cells of the liver. Increased high molecular weight sialylated oligosaccharides are found in patients’ urine 9. Recently, several patients with full-blown manifestations of myoclonus without visual symptoms and no measurable NEU1 activity have been reported 10, 11. This interesting finding suggests that mutations affecting NEU1 activity can exist in the absence of other clinical signs that are characteristic of sialidosis.
Their overall normal appearance and intelligence, the absence of overt skeletal and visceral abnormalities and long survival explain why patients with type I sialidosis are often overlooked. The diagnosis of type I cases may be guided by either their ophthalmologic presentation combined with sialyl-oligosacchariduria, or by whole genome sequencing 8, 10, 12. The penetrance and degree of severity of the symptoms in these patients correlate closely with the type of NEU1 mutations involved and, in turn, the levels of residual enzyme activity (see below).
1.2 Type II sialidosis
Type II sialidosis is the severe, neuropathic form of the disease which is further classified in three subtypes: congenital or hydropic with onset in utero, infantile with onset between birth and 12 months, and juvenile with onset past 2 years of age. Type II patients with the most acute, congenital form of the disease develop hydrops fetalis, neonatal ascites, or both; they are stillborn or die shortly after birth with a systemic and fulminant condition. Their clinical presentation at birth includes facial edema, inguinal hernias and hepatosplenomegaly 6, 7. All patients with type II disease have, among other symptoms, coarse face, enlargement of spleen and liver, dysostosis multiplex, vertebral deformities, and severe mental retardation. Both the infantile patients with longer survival and the juvenile cases develop macular cherry-red spots and myoclonus, and may also have hearing loss and angiokeratoma 6, 13–15. Their life expectancy can vary greatly depending on the associated mutations and the severity of the symptoms.
2. Galactosialidosis
Severely reduced or deficient NEU1 activity secondary to a primary defect of the lysosomal serine carboxypeptidase, protective protein cathepsin A (PPCA) is characteristic of galactosialidosis (GS) 7, 16, 17. Patients with sialidosis and those with GS share clinical and biochemical features that are attributed at least in part to the loss of NEU1 function in both diseases (see below). Similarly to sialidosis, patients with GS are clinically heterogeneous and differ widely in severity and age of onset of the symptoms. The severe, early onset forms of the disease develop a systemic condition associated with fetal hydrops, skeletal dysplasia, visceromegaly, renal and cardiac failure, variable neurological involvement and early death. These phenotypic aberrations are shared with patients with the early infantile, neurodegenerative form of sialidosis, which is fatal in early childhood.
3. Lysosomal NEU1 and Its Auxiliary Protein PPCA
NEU1 is a lysosomal exo-glycosidase that catalyzes the cleavage of terminal N-acetylated neuraminic acids (sialic acid) linked to the saccharide chains of glycoproteins, glycolipids as well as oligo- and polysaccharides 7. The enzyme belongs to the superfamily of sialidases that have a conserved active site and similar sequence motifs. Their wide distribution in nature largely coincides with that of their target sugar residue, sialic acids. Much like sialic acids18, sialidases play a central role in many biological processes, including cell proliferation/differentiation, clearance of plasma proteins, cell adhesion, catabolism of gangliosides and glycoproteins, immunocyte function, modification of receptors, and the developmental modeling of myelin. The pivotal and diverse functions of these enzymes, many of which remain undiscovered, likely account for the existence of three additional mammalian sialidases, besides NEU1, which are encoded by different genes 19–22. Based on their subcellular localization, these enzymes are defined as cytosolic (NEU2), plasma-membrane (NEU3), and mitochondrial/lysosomal/intracellular membranes (NEU4). The 4 mammalian sialidases have a distinctive tissue distribution, pH optimum, kinetic properties, responses to ions and detergents, and preference for sialic acid linkages 19. There appears to be little overlap in the function of the individual sialidases, despite their shared mechanism of action. This lack of redundancy is reflected in that none of the other three enzymes can apparently compensate for the loss of lysosomal NEU1 in patients with sialidosis.
NEU1 is the most abundant and ubiquitous of the 4 mammalian sialidases. The enzyme has a wide tissue distribution, albeit that its expression levels vary greatly 7, 19. Additionally, NEU1 differs from the other mammalian sialidases in that it is active exclusively in a high molecular weight, multienzyme complex containing at least two other hydrolases: the glycosidase β-galactosidase (β-GAL) and the PPCA. By virtue of their association with PPCA, NEU1 and β-GAL acquire their active and stable conformation in lysosomes. Only a fraction of PPCA and β-GAL activities is found in the NEU1-PPCA-β-GAL complex, which instead contains all of the NEU1 catalytic activity 23. Within the complex, PPCA functions as an indispensable chaperone/transport protein, especially for NEU1. was demonstrated that NEU1, being poorly mannose 6-phosphorylated, depends on its association with PPCA for lysosomal compartmentalization, catalytic activation and stability in lysosomes. An equally valid mode of targeting NEU1 to the lysosomes has been shown, which entails the interaction of NEU1 with the membrane via a short C-terminal internalization signal 24, This model explains the occurrence of NEU1 at the PM, the inner lysosomal membrane and the lysosomal lumen. Whether this targeting mechanism of NEU1 requires or not the association with PPCA is still not fully understood24. The two proteins interact already in an early biosynthetic compartment. The C-terminal portion of PPCA, which includes proline 451 important for this interaction with NEU1, also embeds an essential NEU1 binding domain. The presence of binding sites in NEU1 that have affinity for both PPCA and other NEU1 molecules suggests that heterodimerization of NEU1 with PPCA is critical not only for the routing of the enzyme to the lysosome, but also for preventing premature self-association and aggregation early during biosynthesis 23, 25. It is therefore predictable that NEU1 mutations that affect its interaction with PPCA lead to disease, even though the residues forming the active site of the enzyme are intact.
4. Disease-causing NEU1 Mutations
To date, more than 40 NEU1 disease-causing mutations have been identified in sialidosis patients (Table 1). Most of these are missense mutations that do not affect NEU1 mRNA synthesis or stability, since all patients so far described are mRNA positive. On the basis of their biochemical properties, NEU1 protein variants can be divided into three groups: in the first group, the mutant enzyme is catalytically inactive and does not localize to the lysosomes; in the second group, the mutant enzyme localizes to the lysosomes but is enzymatically inactive; in the third group, the mutant enzyme has residual activity and localizes to the lysosomes.
Table 1.
| Codon number | Type of mutation | Aminoacid change | Patient | References |
|---|---|---|---|---|
| 1 | Missense | MET-ILE | Type II | 63 |
| 8 | Duplication | Duplication THR-ASP | Type II | 2 |
| 15 | Missense | TRP-TERM Code | Type II | 13 |
| 23 | Missense | TRP-TERM Code | Type II | 63 |
| 29 | Missense | TRP-TERM Code | Type II | 64 |
| 54 | Missense | VAL-MET | Type I | 26 |
| 55 | Missense | GLN-TERM Code | Type I | 65 |
| 68 | Missense | GLY-VAL | Type II | 25 |
| 80 | Missense | PRO-LEU | Type II | 27 |
| 91 | Missense | LEU-ARG | Type II | 1 |
| 111 | Missense | LEU-PRO | Type I | 66 |
| 136 | Missense | GLY-GLU | Type I | 66 |
| 135 | Missense | ASP-ASN | Type I | 67 |
| 136 | Missense | GLY-GLU | Type I | 66 |
| 182 | Missense | SER-GLY | Type I | 25 |
| 208 | Deletion | Frameshift/TERM code | Type II | 25 |
| 217 | Missense | VAL-MET | Type I | 68 |
| 219 | Missense | GLY-ALA | Type I | 26 |
| 225 | Missense | ARG-PRO | Type II | 63 |
| 227 | Missense | GLY-ARG | Type II | 25 |
| 231 | Missense | LEU-HIS | Type I | 26 |
| 240 | Missense | TRP-ARG | Type II | 27 |
| 243 | Missense | GLY-ARG | Type I | 68 |
| 260 | Missense | PHE-TYR | Type II | 2 |
| 270 | Missense | LEU-PHE | Type II | 25 |
| 275 | Missense | VAL-ALA | Type I | 66 |
| 280 | Missense | ARG-TERM code | Type II | 26 |
| 294 | Missense | ARG-CYS | Type I | 26 |
| 298 | Missense | ALA-VAL | Type II | 25 |
| 316 | Missense | PRO-SER | Type I | 27 |
| 319 | Missense | ALA-VAL | Type I | 65 |
| 328 | Missense | GLY-SER | Type I | 25 |
| 335 | Missense | PRO-GLN | Type II | 26 |
| 340 | Splicing | Type II | 13, 29 | |
| 341 | Missense | ARG-GLY | Type II | 63 |
| 345 | Missense | THR-ILE | Type I | 66 |
| 347 | Missense | ARG-TERM code | Type II | 66 |
| 363 | Missense | LEU-PRO | Type II | 2 |
| 370 | Missense | TYR-CYS | Type II | 26 |
| 377 | Missense | GLU-TERM code | Type I | 1 |
| 397 | Deletion | Frameshift/longer ORF | Type I | 30 |
| 399 | Duplication | Duplication HIS-TYR | Type I | 26 |
| 445 | Deletion | Frameshift/longer ORF | Type II | 1 |
The 3D structure of NEU1 has not been solved yet, but investigators have used the crystal structures of bacterial sialidases as templates to model several of the NEU1 amino acid substitutions associated with different clinical phenotypes. Interestingly, the majority of those substitutions appear to be located at the core surface of the molecule, suggesting that they could affect the interaction of NEU1 with its chaperone PPCA (see above). In fact, three pathogenic mutations, F260Y, L270F and A298V, clustered at the surface of the bacterial sialidases, were shown to be correctly synthesized but rapidly degraded because the resulting proteins were unable to associate with PPCA 25. By analyzing the hydrodynamic properties of NEU1 and PPCA, a putative region of interaction between the two proteins was identified 16, 26. This region spanning residues 23–250 was shown to be important for NEU1 binding to the precursor form of PPCA. Thus, NEU1 mutations affecting amino acids within this domain will likely affect the stability of the enzyme and its PPCA-mediated trafficking to the lysosomes. Importantly, a few missense NEU1 mutations have been found in both type I and type II patients (Table 1). In these cases, if the patient is compound heterozygote for a “mild” and a “severe” mutation the level of residual enzyme activity and, in turn, the degree of severity of the symptoms will depend on how the individual amino acid substitutions impact on the overall biochemical properties of NEU1 8, 13, 14, 25–30. In addition, environmental factors, including diet and life style, as well as other genetic factors could influence the penetrance of specific phenotypic alterations and the manifestations of the clinical symptoms. In this respect, a recent publication described the identification of 9 individuals carrying NEU1 mutations found in type II severe cases of sialidosis, but with an atypical presentation of type I disease, limited to mild or severe myoclonus without macular cherry-red spot and oligosacchariduria 10, 11. This finding suggests that NEU1 deficiency does not necessarily result in a life threatening condition 10, 1110, 1110, 1110, 1110, 1110, 119, 109, 109, 10.
5. Mouse Models with NEU1 Deficiency
5.1 Neu1 knockout model
Complete loss of Neu1 in mice is compatible with life, but Neu1−/− mice develop a systemic and neurodegenerative condition that closely resembles the early onset, type II form of sialidosis and die prematurely 31. Mutant mice present signs of the disease at birth, with progressive oligosacchariduria and expansion of the lysosomal compartment in cells of virtually all systemic organs and the nervous system, as well as bone, muscle and cartilage. The primary affected cells are epithelia, reticulo-endothelia and histiocytes. Symptoms include growth retardation, hepatosplenomegaly, noticeable edema of the limbs and eyelids, kyphosis of the lumbar spine, lordosis of the cervical and thoracic spine, neurological impairment and degeneration. At the end of their life span (6 to 7 months of age) mutant mice suffer from dyspnea, severe loss of weight, diffuse edema, gait abnormalities and tremor 31.
The pathological manifestations in Neu1−/− mice that more closely recapitulate those in patients are discussed in detail below. They include time-dependent enlargement of the spleen, which is associated with extramedullary hematopoiesis (EMH). In the kidney, the site with the highest NEU1 mRNA and protein expression in normal mice, pronounced lysosomal vacuolization of the renal tubular epithelium is noticeable already in the neonatal period and worsen with age. Impaired kidney function is likely one of the main causes of the progressive and diffuse edema characteristic of Neu1−/− mice and patients. Altered homeostasis of muscle connective tissue is responsible for the muscle atrophy and hypotonia 31, 32. In the brain, these mice show extensive vacuolization of the epithelial cells of the choroid plexus, and the endothelial cells of the ependymal layer 31, 33. Microglia and perivascular macrophages are among the most affected cells, generally located juxtaposed to the degenerating neurons. This is particularly evident in the dentate gyrus and the hippocampus, but these cells are also scattered throughout the cortex and in the cerebellum, triggering a widespread microgliosis.
A common molecular pathway has been identified that is at the basis of all these phenotypic alterations in the Neu1−/− mice (see below), albeit that the pathogenic effects downstream of this pathway depend on the physiological characteristics and functions of the individual organs (Figure 1).
Figure 1.
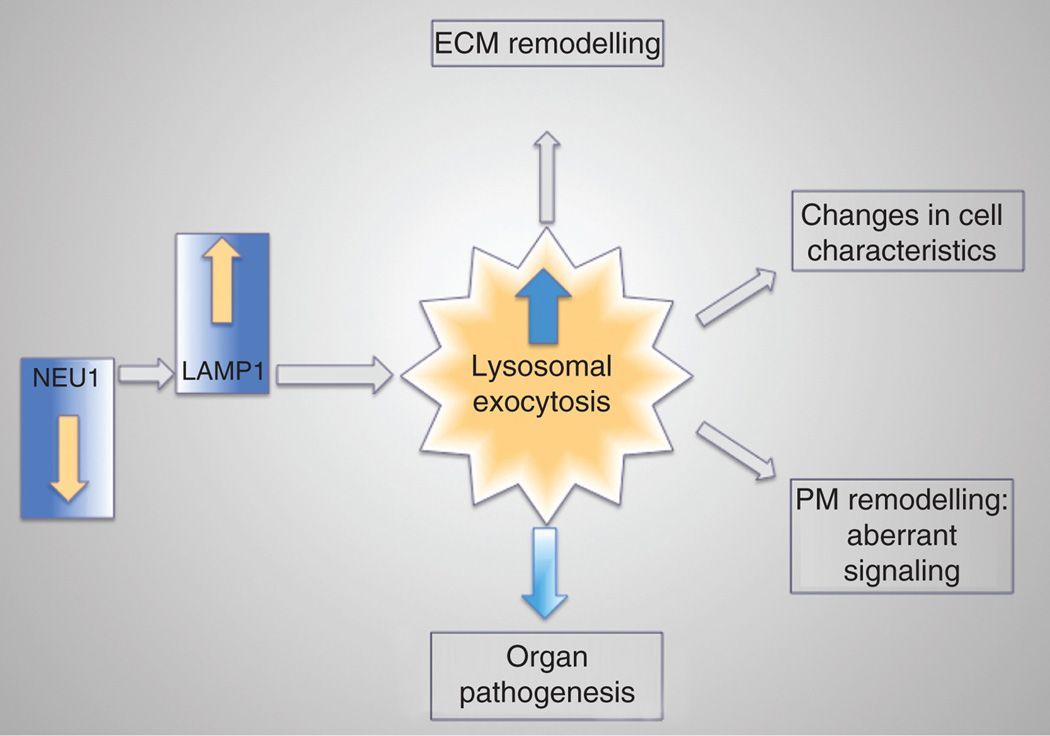
Schematic representation of the events that lead to organ pathogenesis downstream of NEU1 deficiency. In absence of NEU1, unprocessed LAMP1 accumulation leads to increased number of lysosomes at the PM resulting in exacerbated lysosomal exocytosis. The abnormal release of lysosomal content extracellularly causes ECM and PM remodeling, changes cell characteristics and ultimately causes organ pathogenesis.
5.2 Type I sialidosis model: Neu1−/−;NEU1V54M
Recently, a transgenic mouse model of the attenuated, type I form of sialidosis has been genetically engineered to express a NEU1 variant carrying the aminoacid substitution V54M, previously identified in a non-neuropathic adult patient 26. A NEU1V54M transgenic founder was crossed into the Neu1−/− background to generate mice with residual NEU1 enzyme activity 34. These mice are fertile and viable with no apparent early signs of disease. They have no neurological involvement and a normal lifespan. Between 1 and 2 years of age, these mice develop edema, enlargement of the kidneys with vacuolization of the tubular epithelium, and oligosacchariduria 34. NEU1 activity is reduced in all the Neu1−/−;NEU1V54Morgans, although activity levels may vary among different tissues. The lowest residual enzyme activity is measured in the bone marrow, liver, salivary glands and kidney, while in the heart, spleen, lung and brain the residual activity ranges between 30% and 80% of control values. No overt morphological changes are visible in the brain or in most of the visceral organs until late in life (1 year), making these mice a truthful model of type I sialidosis.
5.3 PPCA−/− with secondary deficiency of Neu1
Mice homozygous for a null mutation at the Ppca (Ctsa) locus have no cathepsin A activity and severe secondary deficiency of Neu1 35. Again, these mice have a clinical and pathological presentation that closely recapitulates the early onset forms of both GS and sialidosis 35. They develop progressive nephropathy, time-dependent splenomegaly, heart involvement, and have shortened lifespan. They are also infertile as homozygous knockouts because of structural changes to the blood-epididymal barrier, resulting in altered sperm motility 36. The secondary loss of Neu1 activity explains why many phenotypic abnormalities in Ppca−/− mice are similar to those seen in the Neu1−/− model, although on close examination features that are unique for one or the other disease model have been identified 31. The most overt difference is seen in the cerebellum; early in life Ppca−/− mice acquire acute and progressive ataxia that is associated with regional loss of cerebellar Purkinje cells and impaired motor coordination 31. This phenotype is not observed in the Neu1−/− mice, at least not until the end of their lifespan. Because the expression levels of PPCA in Purkinje cells are greater than those of Neu1, it is possible that these neurons are more sensitive to the loss of cathepsin A than of Neu1 activity, but more rigorous testing is needed to corroborate this hypothesis.
5.4 CathAS190A–Neo
CathAS190A–Neo mice were generated by homologous recombination in murine ES cells, using a targeting construct carrying a point mutation that resulted in serine 190 to alanine amino acid substitution at the catalytic site of the Ppca protein, followed by a PGK-Neo cassette inserted in intron 7 of the Ppca gene 37. Although the mutation targeted the Ppca locus, the CathAS190A-Neo mice displayed a drastically reduced activity of Neu1 in most tissues, due to destabilization of Ppca mRNA by the Neo cassette. Contrary to the Ppca−/− mice, the CathAS190A-Neo mice are apparently vital and fertile, develop normally and have a normal lifespan, suggesting that 10% NEU1 activity is sufficient to promote normal development and growth 37, 38. Upon removal of the Neo cassette from intron 7, the same authors successfully generated mice (CathAS190A) with an isolated deficiency of cathepsin A, but with intact protective properties toward NEU1; CathAS190A have normal Neu1 activity 37.
6. Mechanisms of Pathogenesis in Mouse Models of Sialidosis
6.1 Neu1 as negative regulator of lysosomal exocytosis
Studies of the molecular mechanism(s) underlying some of the canonical phenotypes of sialidosis in the Neu1−/− mice revealed a new role of NEU1 as negative regulator of lysosomal exocytosis 39. This calcium-dependent process was initially characterized in specialized secretory cells containing the so called “secretory lysosomes”, such as platelets, mast cells, neutrophils, cytotoxic T cells, melanocytes and macrophages 39–42. It was later established that lysosomal exocytosis occurs in virtually all cell types, including fibroblasts, epithelial cells and neurons 33, 42, and is activated under physiological or pathological conditions for the repair and renewal of damaged plasma membrane (PM), the removal of pathogenic bacteria, the release of human immunodeficiency virus from infected cells, and the remodeling of the extracellular matrix (ECM).
Lysosomal exocytosis begins with the recruitment of a selected pool of lysosomes to the actin cytoskeleton followed by docking of the organelles at the PM. Upon changes of intracellular calcium concentration, docked lysosomes fuse with the PM and release their luminal contents into the ECM 39, 43, 44. Considering its many physiological roles, it is predictable that the extent of lysosomal exocytosis in any cell type would greatly affect the characteristic and composition of their PM and the surrounding ECM with overall loss of tissue homeostasis. Several gene defects have been identified that result in impaired lysosomal exocytosis and lead to immune deficiency, bleeding disorders and albinism 40, 44–46. Characterization of these genetic defects has enabled the identification of key proteins required for the movement of lysosomes along the cytoskeletal network and for their fusion with the PM 45, 46.
It is now established that NEU1 negatively regulates lysosomal exocytosis by influencing the levels of one of its substrates, the Lysosomal Associated Membrane Protein 1 (LAMP1) 39. LAMP1 is an integral membrane protein with a single transmembrane domain, a small cytoplasmic tail of 12-amino-acids and a highly glycosylated and sialylated N-terminal portion facing the lumen of the lysosomes 39, 47. LAMP1 plays an active role in the docking of lysosomes at the PM, although the molecular mechanism is still not known 39, 48. Defective or deficient NEU1 activity results in the impaired processing of the sialic acids on LAMP1, which consequently accumulates in an oversialylated state, and has a prolonged half-life. For as yet unknown reasons, accumulation of oversialylated LAMP1 increases the number of LAMP1-marked lysosomes that dock at the PM, poised to engage in lysosomal exocytosis upon calcium influx (Figure 2a) 39. LAMP1’s key function in the docking of lysosomes to the PM has been further validated by the demonstration that silencing LAMP1 in NEU1-deficient cells normalizes the number of lysosomes docked at the PM and decreases the extent of lysosomal exocytosis 39. Thus, the ultimate consequence of NEU1 loss of function is the excessive extracellular release of lysosomal luminal contents from deficient cells of different tissues and organs. Excessive lysosomal exocytosis has now been linked to several pathological manifestations characteristic of sialidosis, examples of which are discussed below 32, 33, 39, 49.
Figure 2.
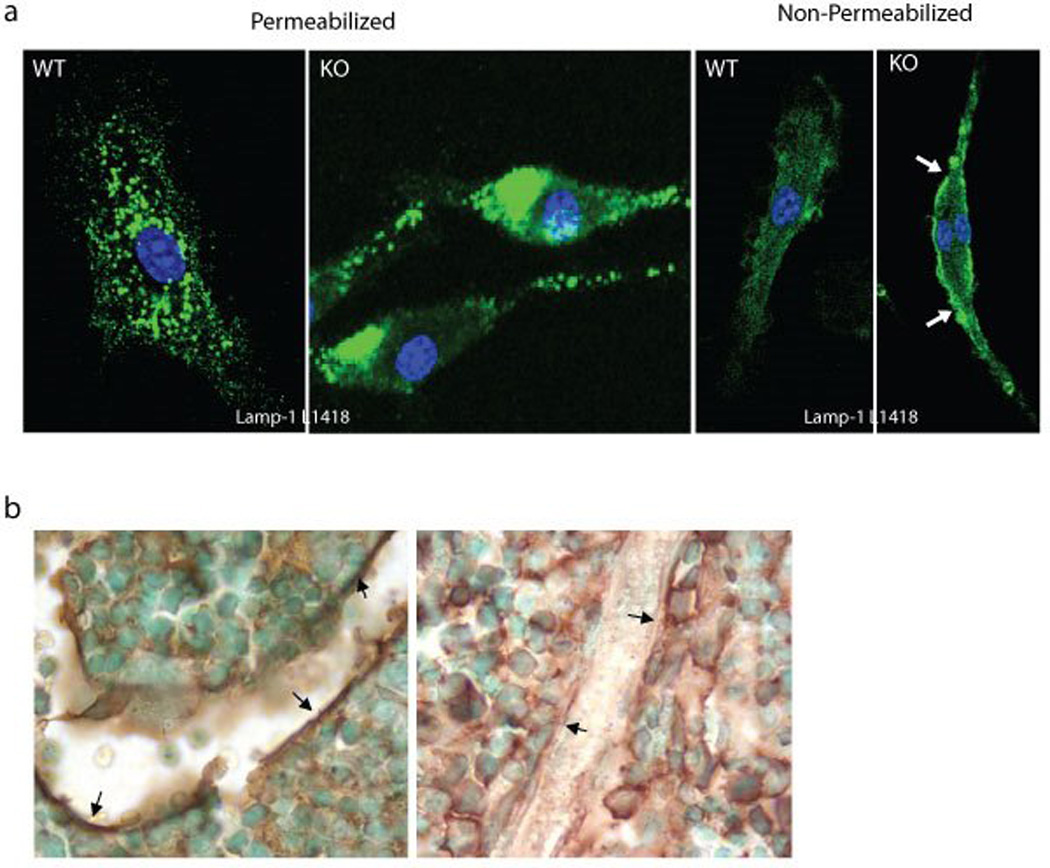
a) Comparison of immunofluorescence staining of permeabilized and non-permeabilized WT and Neu1−/− macrophages with LAMP1 antibodies against either the C-terminal cytosolic domain (L1418) or the glycosylated luminal domain (1D4B) of LAMP1; b) Bone sections from 2 months old Neu1−/− and control mice were stained with the anti-VCAM-1; Strong immunoreactivity was observed only in the sections from the control in the region lining the bone (arrows). Adapted from Yogalingam et al Dev. Cell 2008, with permission of Elsevier.
7. Aspects of Pathogenesis Attributed to Excessive Lysosomal exocytosis
7.1 Extramedullary hematopoiesis and splenomegaly
A fully penetrant phenotype in the Neu1−/− mice that recapitulates one of the typical clinical manifestations of type II sialidosis in children is splenomegaly 6. Histological and cellular characterization of the spleen in mutant mice of increasing age identified a time-dependent expansion of total splenic cell counts, and increased number of erythroid precursors and megakaryocytes. These alterations in the spleen were paralleled by increased number of hematopoietic progenitors in the peripheral blood and by an overall lower number of these cells in the bone marrow (BM). Together these features were indicative of EMH 31. In addition, transplantation of Neu1−/− mice with genetically modified BM cells expressing NEU1, as potential therapy for sialidosis, was unsuccessful because sub-lethally irradiated mice failed long-term engraftment 39. The search for a potentially common molecular cause that would explain both time-dependent EMH and lack of BM engraftment in Neu1−/− mice uncovered for the first time the role of NEU1 as a negative regulator of lysosomal exocytosis.
Excessive exocytosis of lysosomal contents in the Neu1−/− BM impacts the composition of both the BM cells and the BM microenvironment, which ultimately redirects hematopoiesis to the spleen. Increased levels of active proteases, such as neutrophil elastase and cathepsin G, exocytosed by Neu1−/− BM neutrophils and macrophages into the extracellular fluid, results in the inactivation of serpina-1 and serpina-3, and premature degradation of vascular cell adhesion molecule 1 (VCAM-1) at the surface of the BM stromal cells; consequently, these cells loose their ability to retain progenitors in the BM niche and hematopoiesis resumes in the spleen (Figure 2b) 39. These original findings identify sialidosis as the only genetic disease known to date, in which lysosomal exocytosis is exacerbated rather than impaired, and suggest that other phenotypic alterations in the sialidosis model are linked to this process.
7.2 Muscle atrophy
One such phenotype in Neu1 mutant mice that also occurs in patients with type II sialidosis is muscle atrophy, associated with hypotonia, and osteoskelatal deformities 6, 32, 50. Progressive degeneration of muscle fibers in Neu1−/− mice is not cell autonomous, but rather it is contingent upon unique alterations in the supporting connective tissue. Histopathological examination of the skeletal muscle revealed an abnormal expansion of the epimysium and perimysium with increased number of proliferating cells (Figure 3). The latter cells stained positive for transcription factor 4, an early marker of the developing muscle connective tissue, identifying them as myofibroblasts. Their continuous proliferative activity explains the concomitant upregulation and massive deposition of collagens (I, III, IV and VI) and other ECM components (vimentin, fibronectin), coupled to the activation of matrix metalloproteinases (MMP2 and MMP9) and their inhibitors (TIMP1 and TIMP2) 32. In addition, Neu1−/− primary myofibroblasts cultured in vitro have increased levels of oversialylated Lamp1 and, in turn, excessive exocytosis of lysosomal enzymes, including cathepsin B and cathepsin L 32. These combined features enable the ever-expanding connective tissue to gradually infiltrate and invade the adjacent muscle fibers, which become increasingly fragmented and eventually degenerate (Figure 3) 32. Thus, as it is the case for the Neu1−/− BM, the progressive muscle atrophy in the Neu1−/− mice may also be explained by the occurrence of excessive lysosomal exocytosis from cells of the supportive connective tissue, which likely facilitates the aberrant infiltration of muscle fibers and their degeneration. Notably, muscle damage is apparently not associated with necrosis or inflammation, particularly in the early phases of fiber degeneration. Infiltration and activation of tissue macrophages ensues with the progression of the degenerative process. Overall, the muscle phenotype in Neu1 mutant mice is unique compared with other known myopathies, and may reenact in mice the neuromuscular manifestations seen in the type II sialidosis.
Figure 3.
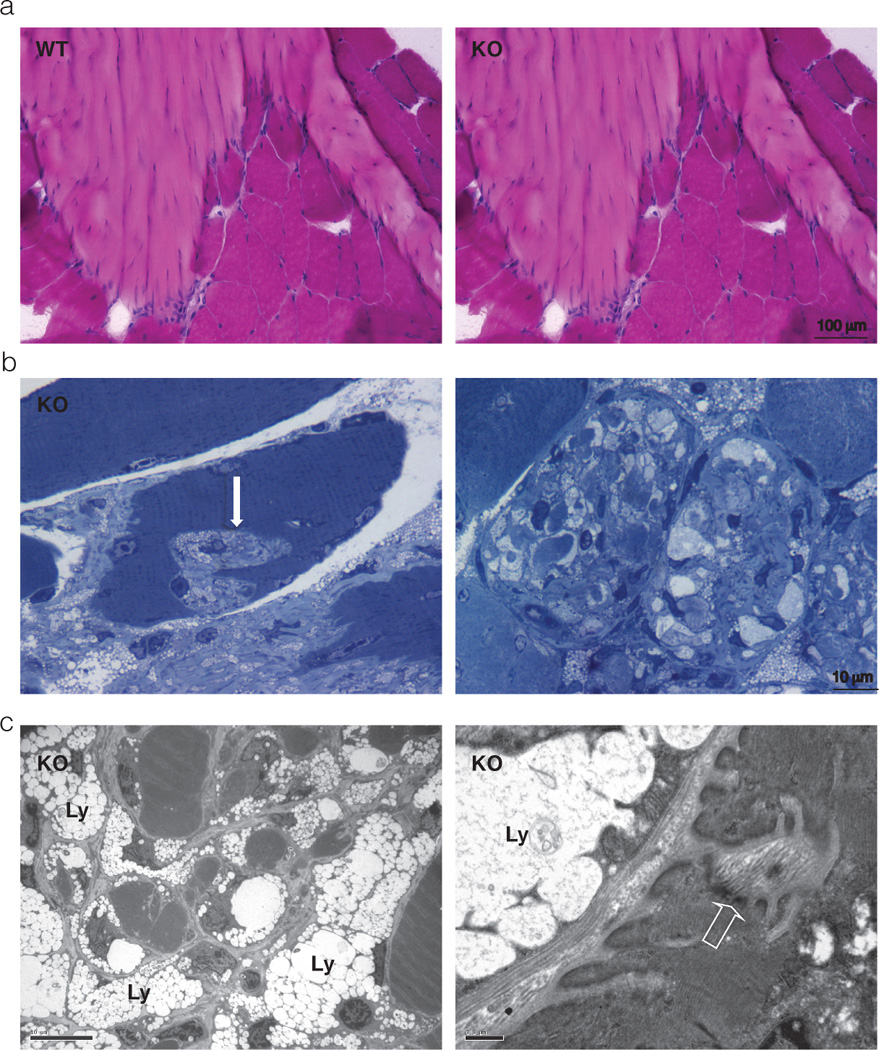
a) Hematoxilin & Eosin staining of transverse sections of WT and Neu1−/− gastrocnemius muscle; large areas of connective tissue infiltrates and increased number of cells in the Neu1−/− myotendinous junction are observed when compared to the WT tissue; b) Toluidine blue-stained semithin Neu1−/− gastrocnemius muscle sections; invagination of the sarcolemma with infiltration of the muscle fibers by ECM components is observed (arrow on the left panel); in an advanced stage, ECM invagination results in severe fragmentation of the cytosol and complete disruption of the muscle cytoarchitecture as observed in a transverse section (right panel); c) Electron microscopy ultrastructural analysis of Neu1−/− gastrocnemius muscle, showing cytosolic fragmentation and infiltration of muscle fibers by fibroblast-like cells whose cytosol is filled with storage lysosomes (Ly) and thickening of the sarcolemma (open arrow). Adapted from Zanoteli et al. Biochim, Biophys Acta 2010, with permission of Elsevier.
7.3 Hearing loss
Morphological abnormalities consistent with progressive hearing loss are evident in the external, middle, and inner ear of Neu1−/− mice 49. This phenotype is again consistent with what has been reported in type II patients 6. Histopathological and ultrastructural analyses revealed thickened cerumen occlusion in the external auditory canal and severe otitis media; infiltration of connective tissue in the middle ear ossicles that also showed signs of chronic inflammation; extensive vacuolization in many cells of the cochlea, most prominent in the marginal cells of the stria vascularis. Some of the ultrastructural features, such as increased invaginations and cavities, in the apical surface of the marginal cells of the stria vascularis, could be the result of excessive lysosomal exocytosis into the endolymph. In fact, numerous lysosomes docking at the apical surface of the marginal cells marked positive for Lamp1 and Lamp2 49. Exacerbated lysosomal exocytosis into the endolymph is probably responsible for the changes in endolymphatic potential, defective sound transduction in the sensorial hair cells and ultimately hearing loss 49.
7.4 Neurodegeneration and links with Alzheimer’s Disease
Through studies of mouse models of LSDs, it is becoming increasingly apparent that these pediatric conditions develop pathological signs, especially in the CNS, which resemble adult neurodegenerative diseases, like Alzheimer’s disease (AD) and Parkinson disease 51 52–56. Also the Neu1−/− mice exhibit early in life a brain phenotype with signs of early aging. However, what makes the Neu1−/− mouse model unique compared to other LSD models is the presence of amyloid deposits resembling the plaques characteristic of AD 33. Histopathological analyses of the Neu1−/− brain identified numerous multifocal, eosinophilic deposits, heterogeneous in size and shape, especially in the CA3 region of the hippocampus and the fimbria. These deposits contain proteinaceous material that stained positive with the Congo red/Chrysamine-G derivative Methoxy-X04, a compound with high affinity for amyloid. At the ultrastructural level, the same region of the Neu1−/− brain showed many dystrophic neurites, filled with electron dense vacuoles, likely representing autophagosomes or autophagolysosomes. Several lines of evidence linked these phenotypic alterations directly to Neu1 loss of function: 1) in absence of Neu1 APP is oversialylated, hence it is a natural substrate of the enzyme; 2) in Neu1−/− hippocampal neurons the amyloid precursor protein (APP) accumulates in lysosomal puncta as early as 1 month of age; 3) in older mutant mice increased levels of APP and proteolytically processed Aβ peptides are detected in purified lysosomes from hippocampal extracts and in the amyloid deposits (Figure 4); 4) combined accumulation of APP and Aβ peptides promotes the formation of the toxic amyloid peptide Aβ42; 5) the levels of secreted Aβ42 are higher in Neu1−/− cerebrospinal fluid and in the medium of Neu1−/− hippocampal neurosphere cultures than in the corresponding control samples 33. Finally, the demonstration that in absence of Neu1 Aβ is released via lysosomal exocytosis identifies this process as a novel mechanism for the extracellular deposition of this toxic peptide. Indeed, deletion of Neu1 in a transgenic mouse model of AD accelerates APP accumulation and plaque formation. Conversely, exogenous expression of Neu1 in the brain of these AD mice largely prevents plaque formation (Figure 4). Combined these observations unequivocally link Neu1 loss of function to the occurrence of an AD-like phenotype in the sialidosis mice, and identify NEU1 enzyme activity as a risk factor for the development of this disease 33.
Figure 4.
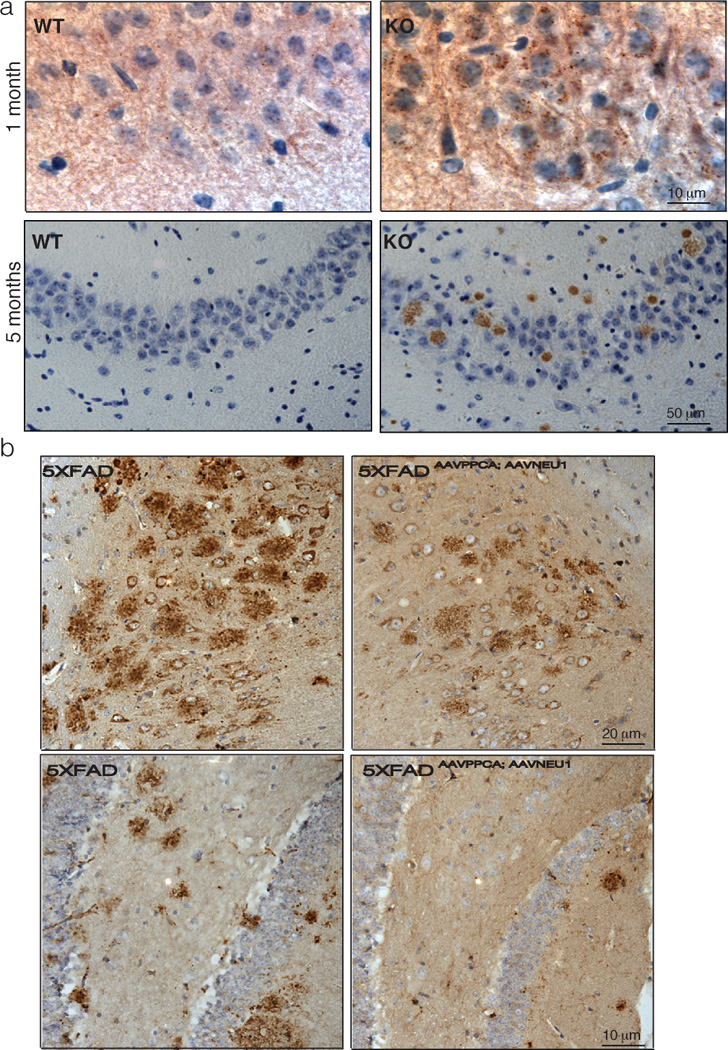
a) Pathological abnormalities in the Neu1−/− hippocampus compared to control; in the Neu1−/− hippocampus APP starts to accumulate intracellularly as early as 1 month of age, as shown by immunohistochemistry analysis with anti-APP N-terminal antibody (brown; upper panel); amyloid deposits are observed in 5-month-old Neu1−/− brain stained positive for APP (N-terminal antibody; lower panel); b) Injection of an AAV containing human NEU1 and PPCA in the hippocampus of 4- (upper panel) and 6-month-old (lower panel) 5XFAD transgenic model (familial AD mouse model), showing reduction of the number of amyloid plaques compared with that seen in the 5XFAD animals injected only with carrier solution (plaques were identified by 4G8 immunostaining in brown). Adapted from Annunziata et al 2013 Nat Commun, with permission of Nature Publishing group.
8. Additional Pathogenic Pathways Downstream of Neu1 Loss of Function
8.1 NEU1- dependent regulation of phagocytosis in macrophages
Pshezhetsky and coworkers have described other cellular pathways that depend on Neu1 activity 38, 57. Macrophages isolated from mice carrying homozygous CathAS190A–Neo show reduced phagocytosis 57. This feature is characteristic of these mice but not of the CathAS190A mice (see above), suggesting that secondary deficiency of Neu1 underpins this specific phenotype. Increased sialylation of multiple cellular proteins is observed in macrophages isolated from CathAS190A–Neo mice. In addition, it was demonstrated that the Fcγ receptor is a substrate of Neu1. Treatment of cells with exogenous Neu1 reduces overall sialylation of cellular membrane proteins and restores the phagocytic capacity of macrophages.
8.2 NEU1 regulation of insulin signaling
The same group also showed that Neu1 potentiates the proliferative response to insulin in cultured myoblasts. CathAS190A–Neo mice with secondary Neu1 deficiency develop glucose intolerance and insulin resistance when fed with high fat diet, due to decreased insulin resistance 38. CathAS190A–Neo mice produce normal insulin levels but have reduced insulin sensitivity. When insulin signaling was studied in the muscle and liver, the major insulin target tissues, it was demonstrated that intraperitoneal injections of insulin induces the phosphorylation of the insulin receptor kinase (IRK) in control animals but to a much lower extent in CathAS190A–Neo mice. Insulin signaling was analyzed in fibroblasts from sialidosis patients and showed reduced insulin-dependent AKT phosphorylation compared with control fibroblasts, thereby confirming impaired insulin signaling when Neu1 is reduced or absent 38. IRK was also shown to be a target of Neu1 and to remain oversialylated when Neu1 activity is reduced. It has been postulated that insulin binding to IRK induces its desialylation by NEU1, which in turn stimulates its activation 38. It still remains to be determined if primary deficiency of PPCA in this mouse model is in part responsible for the reduced insulin signaling.
9. Therapy for Sialidosis
Enzyme replacement therapy (ERT) is the canonical therapeutic approach for the treatment of non-neuropathic patients with lysosomal storage disorders 58. A short-term ERT was also attempted in Neu1−/− mice using a recombinant Neu1 enzyme purified from overexpressing insect cells 59. Increased levels of Neu1 enzyme activity and extensive correction of the pathology in most of the systemic organs were achieved by this treatment. Unfortunately, the recombinant enzyme was highly immunogenic in the mutant mice and elicited a severe immune response, limiting the therapeutic use of this approach 60. NEU1 immunogenicity, its tendency to aggregate, and its strict dependence on PPCA for lysosomal localization and catalytic activation, complicate the development of an effective therapy for sialidosis. These caveats are problematic especially for non-neuropathic type I patients, who may be more numerous than expected, and would greatly benefit from therapeutic means aimed at increasing their endogenous residual NEU1 activity.
With this idea in mind, a pharmacologic chaperone-mediated therapy has been recently tested successfully in the mouse model of type I sialidosis, Neu1−/−;NEU1V54M (see above). This therapeutic approach is based on the observation that both wild-type and mutant NEU1 activities can be increased by titrating the levels of available PPCA in vitro 16, 61. It was therefore hypothesized that this PPCA-mediated approach would be feasible in vivo to increase the activity of mutant NEU1 by improving its stability, folding and lysosomal localization.
A self-complementary adeno-associated viral vector expressing PPCA under the control of a liver specific promoter (scAAV2/8LP1-PPCA) was injected systemically in Neu1−/−;NEU1V54M mice 34. The same vector was used successfully for a comprehensive, preclinical gene therapy trial in the Ppca−/− mice, the mouse model of GS 62. One-year-old Neu1−/−;NEU1V54Mmice, a time when clear signs of disease pathology appear, were treated with a single dose injection of the recombinant AAV vector and sacrificed 1 month later. High expression of the PPCA enzyme in the liver of the injected mice resulted in about 3 fold increase of the NEU1V54M basal activity in all tissues tested (Figure 5), improved tissue pathology and decreased levels of high molecular weight sialyl-oligosaccharides in the urine compared with not injected mice 34. Considering that the majority of NEU1 mutations so far identified do not involve the catalytic site of the enzyme, this pharmacologic, chaperone-mediated therapy may be effective for other NEU1 mutations found in patients with type I sialidosis.
Figure 5.
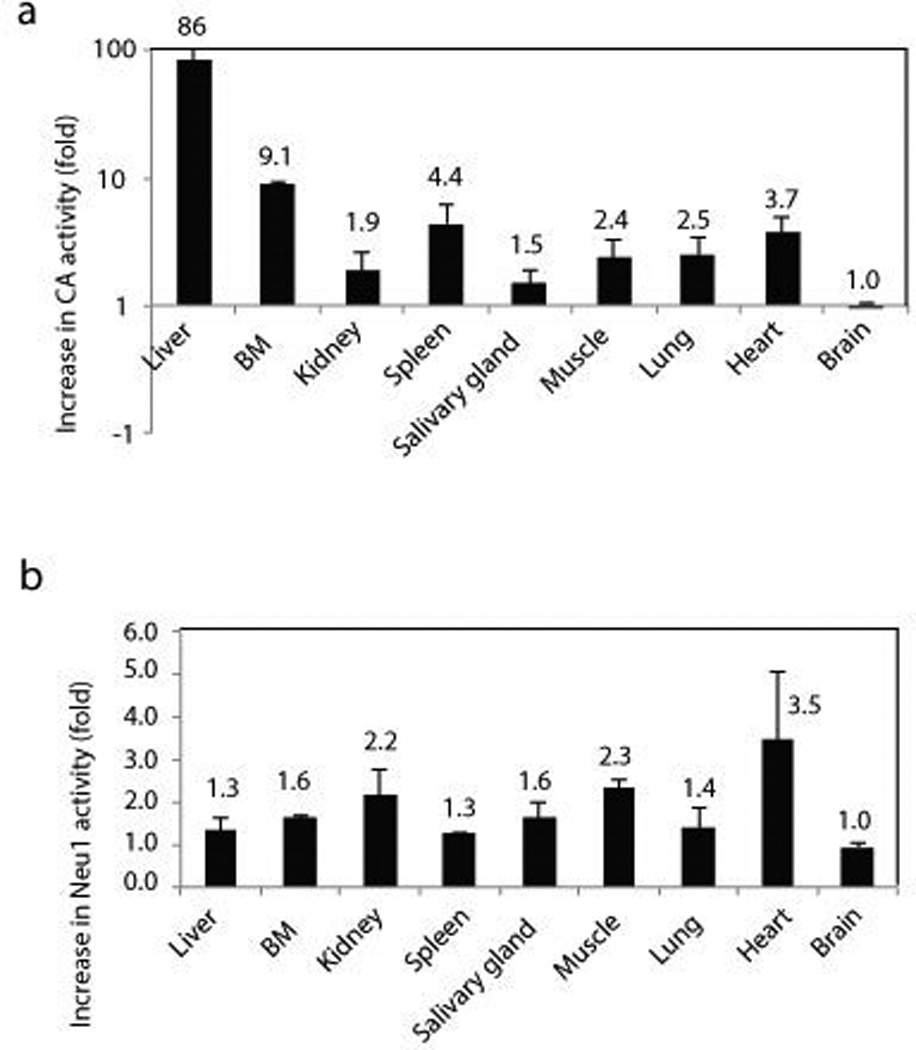
Cathepsin A and NEU1 activities were measured in the tissue homogenates of scAAV2/8LP1-PPCA-treated Neu1−/−;NEU1V54M mice (n=4) and untreated mice (n=4); a) Cathepsin A and, b) NEU1 activities were higher in tissues from rAAV-treated Neu1−/−;NEU1V54M mice than in the corresponding untreated mice; data are represented as mean ± s.d. (error bars). Adapted from Bonten et al. Biochim, Biophys Acta 2013, with permission of Elsevier.
10. Expert Opinion
The fundamental role of the lysosomal sialidase NEU1 in maintaining cell and tissue homeostasis is documented by the systemic, clinical manifestations associated with defective or deficient enzyme activity. Genetic mutations in NEU1 give rise to two rather different diseases: patients with sialidosis type II have an early onset of the symptoms, a catastrophic course and a dismal prognosis, while patients with type I sialidosis present later in life with a mild disease that is mostly confined to ophthalmologic problems, myoclonus and minor or absent neurologic manifestations. Because of their mild clinical course type I patients are sometime difficult to diagnose. In addition, several individuals have been recently recognized as having the type I form of sialidosis only after whole genome sequencing identified mutations in NEU1. These atypical patients developed myoclonus but not the other signs characteristic of the disease, namely macular cherry-red spot and sialyloligosacchariduria. These findings suggest that the number of patients with type I sialidosis may be underestimated and that this form of the disease may be more common than originally predicted.
Taking into consideration the potentially higher number of patients, the late presentation of the disease and the mild symptoms, therapeutic approaches to be sought for sialidosis should be tailored for type I patients. This group of patients will be the most likely to benefit from the treatment, and have a significant improvement of their quality of life and possibly longer survival. In contrast, type II patients who present with an early onset, systemic condition and severe neurological involvement will be a difficult group to treat, unless an in utero approach will become feasible in the future.
The development of suitable therapies for sialidosis has been limited by the nature and biochemical characteristics of NEU1. The enzyme is prone to aggregate, it is highly immunogenic and it is catalytically active only when bound to its auxiliary chaperone PPCA. Moreover, NEU1 by itself is very unstable and difficult to purify because it tends to come out of solution unless it is in complex with PPCA. However, the strict dependence of the enzyme on PPCA for catalytic activation may turn advantageous for therapeutic purposes. This is because NEU1 residual activity in fibroblasts from several type I patients could be increased indirectly by increasing the levels of PPCA; hence, this approach may be conceivable for patients carrying NEU1 missense mutations that permits its interaction with PPCA. In this regard, the AAV-PPCA–mediated gene therapy that will be applied for the treatment of patients with galactosialidosis could become suitable also for type I sialidosis. If this will be the case, the successful identification and enrollment of eligible patients will require the coordinated effort of expert clinicians, basic scientists, pharmaceutical companies, and patient advocacy groups.
In conclusion, despite the rarity and complexity of this lysosomal storage disease, over the last decade remarkable progress has been made towards the understanding of the molecular pathway(s) underlying pathogenesis in sialidosis. However, only a handful of NEU1 substrates have been identified until now. It is predictable that future studies will uncover other NEU1 targets that may implicate NEU1 in additional biological processes and in turn offer novel therapeutic options. Lastly, if specific therapies, for rare diseases like sialidosis, will eventually prove useful for more common adult conditions, pharmaceutical companies may become more interested in investing into large-scale therapeutics for orphan diseases.
Aknowledgment
We apologize that we could not cite all the outstanding contributions to this field of research because of space constraints. A.d’A. holds the Jewelers for Children Endowed Chair in Genetics and Gene Therapy. This work was funded in part by NIH grants GM60905, DK52025 and NIH RO1 GM104981, the Assisi Foundation of Memphis, the American Lebanese Syrian Associated Charities (ALSAC) and the National Tay-Sachs & Allied Disease Association (NTSAD). A.d’A. and I.A. are named as co-inventors on the patent application “Methods and compositions to detect the level of lysosomal exocytosis activity and methods of use”, number PCT/US2012/052629 based, in part, on the research reported herein.
Abbreviations
- NEU1
Neuraminidase 1
- PPCA
Protective protein cathesin A
- β-GAL
β-galactosidase
- GS
galactosialidosis
- EMH
extramedullary hematopoiesis
- PM
plasma membrane
- ECM
extracellular matrix
- BM
bone marrow
- AD
Alzheimer’s disease
Footnotes
Financial and competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1. Bonten E, van der Spoel A, Fornerod M, Grosveld G, d'Azzo A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes & development. 1996 Dec 15;10(24):3156–3169. doi: 10.1101/gad.10.24.3156. ** This paper describes the cloning of the human lysosomal neuraminidase NEU1.
- 2.Pshezhetsky AV, Richard C, Michaud L, Igdoura S, Wang S, Elsliger MA, et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nature genetics. 1997 Mar;15(3):316–320. doi: 10.1038/ng0397-316. [DOI] [PubMed] [Google Scholar]
- 3. Durand P, Gatti R, Cavalieri S, Borrone C, Tondeur M, Michalski JC, et al. Sialidosis (mucolipidosis I) Helvetica paediatrica acta. 1977 Nov;32(4–5):391–400. * This is the first report referring to NEU1 deficiency as sialidosis.
- 4.Cantz M, Gehler J, Spranger J. Mucolipidosis I: increased sialic acid content and deficiency of an alpha-N-acetylneuraminidase in cultured fibroblasts. Biochemical and biophysical research communications. 1977 Jan 24;74(2):732–738. doi: 10.1016/0006-291x(77)90363-1. [DOI] [PubMed] [Google Scholar]
- 5.Lowden JA, O'Brien JS. Sialidosis: a review of human neuraminidase deficiency. American journal of human genetics. 1979 Jan;31(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas G. The Online Metabolic & Molecular Bases of Inherited Diseases. New York: McGraw-Hill Publishing Co.; 2013. [Google Scholar]
- 7.d'Azzo A, Bonten E. Molecular mechanisms of pathogenesis in a glycosphingolipid and a glycoprotein storage disease. Biochem Soc Trans. 2010 Dec;38(6):1453–1457. doi: 10.1042/BST0381453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Caciotti A, Di Rocco M, Filocamo M, Grossi S, Traverso F, d'Azzo A, et al. Type II sialidosis: review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. Journal of neurology. 2009 Nov;256(11):1911–1915. doi: 10.1007/s00415-009-5213-4. * This is a recent comprehensive review of mutations occurring in Type II sialidosis.
- 9.Takahashi Y, Nakamura Y, Yamaguchi S, Orii T. Urinary oligosaccharide excretion and severity of galactosialidosis and sialidosis. Clinica chimica acta; international journal of clinical chemistry. 1991 Dec 16;203(2–3):199–210. doi: 10.1016/0009-8981(91)90292-k. [DOI] [PubMed] [Google Scholar]
- 10. Canafoglia L, Robbiano A, Pareyson D, Panzica F, Nanetti L, Giovagnoli AR, et al. Expanding sialidosis spectrum by genome-wide screening: NEU1 mutations in adult-onset myoclonus. Neurology. 2014 Jun 3;82(22):2003–2006. doi: 10.1212/WNL.0000000000000482. ** This paper shows that sialidosis should be suspected in adult patients with isolated action myoclonus carrying NEU1 mutations without additional clinical signs of sialidosis.
- 11.Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nature genetics. 2014 Nov 17; doi: 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobral I, Cachulo Mda L, Figueira J, Silva R. Sialidosis type I: ophthalmological findings. BMJ case reports. 2014:2014. doi: 10.1136/bcr-2014-205871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loren DJ, Campos Y, d'Azzo A, Wyble L, Grange DK, Gilbert-Barness E, et al. Sialidosis presenting as severe nonimmune fetal hydrops is associated with two novel mutations in lysosomal alpha-neuraminidase. Journal of perinatology : official journal of the California Perinatal Association. 2005 Jul;25(7):491–494. doi: 10.1038/sj.jp.7211335. [DOI] [PubMed] [Google Scholar]
- 14.Lee BH, Kim YM, Kim JH, Kim GH, Lee BS, Kim CJ, et al. Histological, biochemical, and genetic characterization of early-onset fulminating sialidosis type 2 in a Korean neonate with hydrops fetalis. Brain & development. 2014 Feb;36(2):171–175. doi: 10.1016/j.braindev.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg R, Halimi E, Mention-Mulliez K, Cuisset JM, Holder M, Defoort-Dhellemmes S. Five year follow-up of two sisters with type II sialidosis: systemic and ophthalmic findings including OCT analysis. Journal of pediatric ophthalmology and strabismus. 2013;50:e33–e36. doi: 10.3928/01913913-20130625-02. Online. [DOI] [PubMed] [Google Scholar]
- 16.Bonten EJ, Annunziata I, d'Azzo A. Lysosomal multienzyme complex: pros and cons of working together. Cellular and molecular life sciences : CMLS. 2014 Jun;71(11):2017–2032. doi: 10.1007/s00018-013-1538-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. d'Azzo A, Andria G, Bonten E, Annunziata I. The Online Metabolic & Molecular Bases of Inherited Diseases. New York: McGraw-Hill Publishing Co.; 2013. * This chapter includes the most up to date information on the clinical,molecular and biochemical aspects of Galactosialidosis.
- 18.Varki A, Schauer R. Sialic Acids. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al., editors. Essentials of Glycobiology. 2nd ed. NY: Cold Spring Harbor; 2009. [PubMed] [Google Scholar]
- 19.Monti E, Bonten E, D'Azzo A, Bresciani R, Venerando B, Borsani G, et al. Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Advances in carbohydrate chemistry and biochemistry. 2010;64:403–479. doi: 10.1016/S0065-2318(10)64007-3. [DOI] [PubMed] [Google Scholar]
- 20. Miyagi T, Yamaguchi K. Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology. 2012 Jul;22(7):880–896. doi: 10.1093/glycob/cws057. References 19 and 20 are comprehensive review describing the properties and functions of mammalian sialidases
- 21.Giacopuzzi E, Bresciani R, Schauer R, Monti E, Borsani G. New insights on the sialidase protein family revealed by a phylogenetic analysis in metazoa. PloS one. 2012;7(8):e44193. doi: 10.1371/journal.pone.0044193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smutova V, Albohy A, Pan X, Korchagina E, Miyagi T, Bovin N, et al. Structural basis for substrate specificity of mammalian neuraminidases. PloS one. 2014;9(9):e106320. doi: 10.1371/journal.pone.0106320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bonten EJ, Campos Y, Zaitsev V, Nourse A, Waddell B, Lewis W, et al. Heterodimerization of the sialidase NEU1 with the chaperone protective protein/cathepsin A prevents its premature oligomerization. J Biol Chem. 2009 Oct 9;284(41):28430–28441. doi: 10.1074/jbc.M109.031419. ** This paper identifies binding sites on NEU1 and PPCA responsible for their interaction and proposes a novel mechanism of binding between the two proteins.
- 24. Lukong KE, Seyrantepe V, Landry K, Trudel S, Ahmad A, Gahl WA, et al. Intracellular distribution of lysosomal sialidase is controlled by the internalization signal in its cytoplasmic tail. J Biol Chem. 2001 Dec 7;276(49):46172–46181. doi: 10.1074/jbc.M104547200. * This paper identofies a C-terminal peptide in NEU1 that targets the protein to the membrane.
- 25.Lukong KE, Elsliger MA, Chang Y, Richard C, Thomas G, Carey W, et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum Mol Genet. 2000 Apr 12;9(7):1075–1085. doi: 10.1093/hmg/9.7.1075. [DOI] [PubMed] [Google Scholar]
- 26.Bonten EJ, Arts WF, Beck M, Covanis A, Donati MA, Parini R, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9(18):2715–2725. doi: 10.1093/hmg/9.18.2715. [DOI] [PubMed] [Google Scholar]
- 27.Itoh K, Naganawa Y, Matsuzawa F, Aikawa S, Doi H, Sasagasako N, et al. Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes. Journal of human genetics. 2002;47(1):29–37. doi: 10.1007/s10038-002-8652-7. [DOI] [PubMed] [Google Scholar]
- 28.Lee YJ, Son SK, Park JH, Song JS, Cheon CK. NEU1 Mutation in a Korean Infant with Type 2 Sialidosis Presenting as Isolated Fetal Ascites. Pediatrics and neonatology. 2014 Sep 12; doi: 10.1016/j.pedneo.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Penzel R, Uhl J, Kopitz J, Beck M, Otto HF, Cantz M. Splice donor site mutation in the lysosomal neuraminidase gene causing exon skipping and complete loss of enzyme activity in a sialidosis patient. FEBS letters. 2001 Jul 20;501(2–3):135–138. doi: 10.1016/s0014-5793(01)02645-x. [DOI] [PubMed] [Google Scholar]
- 30.Ranganath P, Sharma V, Danda S, Nandineni MR, Dalal AB. Novel mutations in the neuraminidase-1 (NEU1) gene in two patients of sialidosis in India. The Indian journal of medical research. 2012 Dec;136(6):1048–1050. [PMC free article] [PubMed] [Google Scholar]
- 31. de Geest N, Bonten E, Mann L, de Sousa-Hitzler J, Hahn C, d'Azzo A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum Mol Genet. 2002 Jun 1;11(12):1455–1464. doi: 10.1093/hmg/11.12.1455. ** This paper describes the first mouse model of sialidosis.
- 32.Zanoteli E, van de Vlekkert D, Bonten EJ, Hu H, Mann L, Gomero EM, et al. Muscle degeneration in neuraminidase 1-deficient mice results from infiltration of the muscle fibers by expanded connective tissue. Biochim Biophys Acta. 2010 Jul-Aug;1802(7–8):659–672. doi: 10.1016/j.bbadis.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Annunziata I, Patterson A, Helton D, Hu H, Moshiach S, Gomero E, et al. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-beta secretion via deregulated lysosomal exocytosis. Nature communications. 2013;4:2734. doi: 10.1038/ncomms3734. ** This paper describes the realease of amyloidogenic peptides via lysosomal exocytosis in the mouse model of sialidosis.
- 34. Bonten EJ, Yogalingam G, Hu H, Gomero E, van de Vlekkert D, d'Azzo A. Chaperone-mediated gene therapy with recombinant AAV-PPCA in a new mouse model of type I sialidosis. Biochim Biophys Acta. 2013 Oct;1832(10):1784–1792. doi: 10.1016/j.bbadis.2013.06.002. ** This report suggests that at least some of the NEU1 mutations in type I sialidosis patients respond to treatment with PPCA.
- 35.Zhou XY, Morreau H, Rottier R, Davis D, Bonten E, Gillemans N, et al. Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes & development. 1995 Nov 1;9(21):2623–2634. doi: 10.1101/gad.9.21.2623. [DOI] [PubMed] [Google Scholar]
- 36.Hermo L, Korah N, Gregory M, Liu LY, Cyr DG, D'Azzo A, et al. Structural alterations of epididymal epithelial cells in cathepsin A-deficient mice affect the blood-epididymal barrier and lead to altered sperm motility. Journal of andrology. 2007 Sep-Oct;28(5):784–797. doi: 10.2164/jandrol.107.002980. [DOI] [PubMed] [Google Scholar]
- 37.Seyrantepe V, Hinek A, Peng J, Fedjaev M, Ernest S, Kadota Y, et al. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation. 2008 Apr 15;117(15):1973–1981. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- 38. Dridi L, Seyrantepe V, Fougerat A, Pan X, Bonneil E, Thibault P, et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes. 2013 Jul;62(7):2338–2346. doi: 10.2337/db12-1825. ** The results described in this paper propose Neu1 as a novel component of the signaling pathways of energy metabolism and glucose uptake.
- 39. Yogalingam G, Bonten EJ, van de Vlekkert D, Hu H, Moshiach S, Connell SA, et al. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev Cell. 2008 Jul;15(1):74–86. doi: 10.1016/j.devcel.2008.05.005. ** This paper uncovers the role of NEU1 in regulating lysosomal exocytosis and links exacerbation of this process to the pathogenesis of sialidosis.
- 40.Griffiths GM, Tsun A, Stinchcombe JC. The immunological synapse: a focal point for endocytosis and exocytosis. The Journal of cell biology. 2010 May 3;189(3):399–406. doi: 10.1083/jcb.201002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samie MA, Xu H. Lysosomal exocytosis and lipid storage disorders. Journal of lipid research. 2014 Mar 25;55(6):995–1009. doi: 10.1194/jlr.R046896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. The Journal of cell biology. 1997 Apr 7;137(1):93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. The Journal of cell biology. 2002 Nov 25;159(4):625–635. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stinchcombe J, Bossi G, Griffiths GM. Linking albinism and immunity: the secrets of secretory lysosomes. Science. 2004 Jul 2;305(5680):55–59. doi: 10.1126/science.1095291. [DOI] [PubMed] [Google Scholar]
- 45.Luzio JP, Hackmann Y, Dieckmann NM, Griffiths GM. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harbor perspectives in biology. 2014 Sep;6(9):a016840. doi: 10.1101/cshperspect.a016840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holt OJ, Gallo F, Griffiths GM. Regulating secretory lysosomes. Journal of biochemistry. 2006 Jul;140(1):7–12. doi: 10.1093/jb/mvj126. [DOI] [PubMed] [Google Scholar]
- 47.Schwake M, Schroder B, Saftig P. Lysosomal membrane proteins and their central role in physiology. Traffic. 2013 Jul;14(7):739–748. doi: 10.1111/tra.12056. [DOI] [PubMed] [Google Scholar]
- 48.Kima PE, Burleigh B, Andrews NW. Surface-targeted lysosomal membrane glycoprotein-1 (Lamp-1) enhances lysosome exocytosis and cell invasion by Trypanosoma cruzi. Cellular microbiology. 2000 Dec;2(6):477–486. doi: 10.1046/j.1462-5822.2000.00071.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu X, Steigelman KA, Bonten E, Hu H, He W, Ren T, et al. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim Biophys Acta. 2010 Feb;1802(2):259–268. doi: 10.1016/j.bbadis.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.d’Azzo A, Kolodny EH, Bonten E, Annunziata I. Storage disease of the reticuloendothelial system. 7th edition ed. New York: Nathan and Oski’s; 2009. [Google Scholar]
- 51.Bellettato CM, Scarpa M. Pathophysiology of neuropathic lysosomal storage disorders. Journal of inherited metabolic disease. 2010 Aug;33(4):347–362. doi: 10.1007/s10545-010-9075-9. [DOI] [PubMed] [Google Scholar]
- 52.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. The Lancet Neurology. 2012 Nov;11(11):986–998. doi: 10.1016/S1474-4422(12)70190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R, et al. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012 Apr 11;32(15):5223–5236. doi: 10.1523/JNEUROSCI.4860-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martins C, Hulkova H, Dridi L, Dormoy-Raclet V, Grigoryeva L, Choi Y, et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain : a journal of neurology. 2015 Feb;138(Pt 2):336–355. doi: 10.1093/brain/awu355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohmi K, Zhao HZ, Neufeld EF. Defects in the medial entorhinal cortex and dentate gyrus in the mouse model of Sanfilippo syndrome type B. PloS one. 2011;6(11):e27461. doi: 10.1371/journal.pone.0027461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohmi K, Kudo LC, Ryazantsev S, Zhao HZ, Karsten SL, Neufeld EF. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proceedings of the National Academy of Sciences of the United States of America. 2009 May 19;106(20):8332–8337. doi: 10.1073/pnas.0903223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Seyrantepe V, Iannello A, Liang F, Kanshin E, Jayanth P, Samarani S, et al. Regulation of phagocytosis in macrophages by neuraminidase 1. J Biol Chem. 2010 Jan 1;285(1):206–215. doi: 10.1074/jbc.M109.055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ortolano S, Vieitez I, Navarro C, Spuch C. Treatment of lysosomal storage diseases: recent patents and future strategies. Recent patents on endocrine, metabolic & immune drug discovery. 2014 Jan;8(1):9–25. doi: 10.2174/1872214808666140115111350. [DOI] [PubMed] [Google Scholar]
- 59.Bonten EJ, Wang D, Toy JN, Mann L, Mignardot A, Yogalingam G, et al. Targeting macrophages with baculovirus-produced lysosomal enzymes: implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. FASEB J. 2004 doi: 10.1096/fj.03-0941fje. In Press. [DOI] [PubMed] [Google Scholar]
- 60.Wang D, Bonten EJ, Yogalingam G, Mann L, d'Azzo A. Short-term, high dose enzyme replacement therapy in sialidosis mice. Molecular genetics and metabolism. 2005 Jul;85(3):181–189. doi: 10.1016/j.ymgme.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 61.Bonten EJ, d'Azzo A. Lysosomal neuraminidase Catalytic activation in insect cells is controlled by the protective protein/cathepsin A. J Biol Chem. 2000;275(48):37657–37663. doi: 10.1074/jbc.M007380200. [DOI] [PubMed] [Google Scholar]
- 62. Hu H, Gomero E, Bonten E, Gray JT, Allay J, Wu Y, et al. Preclinical dose-finding study with a liver-tropic, recombinant AAV-2/8 vector in the mouse model of galactosialidosis. Molecular therapy : the journal of the American Society of Gene Therapy. 2012 Feb;20(2):267–274. doi: 10.1038/mt.2011.227. ** This paper reports the first preclinical studies for the treatment of Galactosialidosis using an in-vivo gene therapy approach.
- 63.Pattison S, Pankarican M, Rupar CA, Graham FL, Igdoura SA. Five novel mutations in the lysosomal sialidase gene (NEU1) in type II sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum Mutat. 2004 Jan;23(1):32–39. doi: 10.1002/humu.10278. [DOI] [PubMed] [Google Scholar]
- 64.Sergi C, Penzel R, Uhl J, Zoubaa S, Dietrich H, Decker N, et al. Prenatal diagnosis and fetal pathology in a Turkish family harboring a novel nonsense mutation in the lysosomal alpha-N-acetyl-neuraminidase (sialidase) gene. Human genetics. 2001 Oct;109(4):421–428. doi: 10.1007/s004390100592. [DOI] [PubMed] [Google Scholar]
- 65.Huang YZ, Lai SC, Lu CS, Weng YH, Chuang WL, Chen RS. Abnormal cortical excitability with preserved brainstem and spinal reflexes in sialidosis type I. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2008 May;119(5):1042–1050. doi: 10.1016/j.clinph.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 66.Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, Pshezhetsky AV. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat. 2003;22(5):343–352. doi: 10.1002/humu.10268. [DOI] [PubMed] [Google Scholar]
- 67.Sekijima Y, Nakamura K, Kishida D, Narita A, Adachi K, Ohno K, et al. Clinical and serial MRI findings of a sialidosis type I patient with a novel missense mutation in the NEU1 gene. Internal medicine. 2013;52(1):119–124. doi: 10.2169/internalmedicine.52.8901. [DOI] [PubMed] [Google Scholar]
- 68.Naganawa Y, Itoh K, Shimmoto M, Takiguchi K, Doi H, Nishizawa Y, et al. Molecular and structural studies of Japanese patients with sialidosis type 1. Journal of human genetics. 2000;45(4):241–249. doi: 10.1007/s100380070034. [DOI] [PubMed] [Google Scholar]