

Abstract
The Janus Kinases (JAKs) and their downstream effectors Signal Transducer and Activator of Transcription proteins (STATs) form a critical immune cell signaling circuit, which is of fundamental importance in innate immunity, inflammation and hematopoiesis and dysregulation is frequently observed in immune disease and cancer. The high degree of structural conservation of the JAK ATP binding pockets has posed a considerable challenge to medicinal chemists seeking to develop highly selective inhibitors as pharmacological probes and as clinical drugs. Here we report the discovery and optimization of 2,4-substituted pyrimidines as covalent JAK3 inhibitors that exploit a unique cysteine (Cys909) residue in JAK3. Investigation of structure-activity-relationship (SAR) utilizing biochemical and transformed Ba/F3 cellular assays resulted in identification of potent and selective inhibitors such as compounds 9 and 45. A 2.9 Å co-crystal structure of JAK3 in complex with 9 confirms the covalent interaction. Compound 9 exhibited decent pharmacokinetic properties and is suitable for use in vivo. These inhibitors provide a set of useful tools to pharmacologically interrogate JAK3-dependent biology.
Keywords: JAK3, Covalent kinase inhibitors, Structure-based design, Structure-activity relationship, Drug discovery
TOC image

Introduction
The Janus Kinases (JAKs) and their downstream effectors Signal Transducer and Activator of Transcription proteins (STATs) form a critical immune cell signaling circuit.1 There are four JAK family members in mammals, JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2), each of which can bind to distinct cytokines and/or growth factor receptors. As the receptors that act upstream of JAKs are dimeric or multimeric, more than one JAK molecule is recruited to each receptor. Ligand binding to the receptor results in a conformation change that brings two JAK molecules close together allowing them to trans-phosphorylate and activate each other. Once active, JAKs catalyze the phosphorylation of tyrosine residues in the receptor allowing the recruitment of specific STATs which are in turn phosphorylated by the JAK. This promotes STAT dimerization which induces translocation to the nucleus and activation of gene regulatory programs.2 JAK/STAT signaling is of fundamental importance in innate immunity, inflammation and hematopoiesis and dysregulation is frequently observed in immune disease and cancer.3 The JAKs have been the subject of extensive drug discovery efforts and numerous small molecule ATP-competitive inhibitors have been developed including ruxolitinib (1, JAK1/2 inhibitor)4 approved as an anti-myelofibrosis drug, tofacitinib (2, pan-JAK inhibitor)5 approved for rheumatoid arthritis, and several others that are currently undergoing clinical investigation.6 The high degree of structural conservation of the JAK ATP binding pockets has posed a considerable challenge to medicinal chemists seeking to develop highly selective inhibitors as pharmacological probes and as clinical drugs.7
Among the JAK family JAK3 exhibits predominant expression in the hematopoietic system in contrast to other JAKs which are broadly expressed in a variety of cell types.2b JAK3 is required for signaling by cytokines, including interleukin 2 (IL-2), IL-4, IL-7, IL-9, IL-15 and IL-21, that act via receptors that contain the common gamma chain (γc) cytokine receptor subunit.8 In addition to the γc subunit, most of these receptors contain a second unique α subunit that determines cytokine specificity. In these receptors, JAK3 binds to the γc subunit and JAK1 to the α subunit. The IL-2 Receptor (IL-2R) and IL-15R are exceptions to this as they can form trimeric complexes through the recruitment of α and β chains. In this case the β subunit binds JAK1; the α subunit, while involved in cytokine binding, does not directly bind to a JAK.9 Signaling via the γc receptors is essential for the normal development and function of the immune system. Loss-of-function mutations in either γc or JAK3 in humans results in severe combined immunodeficiency caused by defective development of T- and NK cells. The role for JAK3 in T and natural killer (NK) cell development was subsequently confirmed by deletion of Jak3 in mice; JAK3 knockouts also lacked normal numbers of B cells indicating a more important role for JAK3 in B cell development in mice than in humans.2b, 10 On the basis of JAK3’s essential function in immune signaling and its restricted expression in hematopoietic tissues, JAK3 has been pursued as a target for the treatment of autoimmune and inflammatory diseases. JAK3 has also been explored as a potential anti-cancer target due to aberrant activation in several lymphoproliferative disorders;11 for example, mutations in JAK3 have been found in leukemias such as T-cell acute lymphoblastic leukemia (T-ALL) and T-cell prolymphocytic leukemia (T-PLL).12 The A572V activating mutation in the kinase domain of JAK3 was discovered in acute megakaryoblastic leukemia and natural killer/T-cell lymphoma, and has been demonstrated to be capable of forming tumors in mice.13 Recently JAK3 mutations were also found to promote programmed death ligand 1 (PD-L1) induction in lung cancer cells an in the tumor immune microenvironments.14
The most obvious strategy to develop small-molecule JAK3 inhibitors is to target the catalytic ATP-binding site of the JAK3 kinase domain (JH1). Numerous ATP-competitive JAK3 inhibitors have been developed (Figure 1A).15 Tofacitinib (2) was developed as a specific JAK3 inhibitor to prevent organ-transplant rejection but subsequent studies revealed that it is also a potent inhibitor of JAK1 and JAK2.5, 16 The combination of compound 2’s excellent potency, selectivity and pharmacological properties have made it a favored inhibitor to interrogate JAK kinase activity in numerous biological models.13b, 17 In our efforts to develop a more selective Jak3 inhibitor, we noted that among JAK family members JAK3 is unique in having a cysteine residue at the gatekeeper-plus-7 (GK+7) position.18 This residue is Cys909 in human JAK, and it is structurally equivalent to cysteine residues in epidermal growth factor receptor (EGFR) and Bruton’s tyrosine kinase (BTK) that have been successfully targeted by covalent kinase inhibitors that are now approved drugs. Afatinib19 (6) targets Cys797 in EGFR and ibrutinib20 (7) targets Cys481 in BTK. We and others have also developed mutant-selective inhibitors of the drug-resistant EGFR T790M that react covalently with Cys797. WZ400221 (8) is a pyrimidine-based inhibitor with an acrylamide “warhead”22 that potently inhibits EGFR T790M. Recently three other groups have also reported the development of covalent JAK3 inhibitors. In 2013 Winssinger et al. reported a covalent probe which selectively labeled JAK3, EGFR and ERBB2.23 Taunton et.al reported the development of ‘reversible-covalent’ JAK3 inhibitors incorporating a cyanoacrylamide warhead which inhibited JAK3 in a biochemical kinase assay with an IC50 of 100 nM while sparing other JAKs up to a concentration of 10 μM.24 Goedken et.al reported several tricyclic covalent JAK3 inhibitors with optimized inhibitors exhibiting biochemical IC50’s of 7 nM, good kinome selectivity and confirmed covalent binding to Cys909 using kinetic and mass-spectrometry studies.25 Here we report a medicinal campaign to derive potent disubstituted pyrimidine-based inhibitors that exploit an acrylamide electrophile to form a covalent bond to Cys909. Optimization was guided by cellular assays using Translocation ETS Leukemia protein (Tel)-JAK26 fusion kinase transformed Ba/F3 cells that provided an efficient and reliable measure of the ability of compounds to inhibit JAK signaling in cellular context.
Figure 1.

Structures of exemplary JAK3 inhibitors (2–5) and lead compounds (9, 45 and 50) in this work.
Results
Identification of Compound 9 as a Potent JAK3 Inhibitor
Amongst the 11 kinases that possess a cysteine at the GK+7 position only JAK3 and mitogen-activated protein kinase kinase 7 (MAP2K7) have a methionine gatekeeper, which is also present in the drug-resistant EGFRT790M that is targeted by the pyrimidine-based inhibitor WZ4002 (8). Compound 8 is a much less potent inhibitor of cellular JAK3 kinase activity (JAK3 Kd = 150 nM, TEL-JAK3 Ba/F3 IC50 = 2.82 μM) compared to T790M EGFR (EGFRL858R/T790M Ba/F3 IC50 = 8 nM).21 Through the syntheses of a small collection of analogs of 8 we discovered that relatively subtle changes could result in a dramatic improvement in JAK3 inhibitory potency relative to EGFRT790M. For example, 9 where the ether linkage of 8 is replaced with an aminomethylene (-NHCH2-) linkage resulted in a great improvement in JAK3 inhibition in a fixed time-point Z’-lyte enzymatic assay (Life Technology, SelectScreen, IC50 = 4.8 nM) (Scheme 1A).
Scheme 1.

α i) DIEA, 1,4-dioxane, RT; ii) TFA, 2-BuOH, 100 °C; iii) Raney nickel, H2, MeOH; iv) acryloyl chloride, sat. NaHCO3, THF, 0 °C~RT.
To broadly assess its kinase selectivity, 9 was profiled against a diverse panel of 456 kinases (DiscoveRX, KinomeScan27) using an in vitro ATP-site competition binding assay at a concentration of 1.0 μM. Compound 9 exhibited good overall kinase selectivity with an S(5) selectivity score, defined as the percentage of kinases with scores less than 5 (S(5))27b, of 0.02. The results suggested that 9 most potently inhibits JAK3 and identified fms-related tyrosine kinase 3 (FLT3) and several tyrosine protein kinase (TEC)-family kinases as being potential off-targets (Figure 2). Enzymatic assays using the Z’-lyte or LanthaScreen28 formats confirmed enzymatic inhibition of FLT3 (IC50 = 13 nM), TTK protein kinase (TTK, IC50 = 49 nM), BLK proto-oncogene (BLK, IC50 = 157 nM) and tyrosine protein kinase TXK (TXK, IC50 = 36 nM). Compound 9 showed very low inhibition scores for other JAKs and wild-type (WT) EGFR, which is consistent with the over 180-fold higher IC50s against JAK1, JAK2, TYK2 and EGFRWT (IC50s = 896, 1050, > 10000 and 409 nM respectively). As BTK and ITK have important functions in B-cell and T-cell signaling pathways, we confirmed that compound 9 possesses over 165-fold higher IC50s for BTK or ITK (IC50s = 794 and 1070 nM respectively) (Table S1).
Figure 2.

KinomeScan kinase selectivity profiles for Compound 9. Compound 9 were profiled at a concentration of 1 μM against a diverse panel of more than 456 kinases and mutants. Scores for primary screen hits were reported as a percent of the DMSO control (% control). The lower the Kd is likely to be, such that scores of zero represent strong hits. Scores are related to the probability of being a hit but are not strictly an affinity measurement.
As enzymatic potencies sometimes do not translate into cellular inhibition, the ability of 9 to inhibit the proliferation of kinase-transformed Ba/F3 cells was evaluated. Ba/F3 cells are a murine pre-B cell that can readily be transformed with activated kinases to allow for growth in the absence of IL-3, and are frequently used to evaluate the activity of compounds against kinases of interest in a cellular context.29 We utilized JAK1, JAK2 and JAK3 dependent Ba/F3 cell lines, where the JH1 domain of each JAK is fused with the oligomerization domain of TEL which results in constitutive tyrosine kinase activity and confers IL-3 independent proliferation.26 In addition, we engineered a TYK2 Ba/F3 cell line whose proliferation is driven by constitutively activated TYK2 (TYK2E957D).30 As a further control, we also used a Ba/F3 cell line transformed by TEL- Abelson murine leukemia viral oncogene homolog (ABL). To enable a direct comparison with the commonly used JAK inhibitors we profiled reported compounds 1–5,4–5, 15b, 15g, 31 and 1232 against this panel of Ba/F3 cells. Compound 1 exhibited the most potent inhibition of JAK1 and JAK2 Ba/F3 cells, 2 exhibited most potent inhibition of JAK3 Ba/F3 cells and 3 exhibited most selective inhibition of JAK3 (Table S2). Overall the potency and selectivity of these inhibitors are consistent with their reported properties. Consistent with the biochemical assays, 9 selectively inhibited the proliferation of JAK3-dependent Ba/F3 cells (IC50 = 69 nM) relative to other JAK-dependent Ba/F3 cells, for which there was no antiproliferative effect at concentrations below 3.0 μM (Table 2). The general antiproliferative activity that appears at concentrations of approximately 3.0 μM could be due to inhibition of other kinases such as TTK (aka Mps1, IC50 = 49 nM) as inhibition of this kinase has been reported to decrease cancer cell viability.33
Table 2.
SAR of R1

| ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | R1 | JAK3 IC50 (nM)a | Ba/F3 cellular IC50s (nM)b | MLM T1/2 (min)c | ||||
| TEL-JAK1 | TEL-JAK2 | TEL-JAK3 | TYK2 (E957D) | TEL-ABL | ||||
| 9 |

|
4.8 | 3124±116 | 3194±619 | 69±13 | 2266±490 | 3047±116 | 4.7 |
| 14 |
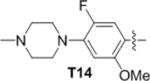
|
46 | 3871 | 6175 | 1305 | 3756 | 5257 | |
| 15 |

|
170 | ||||||
| 16 |

|
1.2 | 513 | 1013 | 11 | 1320 | 2077 | |
| 17 |

|
0.9 | 821 | 993 | 15 | 599 | 1558 | |
| 18 |

|
2.0 | 378 | 692 | 20 | 874 | 1932 | |
| 19 |

|
20 | 1059 | 2685 | 75 | 992 | 3470 | |
| 20 |

|
4.6 | 3182 | 5981 | 125 | 1318 | 2824 | |
| 21 |

|
1.3 | 1857±581 | 3704±410 | 7±0.1 | 2353±201 | 13705±194 | 5.2 |
| 22 |

|
1.4 | 3287 | 2910 | 29 | 1542 | 4494 | 3 |
| 23 |

|
0.9 | 5195±3001 | 9087±4895 | 11±3.3 | 6613±2650 | 22410±4752 | 3 |
| 24 |

|
5.2 | >5E4 | >5E4 | 478 | >5E4 | >5E4 | |
| 25 |

|
20 | >5E4 | >5E4 | 99 | >5E4 | >5E4 | |
| 26 |

|
3.6 | 2004 | 3290 | 23 | 4666 | 8769 | 3.4 |
| 27 |

|
7.4 | 2484 | 4321 | 35 | 4702 | 2326 | |
| 28 |

|
6.2 | 2346 | 2916 | 54 | 4171 | 5477 | |
| 29 |

|
24 | 17440 | 30770 | 5452 | 17300 | 27680 | |
| 30 |

|
99 | 20330 | 30780 | 9802 | 24780 | 17270 | |
| 31 |

|
1600 | 6792 | 9695 | 12430 | 8170 | 11150 | |
| ID | R1 | JAK3 IC50 (nM) | Ba/F3 cellular IC50s (nM) | MLM T1/2 (min) | ||||
|---|---|---|---|---|---|---|---|---|
| TEL-JAK1 | TEL-JAK2 | TEL-JAK3 | TYK2 (E957D) | TEL-ABL | ||||
| 32 |

|
0.6 | 2043 | 3878 | 10 | 1429 | 4055 | 5.1 |
| 33 |

|
1.7 | 5198 | 8487 | 109 | 6502 | 11030 | 8.3 |
| 34 |

|
2.9 | 2792 | 3993 | 114 | 2492 | 3998 | 3.5 |
| 35 |

|
1.4 | 1686 | 3290 | 21 | 994 | 3136 | 10.4 |
| 36 |

|
<0.5 | 1780 | 3164 | 11 | 759 | 4374 | 6.4 |
| 37 |

|
0.7 | 2143 | 2762 | 16 | 5581 | 11820 | 1.5 |
| 38 |
|
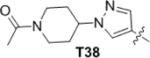
<0.5 | 2797 | 2776 | 28 | 1099 | 5286 | 3 |
| 39 |

|
<0.5 | >5E4 | >5E4 | 143 | >5E4 | >5E4 | 14.3 |
| 40 |

|
0.5 | 3506 | 27680 | 110 | 17260 | 21820 | 3.4 |
| 41 |

|
1.1 | 4553 | 6223 | 67 | 3043 | 10610 | 4.5 |
| 42 |

|
0.6 | 8223 | 19500 | 119 | 4998 | 18660 | 7.2 |
| 43 |

|
<0.5 | 3129 | 6960 | 17 | 1887 | 5398 | 1.8 |
| 44 |
|
<0.5 | 3462 | 6439 | 24 | 2227 | 6830 | 1.9 |
| 45 |

|
<0.5 | 7501±460 | 31133±17012 | 19±8 | >5E4 | >5E4 | 3.3 |
| 46 |

|
<0.5 | 3137±468 | 6211±638 | 22±2 | 2291±1342 | 11169±2688 | 1.8 |
| 47 |
|
<0.5 | 2370±494 | 9081±3534 | 39±5 | 1576±631 | 13157±6948 | 1.3 |
| 48 |
|
0.7 | 23510±13265 | >5E4 | 45±32 | 31496±41386 | >5E4 | 2.6 |
| 49 |

|
0.6 | 3065 | 9098 | 124 | 3227 | 5973 | 1.8 |
| 50 |

|
<0.5 | 3758±1863 | 7177±2847 | 34±7 | 3510±1281 | 8822±126 | 10.8 |
| 51 |

|
0.6 | 5744 | 7287 | 92 | 4090 | 8369 | 4.8 |
Enzymatic IC50s obtained with Z’-Lyte activity assays.
All cellular IC50s were obtained with 8-point titrations and triplicates, the key compounds were tested in at least 3 independent experiments and the standard deviations were shown, compound 9 was always used as a control.
Mouse liver microsomal half-life.
Co-crystal Structure of JAK3-Compound 9 Complex
To investigate the structural basis for achieving selectivity for JAK3 we solved the co-crystal structure of the JAK3 kinase domain in complex with 9 at a resolution of 2.9 Å (Figure 3). In this structure (PDB 4Z16), the anilinopyrimidine moiety of 9 makes the expected bidentate hinge hydrogen bonds with Leu905, and continuous electron density is observed between the acrylamide warhead and Cys909, indicative of covalent bond formation. The kinase exhibits an active conformation, with the DFG-motif in the inward position and both tyrosines 980 and 981 in the activation loop phosphorylated. The orientation and interactions of the anilinopyrimidine portion of the compound closely resembles the interactions observed in the EGFRT790M-8 co-crystal structure (PDB 3IKA)21 (Figure S1). In addition to the hinge hydrogen bonds, the chlorine is directed towards the gatekeeper methionine (Met902 in the present structure) and the methoxy substituent extends toward the sidechain of Tyr904 in the hinge region. Interestingly, the “linker” segment that attaches the acrylamide adopts a distinctly different conformation as compared with the corresponding region of 8 in complex with EGFRT790M. The phenyl group in the linker pivots down and is in van der Waals contact with Leu956 in the floor of the ATP-binding cleft. This orientation is likely enabled by the longer aminomethylene moiety in 9, as compared with the ether linkage in compound 8. In addition, the acrylamide amide is positioned to hydrogen bond with the carbonyl of Arg953, also in the floor of the binding pocket.
Figure 3.

X-ray co-crystal structure of JAK3-9 (PDB 4Z16). Compound 9 (yellow sticks) covalently binds to Cys909 at the GK+7 position of JAK3 (blue ribbons). The hydrogen bonds between the anilinopyrimidine moiety of 9 and Leu905 in the JAK3 hinge region are indicated.
Structure-Activity Relationship (SAR) Studies Based on Compound 9
The syntheses of 9 was readily achieved with high-overall-yield in four steps starting from 2,4,5-trichloropyrimidine. The 4-chloride group was substituted with a 3-nitrobenzyl amine under basic conditions, followed by the substitution of the 2-chloride with a 2-methoxy-4-(4-methylpiperazin-1-yl)aniline under acidic conditions to give 11. The nitro group of 11 was reduced using hydrogenation and the resulted aniline was acrylated to afford 9 (Scheme 1B). In order to elucidate the structural requirements to achieve potent and selective inhibition of JAK3 we prepared approximately 70 analogs. To approach this optimization in a systematic fashion, this chemotype was divided into three moieties: tail (R1), arm (R2) and core (R3), and each of these moieties were varied sequentially. The 2,4-disubstited aniline tail moiety of 9 was replaced with different anilines, aliphatic amines or 4-aminopyrazoles; the 4-acrylamidobenzyl arm of 9 was substituted with functional groups at the double bond, phenyl ring or benzyl position; the pyrimidine core of 9 was modified at 5- or 6-position, or was replaced with a variety of bicyclic cores. All the analogs were profiled with the same JAK3 enzymatic assay and JAKs and ABL dependent Ba/F3 cell line assays. Additionally, the in vitro metabolic stability of some analogs was measured using mouse liver microsomes (MLM) to calculate half-lives (T1/2).34
The first series of analogs focused on modification of the tail moiety (R1) (Table 2). Substitutions at the 5-postion of the aniline (14, 15) resulted in a dramatic loss of potency. In contrast, removing the 2-methoxy group (16) enhanced potency over 5-fold against JAK3, however, it also increased the cytotoxicity toward other transformed Ba/F3 cell lines. Substitutions at the 3-position did not mitigate the cytotoxicity (17–19), nor did introduction of a fluorine at the 2-position (20). Compound 21 with a 4-morpholinylaniline tail potently inhibited JAK3 despite not having the 2-methoxy substitution. Furthermore, 21 showed comparable cellular selectivity and MLM T1/2 as 9 (T1/2 of 4.7 min.). However, 21 only showed moderate selectivity in biochemical assays, with 10~20-fold higher IC50s against JAK1, JAK2 or TTK (Table 1). Adding a 2- or 3-fluoro (22, 23) did not improve the kinase selectivity of 21 and also did not improve the MLM stability. 24 which contains two fluorine substituents at the 2- and 5-positions showed good selectivity but decreased potency against JAK3. 25, with 3,5-difluorine substitutions, exhibited improved potency compared to 24 with good selectivity in cells, even though it showed moderate selectivity in enzymatic assays. Based on 25 several analogs (26–28) were made and exhibited increased potency, but none of them were as selective as 25. Aliphatic tails resulted in complete loss of potency (29–31). 32 with a 1-methyl-1H-pyrazol-3-amine tail showed increased potency, similar selectivity and MLM stability compared with 9; it also showed a 300-fold selectivity window over TTK. Addition of a 4-methyl group to the pyrazole ring (33) enhanced the MLM stability but decreased the potency over 10-fold. The isoxazol-4-amine tail in 34 is disfavored in terms of both potency and stability. Modifications were then focused on the 1-methyl group of the pyrazole tail. Replacement of the 1-methyl with a 1-difluoromethyl (35) or a 1-difluoroethyl (36) maintained the potency and enhanced the MLM stability. However, bulky and hydrophobic substituents resulted in decreased MLM stability (37, 38). 39 with a 2-substituted N-methylacetamide exhibited noticeably improved MLM stability (T1/2 of 14.3 min) and favorable selectivity albeit with only moderate potency against JAK3. Nevertheless, other 2-substituted N-alkylacetamide groups (40–42) failed to improve the potency or stability. Several analogs elaborated with a 2-substituted ethyl group (43–45) showed good potency but decreased MLM stability. To our surprise, 45 with a 2-methoxyethyl exhibited an IC50 of 19 nM against JAK3 Ba/F3 cells, and over 390-fold selectivity over other Ba/F3 cell lines. 45 showed excellent selectivity with JAK3 IC50 of less than 0.5 nM in fixed end point enzymatic assay, and over 70-fold higher IC50 against JAK1 and over 100-fold higher IC50s against JAK2, TYK2, TTK, BTK or ITK (Table 1, S1). 45 was profiled against a diverse panel of 468 kinases (DiscoveRX, KinomeScan27) at a concentration of 100 nM; the compound exhibited excellent overall kinase selectivity with selectivity scores, defined as the percentage of kinases with scores less than 5 (S(5))27b, of 0.01 (Figure 4). The results suggested 45 inhibits JAK3 most potently, and aurora A (AURKA) was likely to be the only off-target which we confirmed to be inhibited by 45 with IC50 of 43 nM. 45 showed relatively poor MLM stability with T1/2 of 3.3 min. Thus, several analogs with different 2-alkoxyethyl groups (46–48) were made to solve this problem. To our disappointment, all changes did not improve the MLM stability. Dimethyl substitution of the 2-methoxyethyl (49) is disfavored with respect to MLM stability as well as potency and selectivity. 50, with a hydroxyl group in the tail, showed 3-fold longer MLM T1/2 compared with 45. Although 50 was not as selective as 45 in Ba/F3 cell lines, it was more potent than 9 with comparable selectivity in Ba/F3 cells. Further modification of 50 failed to improve the selectivity but led to decreased potency and MLM stability (51). In this series of analogs, 45 and 50 stood out with good selectivity or MLM stability, furthermore, we found 50 was actually one of the MLM metabolites of 45 (Figure S2).
Table 1.
Enzymatic IC50s of key compounds a
| Cmpd ID | IC50s (nM) | ||||
|---|---|---|---|---|---|
| JAK1 | JAK2 | JAK3 | TYK2 | TTK | |
| 9 | 896 | 1050 | 4.8 | >10000 | 49 |
| 21 | 24.4 | 17.6 | 1.3 | 28.6 | |
| 25 | 495 | 351 | 20 | 355 | |
| 32 | 49 | 107 | 0.6 | 180 | |
| 45 | 35 | 51 | <0.5 | 407 | 212 |
| 50 | 11 | 32 | <0.5 | 140 | 143 |
| 85 | 743 | 421 | 2.3 | 3120 | |
IC50s against JAK-family kinases were obtained with Z’-Lyte activity assays while IC50s against TTK were obtained with LanthaScreen binding assays.
Figure 4.

KinomeScan kinase selectivity profiles for Compound 45. Compound 45 were profiled at a concentration of 100 nM against a diverse panel of more than 468 kinases and mutants. Scores for primary screen hits are reported as a percent of the DMSO control (% control). The lower the Kd is likely to be, such that scores of zero represent strong hits. Scores are related to the probability of a hit but are not strictly an affinity measurement.
After optimization of the tail moiety, the SAR of the arm (R2) was investigated (Table 3). Replacing the acrylamide with a propionamide (52) resulted in over an 80-fold decrease of potency against JAK3. Installation of a methyl group at α-position of the acrylamide (53) resulted in significant loss of potency and a shorter MLM T1/2. Polar substitutions at the β-position of the acrylamide (54–56) also resulted in loss of potency. Addition of a fluorine ortho to the acrylamide in the phenyl ring (57) also decrease the potency dramatically. Switching the acrylamide from meta- to para- on the phenyl ring decreased the potency considerably in combination with the aniline tail of 9 (58), but did not affect the potency when used in conjunction with other tails (59–62). This change did however decrease the selectivity in some cases (59, 62). Analogs with a para-acrylamide generally showed comparable or diminished MLM stability. Finally, adding a methyl at the benzyl position (63, 64) reduced both potency and selectivity. In summary, the modifications of R2 failed to further improve our JAK3 inhibitors. Several reported inhibitors with similar acrylamide warheads (6, 7, 8 and AZD929135 (65)) were also profiled and compared, none of them showed good inhibition against JAK3 Ba/F3 cells with IC50s over 1.0 μM (Table S2).
Table 3.
SAR of R2

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | R2 | R1 | JAK3 IC50 (nM)a | Ba/F3 cellular IC50s (nM)b | MLM T1/2 (min)c | ||||
| TEL-JAK1 | TEL-JAK2 | TEL-JAK3 | TYK2 (E957D) | TEL-ABL | |||||
| 52 |

|
T9 | 390 | 4948 | 9354 | 7938 | 8870 | 7594 | |
| 53 |

|
T45 | 7.8 | 3825 | 9824 | 947 | 15090 | 23780 | 2.1 |
| 54 |
|
T9 | 245 | ||||||
| 55 |
|
T9 | 64 | 1823 | 17240 | 14650 | 12680 | 26660 | |
| 56 |
|
T9 | 258 | ||||||
| 57 |

|
T32 | 4.4 | 3624 | 2326 | 102 | 1598 | 4123 | |
| 58 |

|
T9 | 83 | 3974 | 4996 | 2363 | 5452 | 4020 | 6 |
| 59 |

|
T21 | 0.87 | 701 | 874 | 17 | 778 | 459 | |
| 60 |

|
T32 | 0.58 | 2008 ±340 | 4733 ±326 | 18±5 | 754± 298 | 2140 ±519 | 3.1 |
| 61 |

|
T39 | 2.0 | 7422 | 10120 | 465 | 6857 | 7811 | 21.3 |
| 62 |

|
T45 | 1.3 | 3117 | 5575 | 131 | 3549 | 3021 | 2.8 |
| 63 |

|
T9 | 6.6 | 5034 | 6039 | 439 | 4215 | 6782 | |
| 64 |

|
T45 | 1.6 | 3173 | 7731 | 57 | 5193 | 9942 | 2.6 |
Enzymatic IC50s obtained with Z’-Lyte activity assays.
All cellular IC50s were obtained with 8-point titrations and triplicates, the key compounds were tested in at least 3 independent experiments and the standard deviations were shown, compound 9 was always used as a control.
Mouse liver microsomal half-life.
Simultaneously we investigated the SAR of the core moiety (R3) (Table 4). We found that a thioether linkage at the 4-postion (66, 67) slightly increased the MLM stability but decreased the potency, and a tertiary amine linkage (-NMe-) decreased the potency even more dramatically (68). Next we investigated the effects of various substituents at the 5-position. We evaluated substituents that possessed electron withdrawing groups such as chlorine (69–72), electron donating groups (73–75) or simply a hydrogen atom (76). However, only 70 with a 5-bromo group showed good potency among these analogs. A 4,6-disubstituted pyrimidine core (77) also led to substantial loss of potency. Next we tried to cyclize the 4,5-substituents into a fused 5-membered or 6-membered ring (78–82), and found that only 78 with a pyrrolo[2,3-d]pyrimidine core showed good potency against JAK3, but with a slightly decreased MLM stability. 81 with a pyrimido[4,5-d]pyrimidinone core possessed a good IC50 against JAK3 in the biochemical assay, but mediocre potency in the TEL-JAK3-transformed Ba/F3 cells. In the end, modifications at the 6-position of the pyrimidine core (83, 84) were fairly disfavored. These results suggested that 5-chlorine is the most suitable, and the secondary amine (-NH-) is a satisfactory linkage at 4-postion; some bicyclic cores did improve the inhibitor modestly, and modifications at 6-position are not tolerated.
Table 4.
SAR of R3

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | R3 | R1 | JAK3 IC50 (nM)a | Ba/F3 cellular IC50s (nM)b | MLM T1/2 (min)c | ||||
| TEL-JAK1 | TEL-JAK2 | TEL-JAK3 | TYK2 (E957D) | TEL-ABL | |||||
| 66 |

|
T9 | 12 | 2324 | 3101 | 433 | 1318 | 2848 | |
| 67 |

|
T32 | 1.2 | 2854 | 3870 | 33 | 1122 | 11040 | 6.9 |
| 68 |

|
T32 | 0.7 | 3490 | 4238 | 112 | 3943 | 5538 | 3.7 |
| 69 |

|
T9 | 30 | 11860 | 11870 | 755 | 5874 | 8890 | |
| 70 |

|
T9 | 2.3 | 2480 | 3005 | 107 | 2005 | 3022 | |
| 71 |

|
T9 | 18 | 3640 | 2426 | 307 | 1836 | 6036 | |
| 72 |

|
T9 | 2570 | ||||||
| 73 |

|
T9 | 31 | 3822 | 4094 | 1089 | 3163 | 6185 | |
| 74 |

|
T9 | 31 | ||||||
| 75 |

|
T9 | 220 | ||||||
| 76 |

|
T9 | 222 | ||||||
| 77 |

|
T16 | 9.9 | 25100 ± 610 | 29895 ± 329 | 1338 ± 61 | 15435 ± 2167 | 32710 ± 976 | |
| 78 |

|
T16 | 0.7 | 1315 | 2733 | 8 | 4458 | 2989 | 3.7 |
| 79 |

|
T16 | 101 | 26720 | >5E4 | 4947 | 24630 | >5E4 | |
| 80 |

|
T16 | 17 | 3457 | 4047 | 210 | 3327 | 2883 | |
| 81 |

|
T16 | 0.9 | 10076 ±1106 | 6728 ±1180 | 159± 11 | 3503 ±81 | 12551 ±4680 | |
| 82 |

|
T16 | 7.0 | 25120 | 8398 | 144 | 5948 | 4922 | |
| 83 |

|
T9 | 499 | 4268 | 5975 | 2563 | 3212 | 5098 | |
| 84 |

|
T9 | 2720 | 7890 | 5306 | 6232 | 4950 | 6671 | |
Enzymatic IC50s obtained with Z’-Lyte activity assays.
All cellular IC50s were obtained with 8-point titrations and triplicates, the key compounds were tested in at least 3 independent experiments and the standard deviations were shown, compound 9 was always used as a control.
Mouse liver microsomal half-life.
Validation of the Target-Engagement and the Covalent Binding
Based on the SAR results above, we selected 9, 45 and 50 as our optimal JAK3 inhibitors, and investigated their inhibitory activity on JAK3-dependent signaling. As STAT5 has been reported as a direct substrate of TEL-JAK3 in Ba/F3 cells,26 we examined the effect of our JAK3 inhibitors on STAT5 phosphorylation in this context. As we expected, STAT5 was constitutively phosphorylated in the cells treated with DMSO (Figure S3). Compound 2 at 300 nM almost fully inhibited the phosphorylation of STAT5 (p-STAT5), while 9 at 300 nM completely abolished p-STAT5, and so did 45 or 50 at 100 nM. These results are consistent with the IC50s in JAK-transformed Ba/F3 proliferation assays. In order to confirm that 45 and 50 are bonafide covalent inhibitors in analogy to compound 9 we performed cellular ‘wash-out’ experiments. JAK3 Ba/F3 cells were treated with these four inhibitors at various concentrations for 3 h, the cells were washed extensively with PBS and then allowed to recover for 4 h. Western blots of the cellular lysates revealed that 9, 45 or 50 were capable of sustained inhibition of p-STAT5 after the washout, whereas the reversible inhibitor 2 was not (Figure 5A). To monitor the degree of JAK3 ‘target engagement’, a biotinylated version of 45 (85) was designed with a biotin tethered via a flexible PEG linker at the tip of 45’s tail moiety (Figure 5C). We confirmed that 85 maintained similar biochemical potency and selectivity for JAK3 (Table 1). 85 showed selective but weak potency against JAK3 Ba/F3 cells with an IC50 of 1.3 μM (Table S2) presumably due to poor membrane permeability. We demonstrated that streptavidin mediated pulldown of 85 in cell lysates allowed for efficient recovery of JAK3 as assessed by western blotting. Consistent with the wash-out results, 85 strongly labeled JAK3 when cells were treated with the reversible inhibitor 2 but not when cells were treated with acrylamide modified inhibitors 9, 45 or 50 (Figure 5B). Compounds 9, 45 and 50 were further demonstrated to be covalent inhibitors using electrospray ionization mass spectrometry, where incubation of recombinant JAK3 kinase domain with inhibitor resulted in addition of the expected molecular weight (Figure 6). Subsequent protease digestion and LC/MS2 experiments identified a single modified peptide LVMEYLPSGC*LR (C*, cysteine labeled by 9, 45 or 50, JAK3 residues 900–911) and localized the site of modification to Cys909 (Figure S4), in agreement with the JAK3/9 co-crystal structure. Cumulatively these results provide strong evidence that 9, 45 and 50 are all irreversible, covalent inhibitors and that Cys909 of JAK3 is the only labeled site.
Figure 5.

Compounds 9, 45, and 50 are covalent, irreversible JAK3 inhibitors. TEL-JAK3 Ba/F3 cells were treated with 2, 9 (1.0 μM), 45 or 50 (300 nM) for 3 h, washed extensively with PBS, allowed to recover for 4 h, then lysed and subjected to western blot for phospho-STAT5 and total STAT5 (A). TEL-JAK3 Ba/F3 cells were treated 2, 9 (1.0 μM), 45 or 50 (300 nM) for 3 h, and the resulting cell lysates were treated with 85 (C) (1.0 μM, 1 h), followed by pull-down with streptavidin beads and immunoblotting with anti-JAK3 antibody (B).
Figure 6.
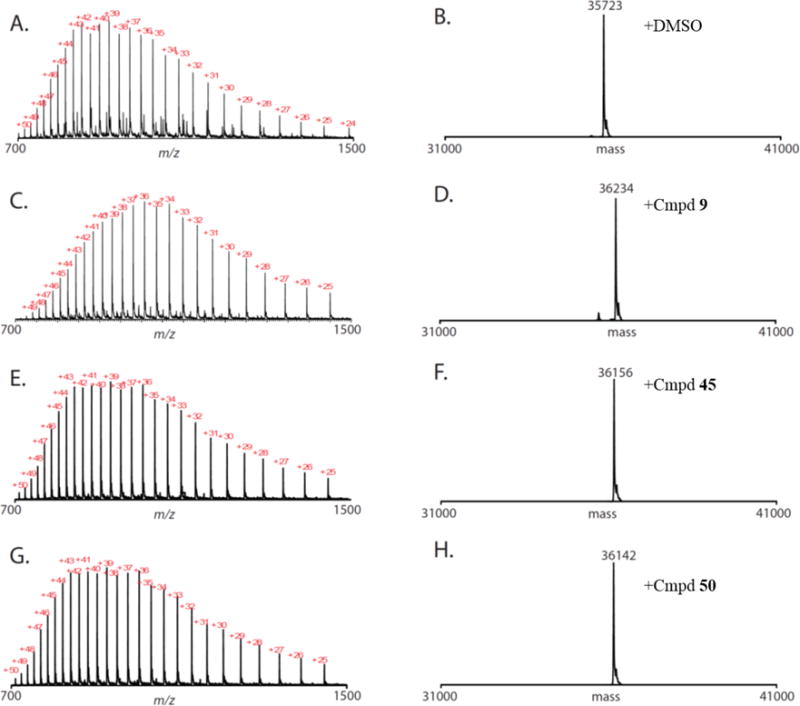
Compound 9, 45, or 50 react quantitatively with JAK3. Raw (A, C, E, G) and deconvoluted (B, D, F, H) mass-spectra obtained for JAK3 after treatment with DMSO (A, B), 9 (C, D), 45 (E, F), or 50 (G, H).
Validation in Cells with Endogenous JAK3
Compounds 9, 45 and 50 were further evaluated for their ability to inhibit JAK3 kinase activity in a variety of other cell types. We evaluated the ability of 9, 45 or 50 to inhibit signaling following stimulation of primary murine bone marrow derived macrophage (BMDMs) with a panel of 4 different cytokines based on their requirement for different cytokines (Figure 7). Among these cytokines, only signaling via IL-4, whose receptor binds JAK1 and JAK3,36 is dependent on JAK3. In contrast, granulocyte-macrophage colony-stimulating factor (GM-CSF), which signals via JAK2, and IL-10 and interferon beta (IFNβ), which signal via JAK1 and TYK2, are JAK3 independent.37 9, 45 or 50 completely inhibited IL-4 induced p-STAT6 at a concentration of 500 nM and only partially inhibited IFNβ-induced p-STAT1 at a concentration of 5.0 μM. None of our inhibitors detectably reduced the p-STAT5 level stimulated by GM-CSF or p-STAT3 induced by IL-10 at a concentration of 5.0 μM. While demonstrating excellent selectivity in Ba/F3 cellular assays, these inhibitors showed decreased but still decent selectivity in primary cells. The washout experiments with 45 or 50 in BMDMs cells exhibited consistent results as in TEL-JAK3 Ba/F3 cells, and suggested that the inhibitory effect lasted for at least 24 h after the washout (Figure S5). In a similar way, 9, 45 and 50 exhibited consistent potency and selectivity for JAK3 in leukemia cancer cell lines: T-ALL1 and OCL-AML5 (Figure 8). In T-ALL1 cells, IL-2 induced STAT5 phosphorylation was completely inhibited by 9 at a concentration of 1.0 μM or by 45 or 50 at a concentration of 100 nM (Figure 8A). However, in OCL-AML5 cells the GM-CSF induced STAT5 phosphorylation was maintained in the presence of the inhibitors up to a concentration of 10 μM (Figure 8B). In contrast, previously reported pan-JAK inhibitors 1 and 2 could abolish p-STAT5 in both stimulated cell lines at 1.0 μM. Based on these results compounds 9, 45 and 50 are capable of efficiently inhibiting JAK3 and specifically blocking the JAK3-dependent signaling pathway in human cells at sub-micromolar concentrations.
Figure 7.

Compound 9, 45 or 50 potently and selectively inhibited JAK3-dependent signaling in BMDMs cells. BMDMs cells were pre-incubated with the indicated concentrations of 9 (A), 45 (B) or 50 (C) for 3 h. Cells were then stimulated with the indicated cytokines (IL-4 at 10 ng/mL, IFNβ at 500 U/mL, GM-CSF at 10 ng/mL, and IL-10 at 100 ng/mL) for 30 min then lysed. The levels of tyrosine phosphorylation on the appropriate STAT protein were determined by immunoblotting. Levels of total ERK1/2 were also examined to show equal loading. The signaling cascades were described in D.
Figure 8.

Compound 9, 45 and 50 potently and selectively inhibited JAK3-dependent p-STAT5 in TALL-1 cells whereas pan-JAK inhibitors 1 and 2 also potently inhibited JAK2-dependent p-STAT5 in OCL-AML5 cells. TALL-1 (A) or OCL-AML5 (B) cells were pre-incubated with the indicated concentrations of indicated inhibitors for 3 h. Cells were then stimulated with 10 ng/mL of IL-2 (A) or 5 ng of GM-CSF (B) for 30 min then lysed. The p-STAT5 levels were determined by immunoblotting. Levels of total STAT5 were also examined to show equal loading. The signaling cascades were described in C.
Mouse Pharmacokinetic Properties and In Vivo Studies
We evaluated the pharmacokinetic properties of 9 in mice following intravenous and oral delivery. Compound 9 demonstrated reasonable pharmacokinetic properties, with moderate T1/2 of 1.4 h, area under the curve (AUC) value of 795 ng*hr/mL following a 10 mg/Kg oral dose and good oral bioavailability of 66% (Table 5). This suggests that 9 may be a suitable probe for future murine efficacy studies. We evaluated the ability of compound 9 to alter immune cell numbers in genetically engineered models (GEM) of lung adenocarcinoma in mice. Immune cell numbers in mice harboring tumors driven by KrasG12D;Lkb1L/L–38 were assessed in tumor-associated (lung) as well as general immune cell populations (spleen) upon treatment with compound 9. After oral administration with 9 (75 mpk, QD) for 8 days, the numbers of B or T lymphocytes in the tumor-bearing lungs and spleens of treated mice was not affected, however, the number of NK cells was reduced (Figure S6). Similarly, the number of tumor-associated macrophages and neutrophils in the lung of treated mice were not affected. During the short duration of this study, we did not detect any effect on the tumor growth.
Table 5.
Pharmacokinetic properties of 9
| ID | route | Dose (mg/kg) | Tmax (h)a | Cmax (ng/mL)b | AUClast (h*ng/mL)c | T1/2 (h)d | CL (mL/min/kg)e | Vss (L/kg)f | F (%)g |
|---|---|---|---|---|---|---|---|---|---|
| 9 | IV | 2 | – | 158.56 | 241.30 | 1.40 | 135.88 | 18.38 | – |
| PO | 10 | 0.5 | 204.25 | 795.29 | – | – | – | 66 |
Time for peak plasma concentrations.
Peak plasma concentrations.
Area under the concentration time curve.
Terminal half-life.
Plasma Clearance.
Volume in steady state.
Bioavailability.
Discussion
Building on our covalent T790M EGFR inhibitor 8 we designed and synthesized new analogs aiming to target the analogous cysteine residue in JAK3. Among those analogs compound 9 was identified as a potent and selective JAK3 inhibitor compared with current reversible JAK inhibitors. We determined a 2.9 Å co-crystal structure of JAK3 with 9, which revealed that 9 covalently binds to Cys909 of JAK3 as expected. With 9 as a lead, we designed and synthesized dozens of analogs to intensively study the SAR with regard to each moiety of this scaffold. These analogs were tested in enzymatic assays for their potency against JAK3 and profiled against a panel of transformed Ba/F3 cell lines, for their potency and selectivity in a cellular context. Based on these studies we discovered that a 2-methoxy group in the aniline tail of 9 is disfavored by JAK3 presumably due to a disfavored interaction with the bulky side chain of Tyr904 located at the GK+2 position. Hydrophobic or less polar tails are better for selectivity for JAK3 presumably because they have weaker non-covalent affinity for the kinase thereby allowing covalent bond formation to be a more important contributor to the potency. The acrylamidobenzyl arm moiety of 9 is significant for the potency against JAK3 and sensitive to changes, its specific length and flexibility are critical to enable the inhibitors to efficiently form a covalent bond with Cys909 of JAK3. The H-bond between the acrylamide of 9 and the carbonyl of Arg953 fixed the conformation of the acrylamide and left no space to accommodate substituents. The chlorine atom in the pyrimidine core is responsible for both potency and selectivity with its interaction with Met902 of JAK3. Modifications at the 6-position of the pyrimidine core were fairly disfavored as they collided with the carbonyl of Glu903 3.3Å away. Some of these analogs were also evaluated for their MLM stability, the results indicated that more lipophilic analogs with aliphatic non-polar substituents tend to have a shorter half-life, maybe resulting from their enhanced binding with phospholipid in microsomes.39
Among these analogs 9, 45 and 50 stood out with overall favorable potency, selectivity and MLM stability. They all exhibited efficient and selective inhibition of JAK-dependent signaling within different contexts. So far 45 is our most selective JAK3 inhibitor with at least 70-fold selectivity over other JAKs in biochemically assays, at least 390-fold selectivity in JAK-transformed Ba/F3 cells, and decent selectivity in primary cells. In addition to the evidence provided by JAK3-9 co-crystal structure, all three inhibitors were demonstrated to covalently modify Cys909 of JAK3 based upon wash-out and pull-down experiments and mass spectrometry. We developed a biotinylated probe 85 which was demonstrated to be a useful reagent for establishing “target-engagement” in cellular assays. We also found that a short-term treatment with high dose of compound 9 in mice with lung tumors resulted in a decrease in the number of NK cells suggesting that the compounds are active in vivo. Further investigations will be required to elucidate the basis for this phenomena and to assess the activity of covalent JAK3 inhibitors in relevant disease models.
In conclusion, the covalent JAK3 inhibitors 9, 45, 50, and the biotinylated probe 85 provide a set of useful tools to pharmacologically interrogate JAK3-dependent biology. Compound 45 is a highly potent and selective biochemical and cellular inhibitor of JAK3 and is an ideal reagent for cell biological studies. Compound 9 combines favorable target profile with good pharmacokinetic properties and is ready to be used in future animal studies. Finally, the structure-activity relationships with respect to JAK3 combined with the co-crystal structure will serve as an excellent foundation for future JAK3 inhibitor development efforts.
Experimental Section
Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 600 MHz (Varian AS600), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), br (broad singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Sunfire C18 column (19 × 50 mm, 5μM) using a gradient of 15–95% methanol in water containing 0.05% trifluoroacetic acid (TFA) over 22 min (28 min run time) at a flow rate of 20 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by reverse-phase HPLC analysis.
2,5-Dichloro-N-(3-nitrobenzyl)pyrimidin-4-amine (10)
2,4,5-Trichloropyrimidine (112 μL, 1.0 mmol), (3-nitrophenyl)methanamine hydrochloride salt (227 mg, 1.2 mmol), and N,N-diisopropylethylamine (DIEA, 530 μL, 3.0 mmol) were combined in dioxane (5 mL) and stirred overnight. The mixture was then diluted with ethyl acetate and washed with water and brine, dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (hexane:ethyl acetate = 1:1) to yield 270 mg (80%) of 10 as a white solid. 1H NMR (600 MHz, CDCl3) δ 8.21 (s, 1H), 8.18 (d, J = 8.4 Hz, 1H), 8.10 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.56 (dd, J = 7.8, 7.8 Hz, 1H), 5.94 (br, 1H), 4.84 (d, J = 6.0 Hz, 1H). MS (ESI) m/z 299 (M+H)+.
5-Chloro-N2-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)-N4-(3-nitrobenzyl)pyrimidine-2,4-diamine (11)
To 10 (150 mg, 0.5 mmol) and 2-methoxy-4-(4-methylpiperazin-1-yl)aniline (166 mg, 0.75 mmol) in sec-butanol (5 mL) was added trifluoroacetic acid (57 μL, 0.75 mmol) and the mixture was stirred overnight at 100 °C. The mixture was then concentrated, neutralized with ammonia in methanol and purified by column chromatography (dichloromethane:methanol = 10:1) to yield 184 mg (76%) of 11 as a pale-yellow solid. 1H NMR (600 MHz, CD3OD) δ 8.23 (s, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.84 (s, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.54 (dd, J = 7.8, 7.8 Hz, 1H), 6.60 (d, J = 3.0 Hz, 1H), 6.36 (dd, J = 9.0, 3.0 Hz, 1H), 4.75 (s, 1H), 3.83 (s, 3H), 3.15 (m, 4H), 2.64 (m, 4H), 2.36 (s, 3H). MS (ESI) m/z 484 (M+H)+.
N-(3-(((5-chloro-2-((2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)amino)methyl)phenyl)acrylamide (9)
To 11 (97 mg, 0.2 mmol) in MeOH (20 mL) was added 1 mL Raney nickel suspension in MeOH. The reaction mixture was stirred for 3 h under 1 atm of hydrogen. The mixture was then filtered with Celite, and the filtrate was concentrated and dried under vacuum to give a crude product as a white solid. To the obtained white solid in DMF (2 mL) was added DIEA (53 μL, 0.3 mmol), the stirred mixture was then cooled to −60 °C, and acryloyl chloride (17.8 μL, 0.22 mmol) was added dropwise. The reaction mixture was stirred at −60 °C for 10 min, allowed to recover to RT (room temperature) gradually in 30 min, and purified by reverse phase HPLC to give 88 mg (TFA salt, 71% for 2 steps) of 9 as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 10.07 (s, 1H), 7.82 (s, 1H), 7.73 (m, 1H), 7.59 (s, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.29 (s, 1H), 7.18 (dd, J = 7.8, 7.8 Hz, 1H), 6.90 (d, J = 7.2 Hz, 1H), 6.48 (s, 1H), 6.44 (dd, J = 16.8, 10.2 Hz, 1H), 6.31 (d, J = 8.4 Hz, 1H), 6.25 (d, J = 16.8 Hz, 1H), 5.75 (d, J = 10.2 Hz, 1H), 4.56 (d, J = 6.0 Hz, 2H), 3.78 (s, 3H), 3.04 (m, 4H), 3.01 (m, 4H), 2.22 (s, 3H). MS (ESI) m/z 508 (M+H)+.
Compound 14–64, 66–85 were synthesized with same procedures as 9, 3 and 8 were synthesized as reported.19, 31 Compound 1, 2, 6, 7, 12, 13 and 65 were from Selleckchem, 4 was from Santa Cruz, and 5 was from Millipore.
Enzymatic assays
The enzymatic activities against JAK1, JAK2, JAK3, TYK2, FLT3, BLK, TXK, BTK, ITK and AURKA were tested in Z’-Lyte assays with ATP concentrations of Km for each kinases, the activities against TTK were tested in LanthaScreen binding assays. All the protocols are available from Life Technologies.40
Protein expression and purification
Human JAK3 kinase domain (residues 811–1124) was expressed and purified as described previously41 except that JAK3 was co-expressed with human c-SRC (residues 86–536) to achieve more consistent phosphorylation of JAK3.
Crystallization and structure determination
Crystals of JAK3 in complex with 9 were prepared using the hanging drop vapor diffusion method; JAK3 at a concentration of 5 mg/mL was incubated for 30 min with 500 μM compound 9, then added to an equal volume of well solution (0.1 M bis-tris pH 6.5, 0.2 M ammonium sulfate, 16% PEG 3350, 5 mM TCEP) and equilibrated over well solution at 4 °C. Crystals were looped into cryoprotectant solution (0.1 M bis-tris pH 6.5, 0.2 M ammonium sulfate, 20 % PEG 3350, 15% glycerol, 5 mM TCEP) for 30–60 sec, then flash-frozen in liquid nitrogen. X-ray diffraction data were collected at APS beamline 24-ID-E (NE-CAT), processed with XDS,42 and scaled with Scala.43 The structure was solved by molecular replacement with Phaser44 using the JAK3 kinase domain as a search model (PDB 1YVJ).41 Manual refitting of the crystallographic model was performed with Coot45 and refinement was performed with Phenix46 and BUSTER.47 Topology and parameter files for compound 9 were generated with PRODRG.48
Mass spectrometry
JAK3 protein (5 μg) was labeled with a 10-fold excess of inhibitor or DMSO for 30 minutes at 4 °C. After labeling, proteins were desalted using 0.5 mL Zeba spin desalting columns (Thermo Fisher Scientific). Analysis of intact protein was performed essentially as described.49 Briefly, ~5 μg protein was injected onto a self-packed reversed-phase column (500 μm inner diameter, 5 cm of POROS 50R2 resin). After washing to remove salts, protein was eluted with an HPLC gradient (0%–100% B in 1 min, A = 0.2 M acetic acid in water, B = 0.2 M acetic acid in acetonitrile, flow rate = 10 μL/min) into a linear ion trap mass spectrometer (LTQ, Thermo Fisher Scientific, San Jose, CA). Data were acquired in profile mode scanning m/z 300–2000. Mass spectra were deconvoluted using MagTran software (version 1.03b2).50 Sites of covalent modification were identified using a “bottom-up” strategy. Desalted proteins were reduced with tris(2-carboxyethyl)phosphine (10 mM, 10 min room temperature), alkylated with iodoacetamide (20 mM, 30 min room temperature in dark), and digested with trypsin for 4 h at 37 °C. Digests were analyzed by nanoLC-ESI-MS as described with modifications.51 Peptides were loaded onto a self-packed pre-column (4 cm POROS10R2), resolved on an analytical column (30 μm I.D., packed with 12 cm C18) and eluted into the mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific) using an HPLC gradient (Waters NanoAcquity, Milford, MA; 0%–35% B in 60 min, A = 0.2 M acetic acid in water, B = 0.2 M acetic acid in acetonitrile, flow rate = ~30 nL/min). The instrument was operated in data dependent mode such that the top 10 most abundant precursors were subjected to MS/MS (electron multiplier detection, relative collision energy 35%, q = 0.25). Raw data files were converted to .mgf using in-house software52 and searched using Mascot 2.2.1 against a forward-reverse human refseq database. Search parameters specified variable oxidation of methionine, variable inhibitor modification (9, 45, or 50) of cysteine, and fixed carbamidomethylation of cysteine (i.e. cysteines are considered carbamidomethylated or inhibitor labeled). To confirm labeling sites, internally calibrated HCD spectra (image current detection, resolution at m/z 200 = 15,000, relative collision energy 35%) were acquired using an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific). For these experiments, LC parameters were similar to those described above, except that the analytical column was packed with 50 cm of C18 and peptides were eluted with a gradient of 0–35% B in 90 min.
Mouse liver microsomal stability
The MLM assays were previously reported and are commercially available from Scripps Florida.34
Ba/F3 cell viability assays
Ba/F3 derivatives expressing various oncogenic fusion kinases, TEL-JAK1, TEL-JAK2, TEL-JAK3 and TEL-ABL were described previously.26 Ba/F3 cells transformed by TYK2E957D were generated as previously described.30 These cells were maintained in RPMI-1640 medium (GIBCO) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich) and penicillin/streptomycin (Invitrogen). For cell viability assays, the cells were plated at a density of 10,000 cells per well in a 96-well white plate and incubated with DMSO or increasing concentrations of drugs. At 72 h after the initiation of treatment, the relative cell viability was determined using the Cell Titer Glo assay (Promega) and reported as a percentage of the DMSO control. Concentration values for 50% inhibition (IC50) of cell viability were determined with GraphPad Prism software.
Immunoblotting analysis and washout experiment
Ba/F3 cells transformed by TEL-JAK3 were incubated with JAK3 inhibitors or DMSO for 3 h. Then whole-cell lysates were prepared in RIPA buffer (Cell Signaling) with FOCUS™, ProteaseArrest™ (G-Biosciences) and Phosphatase Inhibitor Cocktail Set II (EMD Millipore). Western blotting analyses were conducted after separation by SDS-PAGE electrophoresis and transfer to nitrocellulose membranes. Immunoblotting was performed with each of specific antibodies to STAT5, phospho-STAT5 (Tyr694), α-tubulin (Cell Signaling Tech) and JAK3 (Santa Cruz, #sc-513). For washout experiment, cells were incubated with JAK3 inhibitors or DMSO for 3 h, washed with PBS three times, maintained in RPMI-1640 medium with 10% of FBS for 4 h, and then harvested and the resulting lysates were analyzed. Primary bone marrow derived macrophages (BMDMs) were prepared as described.53 Cells were maintained on bacterial grade DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 U/ml of penicillin G, 100 μg/ml of streptomycin, 0.25 μg/ml of amphotericin (Invitrogen), and 5 ng/ml of macrophage colony-stimulating factor (M-CSF; PreProTech) for 7 days. Cells were then replated onto tissue culture grade plastic and used the following day. Following stimulation cells were lsyed into SDS sample buffer and run on 10% polyacrylamide gels according to standard techniques. Proteins were transferred onto nitrocellulose membranes and blotted with antibodies against total ERK1/2, p-Y641 STAT6, p-Y701 STAT1, pY701 STAT3 or pY694 STAT5 (all from Cell Signaling Tech). The experiments in cancer cell lines were done with similar procedures.
Streptavidin pulldown experiment
Ba/F3 cells transformed by TEL-JAK3 were incubated with JAK3 inhibitors or DMSO for 3 h, and then lysed with Pierce IP lysis buffer (Thermo Scientific) with Halt™ protease inhibitor cocktail (Thermo Scientific). The lysates were treated with 85 (5 μM) at 4 °C overnight, and then further incubated at room temperature for 3 h. The solution was mixed with Streptavidin beads (25 μL, Sigma) and incubated with rotation at 4 °C for 2 h. The beads were washed five times using the lysis buffer with 4 M urea (1 mL), and then boiled in the presence of Lammli sample buffer with 2-mercaptoethanol. The eluted proteins were immunoblotted with each of specific antibodies.
In Vivo Pharmacokinetic Studies
Male Swiss albino mice were dosed via tail vein (intravenous, 0.1% v/v Tween 80, 0.5% w/v NaCMC in water at a dose of 10 mg/kg) or via oral gavage (suspensions in 5% NMP, 5% solutol HS in normal saline intravenously via tail vein at a dose of 2 mg/kg). Blood samples were collected at 0.08, 0.25, 0.5, 1, 2, 4, 8 and 24 h (i.v.) and at Predose, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h (p.o.). Plasma samples were separated by centrifugation of whole blood and stored below −70°C until bioanalysis. All samples were processed for analysis by protein precipitation using acetonitrile and analyzed with fit-for-purpose LC/MS/MS method (LLOQ, 1.06 ng/mL). Pharmacokinetic parameters were calculated using the non-compartmental analysis tool of WinNonlin Enterprise software (version 6.3).
Mouse treatment studies
KrasG12D;Lkb1L/L mice were intrathoracically injected with adenovirus expressing Cre recombinase (2.5×107 PFU/10 μl) at 6 weeks of age to initiate tumor formation.38b All experimental mice were maintained on a mixed genetic background (C57Bl/6, Balb-c, and S129). MR imaging and tumor quantification were performed as described previously.38b Compound 9 and vehicle controls were given per os once daily at a 75 mpk dose.
Total mouse lung cell and tumor infiltrating immune cell characterization was performed as previously described.54 Splenocytes were isolated similarly without collagenase treatment. The antibodies used for immune analysis are listed in supplementary methods. Acquisition of eight color samples was performed on a BD Canto II cytometer equipped with Diva software and analyzed using Flowjo. All in vivo experiments performed in the PK studies and this study were approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute.
Supplementary Material
Acknowledgments
We thank Dr. Cai-Hong Yun for his valuable help with creating topology and parameter files for the refinement of the JAK3-9 co-crystal structure. The TEL-JAK1, TEL-JAK2, TEL-JAK3 and TEL-ABL plasmids used in this studies were generously provided by Dr. Richard Moriggl. G.S.H.-S. was supported by the Deutsche Forschungsgemeinschaft (HE 6897/1-1). We thank Dr. Ying Li for data analysis and Dr. Sara Buhrlage for the proof-reading.
Footnotes
Supporting Information Available: Figure S1–6; Table S1–4; Structures and spectral data of 14–64 and 66–85; Complete KinomeScan profiling data of 9 and 45. This material is available free of charge via the Internet at http://pubs.acs.org. X-ray coordinates and structure factors have been deposited in the Protein Data Bank. PDB accession code: 4Z16.
References
- 1.(a) Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]; (b) Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36(4):503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]; (b) Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunological reviews. 2009;228(1):273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Johnston JA, Bacon CM, Riedy MC, O’Shea JJ. Signaling by IL-2 and related cytokines: JAKs, STATs, and relationship to immunodeficiency. Journal of leukocyte biology. 1996;60(4):441–452. doi: 10.1002/jlb.60.4.441. [DOI] [PubMed] [Google Scholar]; (b) Boudny V, Kovarik J. JAK/STAT signaling pathways and cancer. Janus kinases/signal transducers and activators of transcription. Neoplasma. 2002;49(6):349–355. [PubMed] [Google Scholar]; (c) Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26(47):6724–6737. doi: 10.1038/sj.onc.1210757. [DOI] [PubMed] [Google Scholar]; (d) Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. 2015;194(1):21–27. doi: 10.4049/jimmunol.1401867. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annual review of medicine. 2015;66:311–328. doi: 10.1146/annurev-med-051113-024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, Kelley J, Covington M, Shepard S, Rodgers JD, Haley P, Kantarjian H, Fridman JS, Verstovsek S. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flanagan ME, Blumenkopf TA, Brissette WH, Brown MF, Casavant JM, Shang-Poa C, Doty JL, Elliott EA, Fisher MB, Hines M, Kent C, Kudlacz EM, Lillie BM, Magnuson KS, McCurdy SP, Munchhof MJ, Perry BD, Sawyer PS, Strelevitz TJ, Subramanyam C, Sun J, Whipple DA, Changelian PS. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. Journal of medicinal chemistry. 2010;53(24):8468–8484. doi: 10.1021/jm1004286. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tam CS, Verstovsek S. Investigational Janus kinase inhibitors. Expert opinion on investigational drugs. 2013;22(6):687–699. doi: 10.1517/13543784.2013.774373. [DOI] [PubMed] [Google Scholar]; (b) Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. Journal of medicinal chemistry. 2014;57(12):5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]; (c) Rosenthal A, Mesa RA. Janus kinase inhibitors for the treatment of myeloproliferative neoplasms. Expert opinion on pharmacotherapy. 2014;15(9):1265–1276. doi: 10.1517/14656566.2014.913024. [DOI] [PubMed] [Google Scholar]; (d) Mascarenhas JO, Cross NC, Mesa RA. The future of JAK inhibition in myelofibrosis and beyond. Blood reviews. 2014;28(5):189–196. doi: 10.1016/j.blre.2014.06.002. [DOI] [PubMed] [Google Scholar]; (e) Geyer HL, Mesa RA. Therapy for myeloproliferative neoplasms: when, which agent, and how? Blood. 2014;124(24):3529–3537. doi: 10.1182/blood-2014-05-577635. [DOI] [PubMed] [Google Scholar]
- 7.(a) Menet CJ, Rompaey LV, Geney R. Advances in the discovery of selective JAK inhibitors. Progress in medicinal chemistry. 2013;52:153–223. doi: 10.1016/B978-0-444-62652-3.00004-1. [DOI] [PubMed] [Google Scholar]; (b) Dymock BW, Yang EG, Chu-Farseeva Y, Yao L. Selective JAK inhibitors. Future medicinal chemistry. 2014;6(12):1439–1471. doi: 10.4155/fmc.14.92. [DOI] [PubMed] [Google Scholar]; (c) Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert opinion on investigational drugs. 2014;23(8):1067–1077. doi: 10.1517/13543784.2014.918604. [DOI] [PubMed] [Google Scholar]
- 8.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nature reviews Immunology. 2009;9(7):480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annual review of immunology. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36(4):515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornejo MG, Boggon TJ, Mercher T. JAK3: a two-faced player in hematological disorders. The international journal of biochemistry & cell biology. 2009;41(12):2376–2379. doi: 10.1016/j.biocel.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bains T, Heinrich MC, Loriaux MM, Beadling C, Nelson D, Warrick A, Neff TL, Tyner JW, Dunlap J, Corless CL, Fan G. Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia. 2012;26(9):2144–2146. doi: 10.1038/leu.2012.74. [DOI] [PubMed] [Google Scholar]
- 13.(a) Walters DK, Mercher T, Gu TL, O’Hare T, Tyner JW, Loriaux M, Goss VL, Lee KA, Eide CA, Wong MJ, Stoffregen EP, McGreevey L, Nardone J, Moore SA, Crispino J, Boggon TJ, Heinrich MC, Deininger MW, Polakiewicz RD, Gilliland DG, Druker BJ. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer cell. 2006;10(1):65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]; (b) Koo GC, Tan SY, Tang T, Poon SL, Allen GE, Tan L, Chong SC, Ong WS, Tay K, Tao M, Quek R, Loong S, Yeoh KW, Yap SP, Lee KA, Lim LC, Tan D, Goh C, Cutcutache I, Yu W, Ng CC, Rajasegaran V, Heng HL, Gan A, Ong CK, Rozen S, Tan P, Teh BT, Lim ST. Janus kinase 3-activating mutations identified in natural killer/T-cell lymphoma. Cancer discovery. 2012;2(7):591–597. doi: 10.1158/2159-8290.CD-12-0028. [DOI] [PubMed] [Google Scholar]
- 14.Van Allen EM, Golay HG, Liu Y, Koyama S, Wong K, Taylor-Weiner A, Giannakis M, Harden M, Rojas-Rudilla V, Chevalier A, Thai T, Lydon C, Mach S, Avila AG, Wong JA, Rabin AR, Helmkamp J, Sholl L, Carter SL, Oxnard G, Janne P, Getz G, Lindeman N, Hammerman PS, Garraway LA, Hodi FS, Rodig SJ, Dranoff G, Wong KK, Barbie DA. Long-term Benefit of PD-L1 Blockade in Lung Cancer Associated with JAK3 Activation. Cancer immunology research. 2015 doi: 10.1158/2326-6066.CIR-15-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Narla RK, Liu XP, Myers DE, Uckun FM. 4-(3′-Bromo-4′hydroxylphenyl)-amino-6,7-dimethoxyquinazoline: a novel quinazoline derivative with potent cytotoxic activity against human glioblastoma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 1998;4(6):1405–1414. [PubMed] [Google Scholar]; (b) Chen JJ, Thakur KD, Clark MP, Laughlin SK, George KM, Bookland RG, Davis JR, Cabrera EJ, Easwaran V, De B, George Zhang Y. Development of pyrimidine-based inhibitors of Janus tyrosine kinase 3. Bioorganic & medicinal chemistry letters. 2006;16(21):5633–5638. doi: 10.1016/j.bmcl.2006.08.022. [DOI] [PubMed] [Google Scholar]; (c) Soth M, Hermann JC, Yee C, Alam M, Barnett JW, Berry P, Browner MF, Frank K, Frauchiger S, Harris S, He Y, Hekmat-Nejad M, Hendricks T, Henningsen R, Hilgenkamp R, Ho H, Hoffman A, Hsu PY, Hu DQ, Itano A, Jaime-Figueroa S, Jahangir A, Jin S, Kuglstatter A, Kutach AK, Liao C, Lynch S, Menke J, Niu L, Patel V, Railkar A, Roy D, Shao A, Shaw D, Steiner S, Sun Y, Tan SL, Wang S, Vu MD. 3-Amido pyrrolopyrazine JAK kinase inhibitors: development of a JAK3 vs JAK1 selective inhibitor and evaluation in cellular and in vivo models. Journal of medicinal chemistry. 2013;56(1):345–356. doi: 10.1021/jm301646k. [DOI] [PubMed] [Google Scholar]; (d) de Vicente J, Lemoine R, Bartlett M, Hermann JC, Hekmat-Nejad M, Henningsen R, Jin S, Kuglstatter A, Li H, Lovey AJ, Menke J, Niu L, Patel V, Petersen A, Setti L, Shao A, Tivitmahaisoon P, Vu MD, Soth M. Scaffold hopping towards potent and selective JAK3 inhibitors: discovery of novel C-5 substituted pyrrolopyrazines. Bioorganic & medicinal chemistry letters. 2014;24(21):4969–4975. doi: 10.1016/j.bmcl.2014.09.031. [DOI] [PubMed] [Google Scholar]; (e) Gehringer M, Pfaffenrot E, Bauer S, Laufer SA. Design and synthesis of tricyclic JAK3 inhibitors with picomolar affinities as novel molecular probes. ChemMedChem. 2014;9(2):277–281. doi: 10.1002/cmdc.201300520. [DOI] [PubMed] [Google Scholar]; (f) Jaime-Figueroa S, De Vicente J, Hermann J, Jahangir A, Jin S, Kuglstatter A, Lynch SM, Menke J, Niu L, Patel V, Shao A, Soth M, Vu MD, Yee C. Discovery of a series of novel 5H-pyrrolo[2,3-b]pyrazine-2-phenyl ethers, as potent JAK3 kinase inhibitors. Bioorganic & medicinal chemistry letters. 2013;23(9):2522–2526. doi: 10.1016/j.bmcl.2013.03.015. [DOI] [PubMed] [Google Scholar]; (g) Thoma G, Nuninger F, Falchetto R, Hermes E, Tavares GA, Vangrevelinghe E, Zerwes HG. Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. Journal of medicinal chemistry. 2011;54(1):284–288. doi: 10.1021/jm101157q. [DOI] [PubMed] [Google Scholar]; (h) McDonnell ME, Bian H, Wrobel J, Smith GR, Liang S, Ma H, Reitz AB. Anilino-monoindolylmaleimides as potent and selective JAK3 inhibitors. Bioorganic & medicinal chemistry letters. 2014;24(4):1116–1121. doi: 10.1016/j.bmcl.2014.01.001. [DOI] [PubMed] [Google Scholar]; (i) Ross JA, Spadaro M, Rosado DC, Cavallo F, Kirken RA, Pericle F. Inhibition of JAK3 with a novel, selective and orally active small molecule induces therapeutic response in T-cell malignancies. Leukemia. 2014;28(4):941–944. doi: 10.1038/leu.2013.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, McCurdy SP, Kudlacz EM, Conklyn MJ, Elliott EA, Koslov ER, Fisher MB, Strelevitz TJ, Yoon K, Whipple DA, Sun J, Munchhof MJ, Doty JL, Casavant JM, Blumenkopf TA, Hines M, Brown MF, Lillie BM, Subramanyam C, Shang-Poa C, Milici AJ, Beckius GE, Moyer JD, Su C, Woodworth TG, Gaweco AS, Beals CR, Littman BH, Fisher DA, Smith JF, Zagouras P, Magna HA, Saltarelli MJ, Johnson KS, Nelms LF, Des Etages SG, Hayes LS, Kawabata TT, Finco-Kent D, Baker DL, Larson M, Si MS, Paniagua R, Higgins J, Holm B, Reitz B, Zhou YJ, Morris RE, O’Shea JJ, Borie DC. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302(5646):875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]; (b) Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189(6):2784–2792. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, Leonard WJ. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(9):3508–3513. doi: 10.1073/pnas.1301138111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bouchekioua A, Scourzic L, de Wever O, Zhang Y, Cervera P, Aline-Fardin A, Mercher T, Gaulard P, Nyga R, Jeziorowska D, Douay L, Vainchenker W, Louache F, Gespach C, Solary E, Coppo P. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 2014;28(2):338–348. doi: 10.1038/leu.2013.157. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chemistry & biology. 2013;20(2):146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, Rettig WJ, Meyerson M, Solca F, Greulich H, Wong KK. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown JR. Ibrutinib (PCI-32765), the first BTK (Bruton’s tyrosine kinase) inhibitor in clinical trials. Current hematologic malignancy reports. 2013;8(1):1–6. doi: 10.1007/s11899-012-0147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montgomery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, Che Y. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. Journal of medicinal chemistry. 2014;57(23):10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- 23.Zambaldo C, Sadhu KK, Karthikeyan G, Barluenga S, Daguer JP, Winssinger N. Selective affinity-based probe for oncogenic kinases suitable for live cell imaging. Chemical Science. 2013;4(5):2088–2092. [Google Scholar]
- 24.London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermancic P, Bonnet R, Shoichet BK, Taunton J. Covalent docking of large libraries for the discovery of chemical probes. Nature chemical biology. 2014;10(12):1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goedken ER, Argiriadi MA, Banach DL, Fiamengo BA, Foley SE, Frank KE, George JS, Harris CM, Hobson AD, Ihle DC, Marcotte D, Merta PJ, Michalak ME, Murdock SE, Tomlinson MJ, Voss JW. Tricyclic Covalent Inhibitors Selectively Target Jak3 through an Active Site Thiol. The Journal of biological chemistry. 2015;290(8):4573–4589. doi: 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffe M, Mayeux P, Gouilleux F, Berger R, Gisselbrecht S, Ghysdael J, Bernard OA. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood. 2000;95(6):2076–2083. [PubMed] [Google Scholar]
- 27.(a) Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nature reviews Drug discovery. 2008;7(5):391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]; (b) Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, King RW, Lee JD, Herrgard S, Zarrinkar P, Gray NS. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chemistry & biology. 2011;18(7):868–879. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lebakken CS, Riddle SM, Singh U, Frazee WJ, Eliason HC, Gao Y, Reichling LJ, Marks BD, Vogel KW. Development and applications of a broad-coverage, TR-FRET-based kinase binding assay platform. Journal of biomolecular screening. 2009;14(8):924–935. doi: 10.1177/1087057109339207. [DOI] [PubMed] [Google Scholar]
- 29.Warmuth M, Kim S, Gu XJ, Xia G, Adrian F. Ba/F3 cells and their use in kinase drug discovery. Current opinion in oncology. 2007;19(1):55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 30.Sanda T, Tyner JW, Gutierrez A, Ngo VN, Glover J, Chang BH, Yost A, Ma W, Fleischman AG, Zhou W, Yang Y, Kleppe M, Ahn Y, Tatarek J, Kelliher MA, Neuberg DS, Levine RL, Moriggl R, Muller M, Gray NS, Jamieson CH, Weng AP, Staudt LM, Druker BJ, Look AT. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer discovery. 2013;3(5):564–577. doi: 10.1158/2159-8290.CD-12-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A Randomized, Double-Blind, Placebo-Controlled, Twelve-Week, Dose-Ranging Study of Decernotinib, an Oral Selective JAK-3 Inhibitor, as Monotherapy in Patients With Active Rheumatoid Arthritis. Arthritis Rheumatol. 2015;67(2):334–343. doi: 10.1002/art.38949. [DOI] [PubMed] [Google Scholar]
- 32.Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23(8):1441–1445. doi: 10.1038/leu.2009.50. [DOI] [PubMed] [Google Scholar]
- 33.Kwiatkowski N, Jelluma N, Filippakopoulos P, Soundararajan M, Manak MS, Kwon M, Choi HG, Sim T, Deveraux QL, Rottmann S, Pellman D, Shah JV, Kops GJ, Knapp S, Gray NS. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nature chemical biology. 2010;6(5):359–368. doi: 10.1038/nchembio.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, He Y, Ruiz CH, Koenig M, Cameron MD, Vojkovsky T. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug metabolism and disposition: the biological fate of chemicals. 2009;37(6):1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer discovery. 2014;4(9):1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortes JR, Perez GM, Rivas MD, Zamorano J. Kaempferol inhibits IL-4-induced STAT6 activation by specifically targeting JAK3. J Immunol. 2007;179(6):3881–3887. doi: 10.4049/jimmunol.179.6.3881. [DOI] [PubMed] [Google Scholar]
- 37.O’Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Molecular immunology. 2007;44(10):2497–2506. doi: 10.1016/j.molimm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 38.(a) Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, Bao L, Contreras CM, Li D, Chen L, Krishnamurthy J, Koivunen J, Chirieac LR, Padera RF, Bronson RT, Lindeman NI, Christiani DC, Lin X, Shapiro GI, Janne PA, Johnson BE, Meyerson M, Kwiatkowski DJ, Castrillon DH, Bardeesy N, Sharpless NE, Wong KK. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]; (b) Herter-Sprie GS, Korideck H, Christensen CL, Herter JM, Rhee K, Berbeco RI, Bennett DG, Akbay EA, Kozono D, Mak RH, Mike Makrigiorgos G, Kimmelman AC, Wong KK. Image-guided radiotherapy platform using single nodule conditional lung cancer mouse models. Nature communications. 2014;5:5870. doi: 10.1038/ncomms6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eling TE, DiAugustine RP. A role for phospholipids in the binding and metabolism of drugs by hepatic microsomes. Use of the fluorescent hydrophobic probe 1-anilinonaphthalene-8-sulphonate. The Biochemical journal. 1971;123(4):539–549. doi: 10.1042/bj1230539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Z’-Lyte assays: http://www.lifetechnologies.com/content/dam/LifeTech/migration/files/drug-discovery/pdfs.par.60256.file.dat/20130430%20ssbk%20customer%20protocol%20and%20assay%20conditions.pdf; TTK assay: http://tools.lifetechnologies.com/content/sfs/manuals/TTK_LanthaScreen_Binding.pdf
- 41.Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106(3):996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabsch W. Xds. Acta crystallographica Section D, Biological crystallography. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Evans P. Scaling and assessment of data quality. Acta crystallographica Section D, Biological crystallography. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]; (b) Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological crystallography. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER version 2.10.2. Cambridge, United Kingdom: Global Phasing Ltd; 2011. [Google Scholar]
- 48.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta crystallographica Section D, Biological crystallography. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 49.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, Gray NS. Discovery of potent and selective covalent inhibitors of JNK. Chemistry & biology. 2012;19(1):140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. Journal of the American Society for Mass Spectrometry. 1998;9(3):225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- 51.Ficarro SB, Zhang Y, Lu Y, Moghimi AR, Askenazi M, Hyatt E, Smith ED, Boyer L, Schlaeger TM, Luckey CJ, Marto JA. Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Analytical chemistry. 2009;81(9):3440–3447. doi: 10.1021/ac802720e. [DOI] [PubMed] [Google Scholar]
- 52.Askenazi M, Parikh JR, Marto JA. mzAPI: a new strategy for efficiently sharing mass spectrometry data. Nature methods. 2009;6(4):240–241. doi: 10.1038/nmeth0409-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGuire VA, Gray A, Monk CE, Santos SG, Lee K, Aubareda A, Crowe J, Ronkina N, Schwermann J, Batty IH, Leslie NR, Dean JL, O’Keefe SJ, Boothby M, Gaestel M, Arthur JS. Cross talk between the Akt and p38alpha pathways in macrophages downstream of Toll-like receptor signaling. Molecular and cellular biology. 2013;33(21):4152–4165. doi: 10.1128/MCB.01691-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, Reibel JB, Soucheray M, Cohoon TJ, Janne PA, Meyerson M, Hayes DN, Shapiro GI, Shimamura T, Sholl LM, Rodig SJ, Freeman GJ, Hammerman PS, Dranoff G, Wong KK. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer discovery. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.