

Abstract
Mutations in ubiquilin 2 (Ubqln2) is linked to amyotrophic lateral sclerosis and frontotemporal lobar degeneration. A foremost question regarding Ubqln2 pathogenesis is whether pathogenically mutated Ubqln2 causes neuron death via a gain or loss of functions. To better understand Ubqln2 pathobiology, we created Ubqln2 transgenic and knockout rats and compared phenotypic expression in these novel rat models. Overexpression of Ubqln2 with a pathogenic mutation (P497H substitution) caused cognitive deficits and neuronal loss in transgenic rats at the age of 130 days. In the transgenic rats, neuronal loss was preceded by the progressive formation of Ubqln2 aggregates and was accompanied by the progressive accumulation of the autophagy substrates p62 and LC3-II and the impairment of endosome pathways. In contrast, none of these pathologies observed in mutant Ubqln2 transgenic rats was detected in Ubqln2 knockout rats at the age of 300 days. Together, our findings in Ubqln2 transgenic and knockout rats collectively suggest that pathogenic Ubqln2 causes neuron death mainly through a gain of unrevealed functions rather than a loss of physiological functions.
Keywords: Frontotemporal lobar degeneration, FTLD, Amyotrophic lateral sclerosis, ALS, Ubqln2, p62, Rats, Protein aggregation, Autophagy, Knockout
Introduction
Mutation in ubiquilin 2 (Ubqln2) and sequestosome-1 (SQSTM1 or p62) has been linked to amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD), in which protein aggregation is prominent pathology [5, 27, 29]. The occurrence of protein aggregates indicates that protein degradation systems are overloaded with client proteins or functionally impaired [19]. Cells possess two degradation systems—ubiquitin–proteasome and autophagy–lysosome systems—that cooperatively degrade unfolded and misfolded proteins as well as old and damaged organelles [11]. Short-lived proteins are degraded by the ubiquitin–proteasome system, but long-lived proteins, protein aggregates, and old organelles are degraded by the autophagy–lysosome system [18, 20]. Whereas p62 is required for the formation and degradation of polyubiquitin-containing bodies [17], Ubqln2 shuttles between the nucleus and the cytoplasm to perform various functions related to protein degradation via autophagy and proteasomes [3, 16, 23, 36]. A single Ubqln orthologue is identified in yeast [10], but four Ubqln isoforms are found in mammals with distinct expression patterns [3, 36]. Ubqln1 is ubiquitously expressed, Ubqln2 and 4 have somewhat more restricted but still widespread expression, and UBQLN3 is expressed exclusively in testes [3, 36]. Ubqln proteins have limited functional specialization [16, 23]. Ubqln2 contains an ubiquitin-like domain (UBL) at the N-terminus and an ubiquitin-associated domain (UBA) at the C-terminus [16]. Ubqln2-linked disease is transmitted in a dominant fashion with reduced penetrance in females [5]. Because Ubqln2 is an X-linked gene, all affected males are homozygotes and affected females are heterozygotes. If pathogenic mutation mainly causes a loss of function in Ubqln2, the low penetrance in females may result from retention of a normal allele that compensates for functional loss in the mutant allele; however, X-inactivation may complicate the interpretation of the mutation effects, particularly when one of the two alleles on the X chromosome is partially inactivated [24]. As such, expression of mutated Ubqln2 may be less in females than in males, leading to lower penetrance in females when mutated Ubqln2 mainly causes a gain of function. To understand Ubqln2 pathogenesis, we must examine foremost query whether pathogenically mutated Ubqln2 causes neuron death mainly via a gain or loss of functions.
Animal models provide a valuable tool for dissecting the mechanisms of disease-linked mutations. Giving the complexity of central nervous system (CNS) and the species-specific physiologies, diverse model systems are often required for a better understanding of neurodegenerative diseases, as they may provide inputs on the varying aspects of disease mechanisms. Whereas rodents are an ideal animal model for functional genetics, the rat shows advantage over the mouse in terms of its long history of use in physiological and pharmaceutical studies and its large size for easy use in behavioral tests and surgical operation [2, 4, 21, 31]. A success in modeling TDP-43 and FUS pathologies in rats proves transgenic rats a valuable tool for dissecting the mechanisms of neurodegeneration in ALS and FTLD. Using Ubqln2 transgenic and knockout rat models, we examined whether pathogenically mutated Ubqln2 causes neuron death mainly via a gain or a loss of function. Pathogenic mutation of Ubqln2 predisposed it to aggregation and aggregated mutant Ubqln2 entrapped its wildtype counterparts in protein inclusions. Mutant Ubqln2 aggregation preceded neuron death and cognitive deficit and caused progressive accumulation of the autophagy substrate p62 in transgenic rats. None of these pathological changes was observed in Ubqln2 knockout rats. These findings suggest that mutant Ubqln2 causes neuron death via a gain of function rather than a loss of function.
Materials and methods
Production of Ubqln2 transgenic and knockout rats
Transgenic rats were created and maintained on Sprague– Dawley genomic background as described [12, 13, 33]. CaMKα2-tTA transgenic rats have been characterized in previous studies [12, 34]. The open reading frames (ORF) of human Ubqln2 were amplified by PCR from the cDNA pools that were derived from human HEK293 cells. P497H substitution was introduced into human Ubqln2 ORF by PCR-based mutagenesis. The engineered ORF of human Ubqln2 was inserted between the tetracycline-responsive element (TRE promoter) and SV40 late poly (A) sequence as previously described [39]. Transgenic rats were identified by PCR assay with the following primers: 5′-TTGTTTGTGGATCGCTGTGA-3′ (forward) and 5′-GACAAACTTCACGTCAGGGT-3′ (reverse). Copy number of the transgenes was determined by quantitative PCR with the same set of primers and the copy standard was established by mixing transgenic DNA with rat genomic DNA as described [40]. TRE-Ubqln2 transgenic lines were crossed with CaMKα2-tTA transgenic line to produce double-transgenic offspring, in which the Ubqln2 transgene was expressed in the forebrain neurons [12, 34]. Breeding rats were given Doxycycline (Dox) in drinking water (50 μg/ml) to suppress transgene expression during embryonic development. Mutant Ubqln2 transgenic rats and their controls were deprived of Dox at birth to allow transgene expression and disease induction.
Ubqln2 knockout rats were created by TALEN-assisted gene modification in fertilized rat eggs. Following the instruction provided by Doyle and colleagues [6], a pair of transcription activator-like effector nuclease (TALEN) targeting sequences was selected to bind and cleave rat Ubqln2 (left: 5′-TCCTGAAAGATCAAGACACC-3′; right: 5′-CATGATGGACTGACTGTTCA-3′). The mRNA encoding selected TALEN was synthesized and modified for translation in mammals and was injected into fertilized rat single-cell embryos [32]. TALEN-mediated gene modification was identified by PCR assay (forward primer: 5′-CAGCAGTTCAAGGAAGCGAT-3′; reverse primer: 5′-CGTGGTCGTCGTGCTCGTTC-3′). A deletion of 19 nucleotides (deleted sequence: 5′-CCTTGATCCAGCATGGCAT-3′) was identified in a knockout rat whose Ubqln2 was destroyed by a premature stop codon. Ubqln2 knockout rats were crossed with wildtype Sprague–Dawley for 4 generations before they were used for analysis.
Rat's behavior tests
Spatial learning and memory tasks were examined with a Barnes Maze (Med Associates) as described [14]. In brief, rats were given one training session and four test sessions for 5 consecutive days. During training or testing sessions, rats were placed in the same initial orientation inside a transparent cylinder (start box) that was located at the center of the maze disk and the rats remained in the start box for 1 min such that a standard starting context was ensured. Latency to locate the fixed escaping box was calculated from the time testing started to the time when the animal entered, or its four paws touched, the box. Rat's mobility was monitored with Open Field Activity assay (Med Associates), which measured the total distance a rat traveled within 10 min [13].
Cresyl violet staining and stereological cell counting
Total number of neurons in the frontal cortex and dentate gyrus were estimated with stereological cell counting as described [12, 34]. In brief, rat's forebrains were cut into coronal sections (20 μm) with a Cryostat. Tissue sections containing the frontal cortex (from the apical forebrain to the first occurrence of corpus callosum) or dentate gyrus were collected and every 10th section was counted for neurons in defined brain regions. Tissue sections were stained with Cresyl violet and mounted in sequential order (rostral–caudal). The number of targeted neurons was estimated using a fractionator-based unbiased stereology software program (Stereologer), which was run on a PC computer that was attached to a Nikon 80i microscope with a motorized XYZ stage (Prior). The detail of stereological cell counting was previously described [12, 34].
Antibody information
The following primary antibodies were used for immunostaining: mouse monoclonal anti-GAPDH (Abcam), rabbit anti-GFP and anti-LAMP2a (Abcam), rabbit anti-GFAP (DAKO, North America), and rabbit anti-Iba-1 (Wako Chemicals USA), mouse monoclonal anti-FLAG and rabbit polyclonal anti-LC3 (Novus), mouse monoclonal antibody against Lamin A/C (Cell Signaling Technology), mouse monoclonal antibody recognizing both human and rat Ubqln2 (Novus), rabbit polyclonal antibody against p62 (Novus), rabbit polyclonal antibody to EEA1 (Cell Signaling Technology), rabbit polyclonal antibody against phosphor-tau (Sigma), rabbit polyclonal antibody to TDP-43 (Proteintech), and mouse monoclonal anti-beta-actin (Sigma). Primary antibodies were used at the lowest dilutions recommended by manufacturers. Antibodies to human or rat Ubqln2 were produced by immunizing rabbits with synthetic peptides at Genemed: (N-terminal)-LAGA-NAPQLPN (human) and (N-terminal)-TTTTTAAAPPAATTSSAP (rat). Antiserum was affinity-purified with a peptide affinity column (Pierce).
Golgi staining and electromicroscopy
Golgi impregnation method was used to visualize the neurites of rat's cortical neurons and was done with a kit (FD NeuroTechnologies) as previously described [34]. Ultrastructure of cellular organelles was detected by electromicroscopy as described [14, 34, 39]. In brief, fixed tissues were embedded in Epon 812 and cut into thin sections (50 nm). Sections were stained with uranyl acetate and lead citrate and were examined under a transmission electron microscope (EM facility affiliated to the Department of Pathology, Thomas Jefferson University).
Immunofluorescence staining and microscopy
Animals were perfused with 4 % paraformaldehyde under deep anesthesia and animal brains were dissected after perfusion. The brains were further fixed in the same fixative at 4 °C overnight and then dehydrated in 30 % sucrose as described [13, 14]. Coronal sections (15 μm) of rat forebrain were immunostained first with primary antibodies and then with dye-labeled secondary antibodies. Immunoreactivity for specified proteins was observed under Nikon fluorescence microscope and documented with a digital camera. For determining protein colocalization, tissue sections were examined with a confocal microscope (Imaging Facility of Kimmel Cancer Center at Jefferson). Single-layer images were scanned with a Zeiss LSM510 META confocal system.
Immunoblotting, immunoprecipitation and protein solubility
Rat tissues were homogenized in RIPA buffer and proteins in tissue lysates were separated on SDS-PAGE as described [39]. Immunoreactivity for a target protein was detected with specified primary antibodies after resolved proteins were transferred onto nitrocellulose membrane. For immunoprecipitation, plasmids were transfected into HEK293 cells using lipofectamine-2000. Cells were initially lysed in lysis buffer (Promega) and fully broken by sonication as described [37]. Cell lysates were cleared by centrifugation 10 min at 4 °C and 500 μg of total protein per sample was incubated with FLAG binding resin to precipitate tagged proteins and their partners. Bound proteins were eluted with SDS sample buffer and boiled for 10 min to dissociate protein complexes. Eluted proteins were detected by immunoblotting with specified antibodies.
Ubqln2 solubility in rat tissue was examined with a modified protocol [15]. Rat tissues were sonicated in RIPA buffer containing 1 % Triton X-100 and 0.1 % SDS and tissue lysates were centrifuged at 100,000g for 30 min to produce Triton X-100 soluble fraction. The resulting pellets containing protein aggregates were washed twice in cold PBS and were dissolved in lysis buffer containing 1 % SDS. Ubqln2 solubility in cultured cells was examined using the same procedure, but the resulting pellets were dissolved in urea (7 M). A mixture of protease inhibitors was added to lysates during the procedure. Soluble and insoluble Ubqln2 was detected using standard SDS-PAGE.
Statistical analysis
Numbers of neurons in frontal cortex or dentate gyrus were compared between Ubqln2 transgenic rats and control rats using paired t tests. A p value of less than 0.05 was considered statistical significance.
Study approval
Animal use was in accord with NIH guidelines and the animal use protocol was approved by the Institutional Animal Care and Use Committees at Thomas Jefferson University.
Results
Pathogenic mutation predisposes Ubqln2 to aggregation in cultured cells
Mutation in Ubqln2 is linked to ALS and FTLD and one prominent pathology observed in these patients is Ubqln2-positive inclusion [5, 29, 35]. To understand Ubqln2 pathobiology, we first examined the effect of pathogenic mutation on Ubqln2 biochemical properties. Human Ubqln2, with or without a pathogenic mutation (i.e., P497H substitution), was tagged with FLAG and thus tagged Ubqln2 was detected with FLAG antibody. We examined the effect of pathogenic mutation on Ubqln2 solubility using HEK293 cells which showed the high efficiency of transient transfection (Fig. 1a). While mutant Ubqln2 was constantly detected in detergent-insoluble fraction, wildtype Ubqln2 was barely detected in detergent-insoluble fraction (Fig. 1a). Thus, pathogenic mutation increased the propensity of Ubqln2 to aggregate in vitro.
Fig. 1.
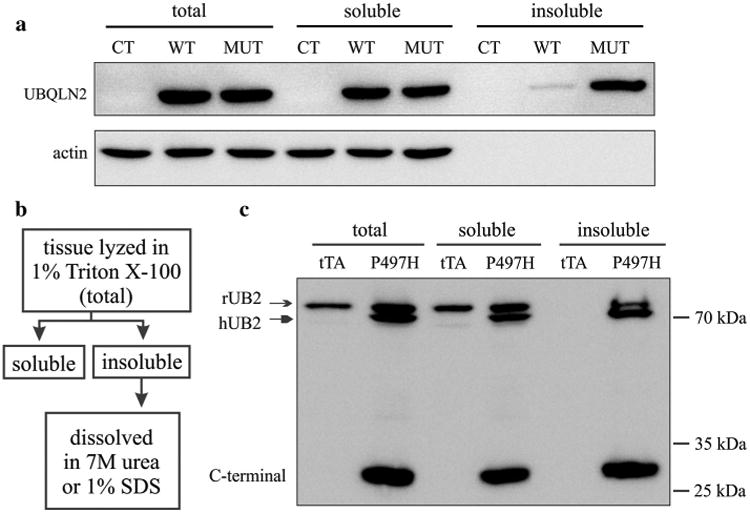
Mutant Ubqln2 was predisposed to aggregation in vitro and in vivo. a Western blotting revealed that mutant Ubqln2 was enriched in Triton X-100 insoluble fraction. HEK293 cells were transiently transfected with plasmids expressing wildtype (WT) or mutant (MUT, P497H substitution) human Ubqln2 that was tagged with FLAG. Triton X-100 soluble and insoluble fractions were isolated as described in “Materials and methods” and displayed in b. b Shows the procedure to separate insoluble from soluble Ubqln2 in cultured cells (dissolved in 7 M urea) and transgenic rats (dissolved in 1 % SDS). c Western blotting revealed that the full-length and C-terminal fragments of Ubqln2 were enriched in Triton X-100 insoluble fraction. Frontal cortex was dissected from CaMKα2-tTA single-transgenic (tTA: the control) and CaMKα2-tTA/TRE-hUBQLN2-P497H double-transgenic rats (P497H) at the age of 80 days. Immunoreactivity was detected with an antibody that recognizes both human and rat Ubqln2
Mutant Ubqln2 aggressively aggregates in transgenic rats
To verify our findings in cellular model, we created transgenic rats and examined the solubility of mutant Ubqln2 in rat's brains. A tetracycline-inducible gene expression system has been developed for transgenic rats in our lab [13, 14, 33, 39]. A single-copy CaMKα2-tTA transgenic line proves to express transgene specifically in rat's forebrain neurons [12, 34]. Taking advantage of existing tTA transgenic lines, we used tetracycline-response element (TRE) to drive Ubqln2 transgene and established two TRE-hUB2-P497H transgenic rat lines (Fig. S1), in which transgene copy was determined by quantitative PCR. Line-2 harbored 2 copies of TRE-hUB2-P497H transgene and Line-20 harbored 20 copies of the transgene (Fig. S1). Individual TRE-hUB2-P497H lines were crossed with the CaMKα2-tTA line to produce double-transgenic offspring, in which mutant Ubqln2 was expressed in the forebrain neurons [12]. Both Ubqln2 transgenic lines expressed human Ubqln2 at moderate levels (Fig. S1a-c). Quantification of immunoblotting confirmed that line-20 expressed human Ubqln2 protein approximately at the two times of that expressed in line-2. We selected line-20 for the following analyses. Using Ubqln2 transgenic rats as an in vivo model, we examined the solubility of mutant Ubqln2 in rat's brains. Tissue lysates were separated into Triton X-100 soluble and insoluble fractions. Ubqln2 immunoreactivity was detected only in the soluble fraction in the control rats, but it was detected in both the soluble and insoluble fractions in mutant Ubqln2 transgenic rats (Fig. 1b, c). Rat endogenous Ubqln2 was detected in the insoluble fraction, suggesting that mutant Ubqln2 entrapped wildtype Ubqln2 in its inclusions. Our finding in transgenic rats was in concert with our observation in cultured cells.
We examined the progression of Ubqln2 aggregation in transgenic rats. Ubqln2 inclusions are observed in the nucleus and cytoplasm of affected neurons in ALS and FTLD [5, 29, 35]. This property of mutant Ubqln2 was recapitulated in transgenic rats (Fig. 2). While Ubqln2 mainly resided in the cytoplasm in wildtype rats (Fig. 2), Ubqln2 aggressively formed inclusions in the cytoplasm and nucleus in mutant Ubqln2 transgenic rats (Fig. 2). Similar Ubqln2 inclusions were observed in the two expressing lines (Fig. 2 and Fig. S1). We examined Ubqln2 distribution first with an antibody that recognized both human and rat Ubqln2 and we found that Ubqln2 aggressively formed inclusions first in the cytoplasm and then in the nucleus (Fig. 2). To examine the effect of mutant Ubqln2 on wildtype Ubqln2 distribution, we generated species-specific antibodies recognizing either human or rat Ubqln2 protein (Fig. S2a). Human-specific antibody detected that mutant Ubqln2 progressively formed inclusions in the cytoplasm and nuclei (Fig. S2). Similar to mutant human Ubqln2, wildtype rat Ubqln2 was also detected as inclusions in the cytoplasm and nuclei (Fig. 2n–p). Immunoprecipitation revealed that mutant human Ubqln2 interacted with both human and rat wildtype Ubqln2 (Fig. 2t–u), suggesting that mutant Ubqln2 induced wildtype Ubqln2 to aggregate in transgenic rats.
Fig. 2.
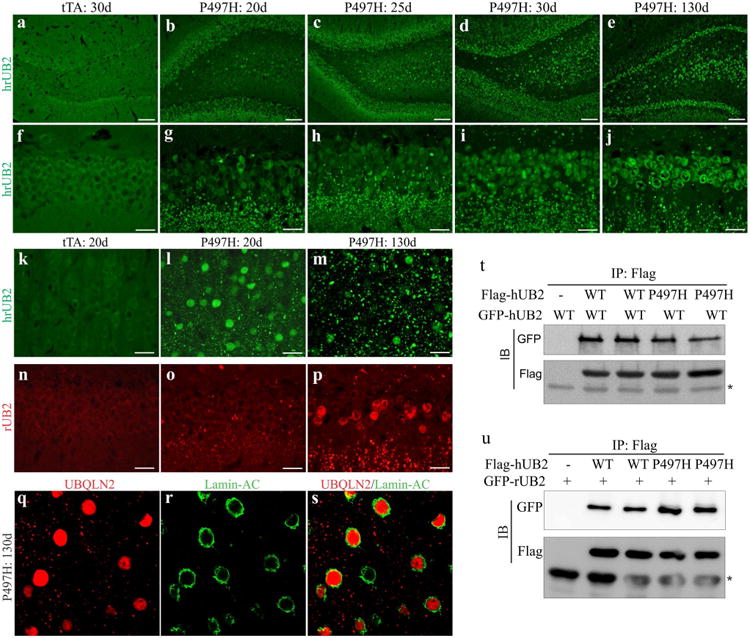
Mutant Ubqln2 aggressively formed inclusions in transgenic rats. a–p Immunostaining revealed that Ubqln2 progressively formed inclusions in the dentate gyrus (a–j, n–p) and cortex (k–m, q–s) in CaMKα2-tTA/TRE-hUBQLN2-P497H double-transgenic rats (P497H), but not in CaMKα2-tTA single-transgenic rats (tTA). Transgenic rats were deprived of Dox at birth to allow transgene expression and were examined at indicated ages (d days). Total Ubqln2 was detected with an antibody that recognized both human and rat Ubqln2 proteins (hrUB2), and rat Ubqln2 was detected with a rat-specific antibody (rUB2). q–s Confocal microscopy revealed that Ubqln2 inclusions were accumulated in the cytoplasm and nucleus of affected cells. Scale bars 100 μm (a–e) and 30 μm (f–p). t–u Immunoprecipitation (IP) followed by immunoblotting (IB) revealed that mutant human Ubqln2 (hUB2: P497H) bound to wildtype (WT) human (hUB2) and rat (rUB2) Ubqln2. Asterisk indicates IgG heavy chain. HEK293 cells were transfected with indicated plasmid and were analyzed 32 h after transfection
Mutant Ubqln2 aggregation precedes neuronal death in transgenic rats
We examined the outcomes of mutant Ubqln2 expression and aggregation in transgenic rats. Behavioral analysis revealed that spatial learning was impaired in Ubqln2 transgenic rats (Fig. 3a), suggesting that mutant Ubqln2 expression in the forebrain caused cognitive deficits. As Ubqln2 is an X-linked gene, disease onset is earlier in male than female patients with Ubqln2 mutation [5]. We did not notice difference in the pathology between male and female transgenic rats. Cresyl violet staining revealed a reduction in neuron density in the dentate gyrus and cortex (Fig. 3b– i), implying that neurons were lost in the brain regions of Ubqln2 transgenic rats. Using unbiased stereological cell counting, we estimated the number of neurons in the cortex and dentate gyrus and found a significant loss of neurons in the brain regions examined (Fig. 3j, k). Intriguingly, neuronal loss was detected only at the late ages (Fig. 3j), but was not detected in rats at the early ages when substantial Ubqln2 aggregates were detected in the brain regions (Fig. 2a–j). Ubqln2 aggregation preceded neuron loss in mutant Ubqln2 transgenic rats. Neuronal impairment was confirmed by Golgi staining (Fig. 3l, m). Compared to wildtype rats (Fig. 3l), mutant rats displayed a distorted structure of cortex, with neurons deprived of neurites (Fig. 3m). In response to neuronal damage, astrocytes and microglia became reactive as revealed by immunostaining (Fig. 3n–q). Overexpression of mutant Ubqln2 in transgenic rats recapitulated the core phenotypes of Ubqln2-linked FTLD.
Fig. 3.
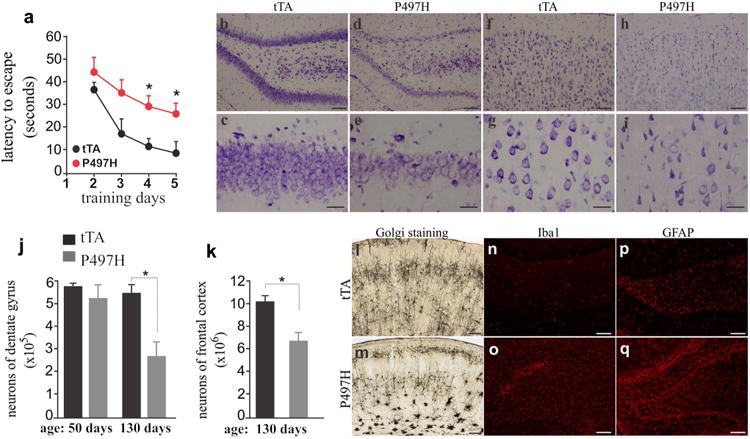
Expression of mutant Ubqln2 in rats caused learning deficiency and neuronal death. a Barnes maze assay revealed spatial learning deficiency in CaMKα2-tTA/TRE-hUBQLN2-P497H double-transgenic rats (P497H) as compared to CaMKα2-tTA single-transgenic rats (tTA) at the age of 130 days. Rats were trained to locate an escape hole in a Barnes maze on the first day and then were examined daily for improvement in finding a fixed escape hole. Data are mean ± SD (n = 5). *p < 0.05. b–i Cresyl violet staining revealed neuronal loss in mutant Ubqln2 transgenic rats (P497H) as compared to the control rats (tTA). Rats of 130 days old were examined for neuronal loss in the dentate gyrus (b–e) and frontal cortex (f–i). j, k Unbiased stereological cell counting confirmed the loss of neurons in the dentate gyrus and frontal cortex of P497H rats. Data are mean ± SD (n = 4). *p < 0.05. l, m Golgi staining detected impaired neurons in P497H rats aging 130 days as compared to age-matched tTA rats. n–q Immunostaining revealed glial activation in P497H rats (o, q) as compared to tTA rats (n, p). The dentate gyrus of transgenic rats was examined for immunoreactivity with microglia marker Iba1 and astrocyte marker GFAP. Scale bars 100 μm (b–h, n–q), 30 μm (c–i), and 200 μm (l–m)
Mutant Ubqln2 induces accumulation of the autophagy substrates p62 and LC3-II
Progressive Ubqln2 aggregation was a prominent feature of mutant Ubqln2 transgenic rats (Figs. 1, 2). Ubiquilin proteins are involved in autophagic degradation [23, 36], and thus we examined the effect of mutant Ubqln2 on autophagy function. We first used a cellular model to determine the effect of pathogenic Ubqln2 on autophagy–proteolytic function (Fig. 4a, b). HEK293 cells were transfected with a plasmid expressing wildtype or a mutant (P497H) human Ubqln2 which was tagged with FLAG. The cells were treated with MG132 which inhibits proteasome to augment the deficit in autophagy–proteolytic activities [30]. Overexpression of wildtype or mutant Ubqln2 in cultured cells caused LC3-II accumulation and this detrimental effect was augmented by pathogenic mutation in Ubqln2 (Fig. 4a, b). The effect of mutant Ubqln2 on autophagic degradation was verified in mutant Ubqln2 transgenic rats (Fig. 3c). Both p62 and LC3-II are degraded via autophagy pathway and they are usually used as measures for the proteolytic activity of autophagy [17]. Expression of mutant Ubqln2 in rats led to progressive accumulation of p62 and LC3-II (Fig. 4c–o), indicating that that autophagic degradation was impaired by mutant Ubqln2 expression in vivo. Mutant Ubqln2 aggregated with p62 which is recently linked to ALS and FTLD [27]. We further examined whether mutant Ubqln2 entraps the other proteins whose encoding genes are associated with ALS and FTLD. We found that TDP-43, FUS, and tau were not detected in Ubqln2 inclusions (Fig. S3). Mutant Ubqln2 selectively entrapped certain proteins in its inclusions (Fig. 4). Together, our findings in cell and animal models suggest that pathogenic Ubqln2 impaired the proteolytic function of autophagy.
Fig. 4.
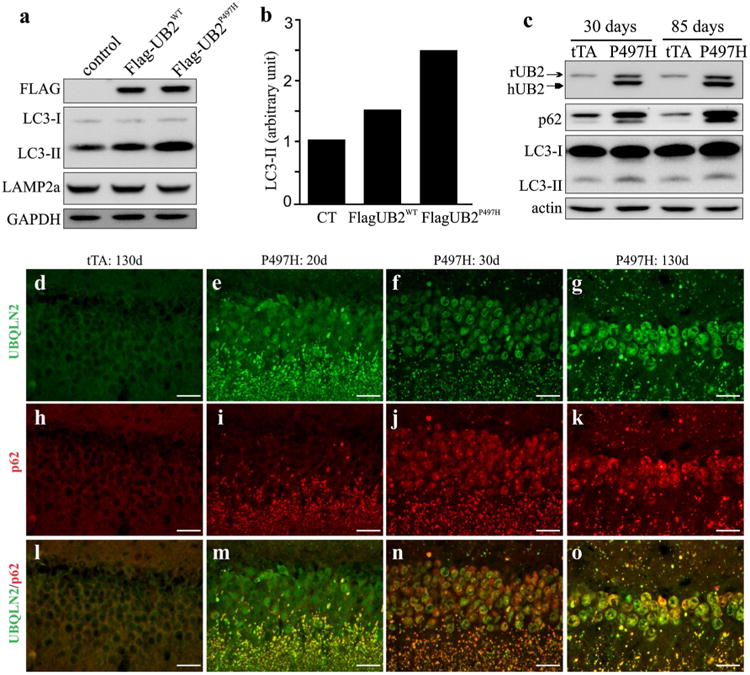
Expression of mutant Ubqln2 led to p62 and LC3-II accumulation in vitro and in vivo. a Immunoblotting revealed that LC3-II accumulated in HEK293 cells expressing mutant Ubqln2. Wildtype and mutant human Ubqln2 proteins were tagged with FLAG. b Immunoblotting density was quantified and calculated as a ratio to the control. HEK293 cells were transfected with empty vector (CT), wildtype Ubqln2-expressing vector (WT), or mutant (P497H substitution) Ubqln2-expressing vector. Cells were treated with the proteasome inhibitor MG132 at 24 h after transfection and were harvested for analysis at 4 h after MG132 treatment. c Immunoblotting revealed accumulation of LC3-II and p62 in mutant Ubqln2 transgenic rats. Frontal cortex was examined for LC3-II and p62 expression. d–o Immunostaining revealed that p62 progressively accumulated in mutant Ubqln2 transgenic rats. All scale bars 30 μm
Mutant Ubqln2 interferes with endosome pathway
Mutant Ubqln2 negatively affected autophagy–proteolytic function (Fig. 4). As multivesicular bodies are required for autophagy maturation [8, 28], we examined the effect of mutant Ubqln2 on endosome pathway. Immunostaining revealed that mutant Ubqln2 expression led to a reduction in early endosome antigen 1 (EEA1) immunoreactivity (Fig. 5a–l). EEA1 is a marker for early endosomes [28], and its reduced immunoreactivity suggests a decrease in early endosomes. By contrast, expression of the lysosomal marker LAMP2a was unaltered in Ubqln2 transgenic rats (Fig. 5m). Electromicroscopy revealed that neurons displayed swollen Golgi complex at early ages (Fig. 5o) and developed vacuoles at later ages (Fig. 5p). Expression of mutant Ubqln2 in rats interfered with endosome pathways.
Fig. 5.
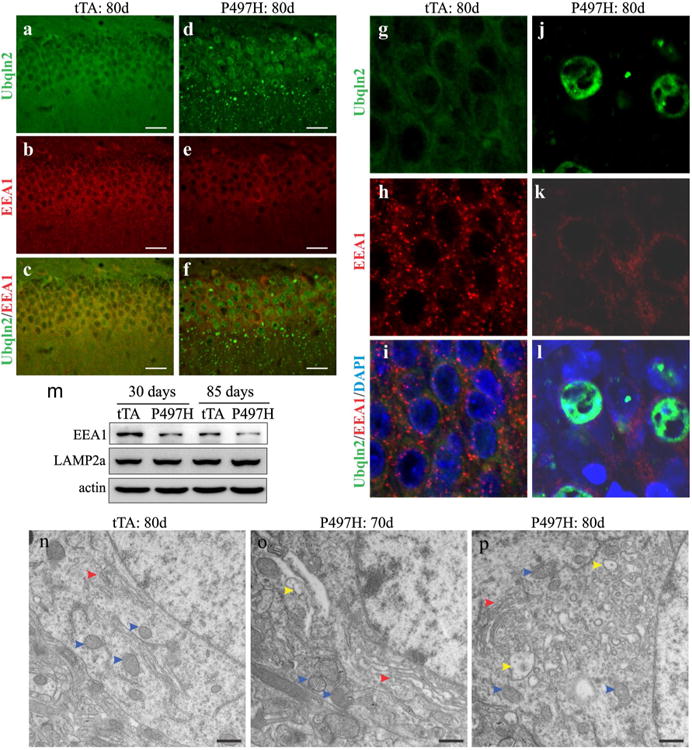
Expression of mutant Ubqln2 in rats interfered with endosome pathways. a–l Immunostaining revealed that early endosomes labeled with EEA1 were decreased in mutant Ubqln2 transgenic rats. CaMKα2-tTA single-transgenic (tTA) and CaMKα2-tTA/TRE-hUBQLN2-P497H double-transgenic rats (P497H) were examined at indicated ages. m Immunoblotting revealed the expression of EEA1 and LAMP2a in mutant Ubqln2 transgenic rats. n–p Electromicroscopy revealed that vacuoles were accumulated in Ubqln2 transgenic rats. The microstructure of cortical neurons was examined for the transgenic control (tTA) and the mutant Ubqln2 transgenic (P497H) rats. Red arrowheads point to Golgi complex, blue arrowheads point to mitochondria, and yellow arrowheads point to vacuoles. Scale bars 30 μm (a–f) and 500 nm (n–p)
Ubqln2 deficiency causes no abnormality in Ubqln2 knockout rats
Mutant Ubqln2 formed inclusions with wildtype Ubqln2 in transgenic rats (Figs. 1, 2), implying that mutant Ubqln2 may induce a self-deficiency to disrupt cellular functions. To examine this possibility, we generated Ubqln2 knockout rats using TALEN-assisted gene modification (Fig. 6). Ubqln2 is mainly expressed in the CNS and its deficiency may affect neuronal functions. A TALEN targeting the rat Ubqln2 induced a deletion of 19 nucleotides near the 5′-end of Ubqln2 open reading frame and thus destroyed the Ubqln2 gene in knockout rats (Fig. 6b). Open Field assay revealed that mobility was unaltered in Ubqln2 knockout rats (Fig. 6c). Overexpression of mutant Ubqln2 caused cognitive deficits and neuronal loss in transgenic rats at the age of 130 days (Fig. 3). At the age of 300 days, Ubqln2 knockout rats did not display cognitive deficit as revealed by Barnes maze assay (Fig. 6d). Overexpression of mutant Ubqln2 caused p62 accumulation and EEA1 reduction in transgenic rats (Figs. 4, 5), but these pathologies were not detected in Ubqln2 knockout rats at advanced ages (Fig. 6e–h). Nissl staining revealed no abnormal structure in the brain of Ubqln2 knockout rats (Fig. 6i, j). Our findings in Ubqln2 knockout rats suggest that Ubqln2 is not essential to neuronal functions at least in the rats.
Fig. 6.
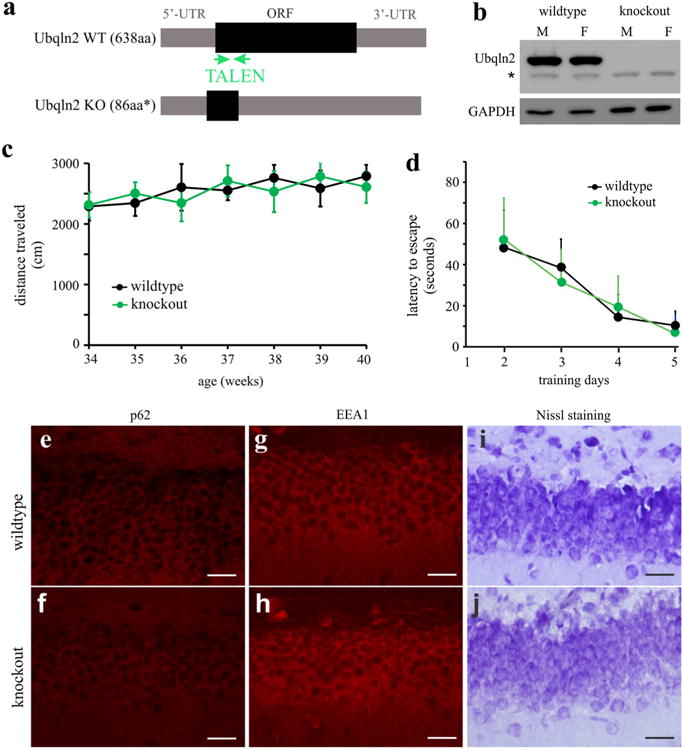
Ubqln2 knockout rats displayed no detectable abnormality. a A strategy that was used to destroy the rat Ubqln2 gene. A pair of TALEN was constructed to cleave rat Ubqln2 within open reading frame (ORF) right after translational start site. TALEN induced a deletion of 19 nt within the ORF and thus introduced a premature stop codon, resulting in a truncated peptide. b Western blotting revealed the complete loss of Ubqln2 in homozygous knockout rats (M male, F female). The symbol asterisk indicates a nonspecific band. c The Open Field assay revealed no abnormality in mobility. Ubqln2 wildtype and knockout rats were examined weekly at indicated ages. Data are mean ± SD (n = 6, equal sex composition). d Barnes maze assay revealed no abnormality in spatial learning in Ubqln2 knockout rats at the age of 280 days. Rats were trained to locate an escape hole in a Barnes maze on the first day and then were examined daily for improvement in finding a fixed escape hole. Data are mean ± SD (n = 6). e–h Immunostaining revealed unaltered p62 and EEA1 expression in Ubqln2 knockout rats. i, j Nissl staining revealed the overall structure of dentate gyrus. All scale bars 30 μm
Discussion
Protein aggregation is a prominent feature of mutant Ubqln2 transgenic rats. As disease was progressing in transgenic rats, mutant Ubqln2 aggressively formed inclusions, which were detected in the cytoplasm (particularly in the neurites) and the nucleus. Mutant Ubqln2 protein was detected in Triton X-100 insoluble fractions, and its reduced solubility in the detergent indicates that mutant Ubqln2 formed protein aggregates in the transgenic rats. Using a cell culture model, we examined whether pathogenic mutation predisposes Ubqln2 to aggregation. In HEK293 cells, transient transgene overexpression predisposed mutant rather than wildtype Ubqln2 to aggregation. Pathogenic mutation affords Ubqln2 the trait of aggregation. Ubqln2 can form dimers [9], and mutant Ubqln2 retains its ability to dimerize with the normal forms of human and rat Ubqln2. Rat endogenous Ubqln2 was indeed detected in mutant Ubqln2 inclusions in the transgenic rats. Ubqln2 is known to shuttle between the nucleus and cytoplasm [16, 23]. Restricting Ubqln2 in protein aggregates likely impairs its normal functions. In ALS and FTLD patients with Ubqln2 mutation, protein inclusions are detected in both the nucleus and the cytoplasm [5, 29, 35]. Thus, Ubqln2 transgenic rats recapitulated a core feature of Ubqln2-linked diseases. In the rat model, protein inclusions were detected at very early ages (20-day-old rats), but neuron loss was detected at late ages (130-day-old rats). Ubqln2 aggregation proceeded neuron death in the transgenic rats, likely contributing to Ubqln2-associated neurotoxicity.
Expression of mutant Ubqln2 in rats caused a deficit in autophagic degradation. As disease was progressing in mutant Ubqln2 transgenic rats, p62 and LC3-II were progressively accumulated in rat's brains. These two substrates of autophagy are widely used as a measure of autophagic degradation and their accumulation indicates the deficiency of the autophagy function [17, 30]. Using a cell culture model, we examined whether pathogenic mutation lends Ubqln2 the ability to interfere with autophagic degradation. Compared to wildtype Ubqln2, mutant Ubqln2 significantly increased LC3-II accumulation when proteasomal function was inhibited. Pathogenic mutation affords Ubqln2 the ability to negatively regulate autophagy. The negative effect on autophagy may not be specific to mutant Ubqln2 as defected autophagy also is observed for other pro-aggregation disease proteins such as alpha-synuclein [30]. As Ubqln2 per se is directly involved in autophagy [26], diverse mechanisms likely underlie autophagy defect in Ubqln2 pathogenesis. The autophagy substrate p62 is essential to neuronal function and survival. p62 binds to poly-ubiquitinated tau and shuttles it to proteasome for degradation [1]. Genetic ablation of p62 suppresses the formation of ubiquitin-positive protein aggregates in neurons [17]. p62 deficiency causes progressive loss of working memory and neurons in knockout mice [25]. Recent studies indicate that mutation of p62 is linked to ALS and FTLD [7, 27]. It is quite intriguing to observe an aberrant interaction between these two disease genes (i.e., p62 and Ubqln2). Altered p62 functions likely relate to Ubqln2 pathogenesis.
Mutant Ubqln2 expression interfered with endosome pathways. Punctate early endosomes marked with EEA1 were significantly reduced in the brain of mutant Ubqln2 transgenic rats. Immunoblotting revealed the decrease of EEA1 protein. Ubqln2 is reported to negatively regulate endocytosis [22]. Mutant Ubqln2 may gain enhanced or retain ability to suppress endosome pathways. Endosomal complexes are required for the maturation of multivesicular bodies which are essential to autophagy maturation [8]. Interrupted endosome pathway is likely associated with autophagy deficiency. As Ubqln2 is involved in endoplasmic reticulum functions [38], we examined the microstructure of endoplasmic reticulum and Golgi complex. These organelles were swollen before abundant vacuoles of varying sizes occurred. Some damaged Golgi complex and endoplasmic reticulum were likely developed into vacuoles in mutant Ubqln2 transgenic rats. Mutant Ubqln2 transgenic rats displayed neuronal death which was preceded by accumulated protein aggregates and was accompanied with interrupted endosome pathways and impaired autophagy. The transgenic rats recapitulated the key features of the diseases related to Ubqln2 mutation and would be useful to the mechanistic study of Ubqln2-linked diseases.
Whereas overexpression of mutant Ubqln2 in transgenic rats caused remarkable proteinopathy and severe neuronal damage, none of these pathological changes were observed in Ubqln2 knockout rats. Deletion of the Ubqln2 caused no abnormality in Ubqln2 knockout rats, which developed no accumulation of the autophagy substrate p62 and no alteration to EEA1 expression. Our findings in Ubqln2 transgenic and knockout rats collectively suggest that pathogenic mutation of Ubqln2 causes neuron death mainly through a gain of unrevealed functions rather than a loss of physiological functions.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NS073829 and NS089701 to H. Z. and NS072113 and NS084089 to X.G.X). The content is the author's responsibility and does not necessarily represent the offcial view of the NIH institutes.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1367-y) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that no conflict of interest exists.
Contributor Information
Qinxue Wu, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
Mujun Liu, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
Cao Huang, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
Xionghao Liu, Department of Neurology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA.
Bo Huang, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
Niansheng Li, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
Hongxia Zhou, Email: Hongxia.zhou@jefferson.edu, Department of Neurology, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA.
Xu-Gang Xia, Email: xugang.xia@jefferson.edu, Department of Pathology, Thomas Jefferson University, JAH506, 1020 Locust Street, Philadelphia, PA 19107, USA.
References
- 1.Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- 2.Bouwknecht JA, Paylor R. Pitfalls in the interpretation of genetic and pharmacological effects on anxiety-like behaviour in rodents. Behav Pharmacol. 2008;19:385–402. doi: 10.1097/FBP.0b013e32830c3658. [DOI] [PubMed] [Google Scholar]
- 3.Conklin D, Holderman S, Whitmore TE, Maurer M, Feldhaus AL. Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene. 2000;249:91–98. doi: 10.1016/s0378-1119(00)00122-0. [DOI] [PubMed] [Google Scholar]
- 4.Cozzi J, Fraichard A, Thiam K. Use of genetically modified rat models for translational medicine. Drug Discov Today. 2008;13:488–494. doi: 10.1016/j.drudis.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 5.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res. 2012;40:W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, Zheng JG, Shi Y, Siddique N, Arrat H, Donkervoort S, Ajroud-Driss S, Sufit RL, Heller SL, Deng HX, Siddique T. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 8.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Isaacs A, Brech A, Stenmark H, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ford DL, Monteiro MJ. Dimerization of ubiquilin is dependent upon the central region of the protein: evidence that the monomer, but not the dimer, is involved in binding presenilins. Biochem J. 2006;399:397–404. doi: 10.1042/BJ20060441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funakoshi M, Sasaki T, Nishimoto T, Kobayashi H. Budding yeast Dsk2p is a polyubiquitin-binding protein that can interact with the proteasome. Proc Natl Acad Sci USA. 2002;99:745–750. doi: 10.1073/pnas.012585199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang C, Tong J, Bi F, Wu Q, Huang B, Zhou H, Xia XG. Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Hum Mol Genet. 2012;21:4602–4614. doi: 10.1093/hmg/dds299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang C, Tong J, Bi F, Zhou H, Xia XG. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122:107–118. doi: 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G, Howley PM. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell. 2000;6:409–419. doi: 10.1016/s1097-2765(00)00040-x. [DOI] [PubMed] [Google Scholar]
- 17.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 18.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59. doi: 10.1016/j.pneurobio.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Jin M, Zhu Z, Wang H, Yu J, Hao Y, Choi A, Ke H, Ma D, Yuan J. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lull ME, Freeman WM, Vrana KE, Mash DC. Correlating human and animal studies of cocaine abuse and gene expression. Ann NY Acad Sci. 2008;1141:58–75. doi: 10.1196/annals.1441.013. [DOI] [PubMed] [Google Scholar]
- 22.N'Diaye EN, Hanyaloglu AC, Kajihara KK, Puthenveedu MA, Wu P, von Zastrow M, Brown EJ. The ubiquitin-like protein PLIC-2 is a negative regulator of G protein-coupled receptor endocytosis. Mol Biol Cell. 2008;19:1252–1260. doi: 10.1091/mbc.E07-08-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.N'Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J, Brown EJ. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009;10:173–179. doi: 10.1038/embor.2008.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollex T, Heard E. Recent advances in X-chromosome inactivation research. Curr Opin Cell Biol. 2012;24:825–832. doi: 10.1016/j.ceb.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 25.Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N, Zhu H, Wooten MC, Diaz-Meco MT, Moscat J, Wooten MW. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 26.Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ. Ubiquilin functions in autophagy and is degraded by chaperonemediated autophagy. Hum Mol Genet. 2010;19:3219–3232. doi: 10.1093/hmg/ddq231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, Grinberg Y, Isaia G, Calvo A, Gentile S, Bruni AC, St George-Hyslop PH, Scarpini E, Gallone S, Pinessi L. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. doi: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Synofzik M, Maetzler W, Grehl T, Prudlo J, Vom Hagen JM, Haack T, Rebassoo P, Munz M, Schols L, Biskup S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging. 2012;33(2949):e13–e17. doi: 10.1016/j.neurobiolaging.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem. 2013;288:15194–15210. doi: 10.1074/jbc.M113.457408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tesson L, Cozzi J, Menoret S, Remy S, Usal C, Fraichard A, Anegon I. Transgenic modifications of the rat genome. Transgenic Res. 2005;14:531–546. doi: 10.1007/s11248-005-5077-z. [DOI] [PubMed] [Google Scholar]
- 32.Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, Gregory PD, Anegon I, Cost GJ. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29:695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 33.Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia XG. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013;32:1917–1926. doi: 10.1038/emboj.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong J, Huang C, Bi F, Wu Q, Huang B, Zhou H. XBP1 depletion precedes ubiquitin aggregation and Golgi fragmentation in TDP-43 transgenic rats. J Neurochem. 2012;123:406–416. doi: 10.1111/jnc.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams KL, Warraich ST, Yang S, Solski JA, Fernando R, Rouleau GA, Nicholson GA, Blair IP. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging. 2012;33:2527.e3–2527.e10. doi: 10.1016/j.neurobiolaging.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Wu S, Mikhailov A, Kallo-Hosein H, Hara K, Yonezawa K, Avruch J. Characterization of ubiquilin 1, an mTOR-interacting protein. Biochim Biophys Acta. 2002;1542:41–56. doi: 10.1016/s0167-4889(01)00164-1. [DOI] [PubMed] [Google Scholar]
- 37.Xia Y, Yan LH, Huang B, Liu M, Liu X, Huang C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J Neurochem. 2014;129:99–106. doi: 10.1111/jnc.12606. [DOI] [PubMed] [Google Scholar]
- 38.Xia Y, Yan LH, Huang B, Liu M, Liu X, Huang C. Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J Neurochem. 2014;129:99–106. doi: 10.1111/jnc.12606. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, Bowser R, Liu YJ, Xia XG. transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010;6:e1000887. doi: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou H, Huang C, Yang M, Landel CP, Xia PY, Liu YJ, Xia XG. Developing tTA transgenic rats for inducible and reversible gene expression. Int J Biol Sci. 2009;2:171–181. doi: 10.7150/ijbs.5.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.