

Abstract
Aims
The specific role of AMPKα1 or AMPKα2 in mediating cardiomyocyte contractile function remains elusive. The present study investigated how AMPK activation modulates the contractility of isolated cardiomyocytes.
Main methods
Mechanical properties and intracellular Ca2+ properties were measured in isolated cardiomyocytes. The stress signaling was evaluated using western blot and immunoprecipitation analysis.
Key findings
AMPK activator, A-769662 induced maximal velocity of shortening (+dL/dt) and relengthening (−dL/dt), peak height and peak shortening (PS) amplitude in both WT and AMPKα2 KO cardiomyocytes, but did not affect time-to-90% relengthening (TR90). AMPK KD cardiomyocytes demonstrated contractile dysfunction compared with cardiomyocytes from WT and AMPKα2 KO hearts. However, the rise of intracellular Ca2+ levels as well as intracellular ATP levels has no significant difference among WT, AMPKα2 KO and AMPK KD groups with and without the presence of A-769662. Besides, WT, AMPKα2 KO and AMPK KD group displayed a phosphorylated AMPK and downstream acetyl-CoA carboxylase (ACC) phosphorylation. Interestingly, A-769662 also triggered troponin I (cTnI) phosphorylation at Ser149 site which is related to contractility of cardiomyocytes. Furthermore, the immunoprecipitation analysis revealed that AMPKα1 of cardiomyocytes was phosphorylated by A-769662.
Significance
This is the first study illustrating that activation of AMPK plays a significant role in mediating the contractile function of cardiomyocytes using transgenic animal models. AMPK activator facilitates the contractility of cardiomyocytes via activating AMPKα1 catalytic subunit. The phosphorylation of cTnI by AMPK could be a factor attributing to the regulation of contractility of cardiomyocytes.
Keywords: Contractile function, AMPK, Troponin I
Introduction
AMP-activated protein kinase (AMPK), a serine/threonine hetero-trimeric kinase, plays an important role in the regulation of energy balance and myocardial signaling in the heart (Morrison and Li, 2011; Moussa and Li, 2012). Previous studies have revealed that AMPK stimulates glucose uptake and glycolysis during stress situations such as exercise, starvation, hypoxia and ischemia (Costa et al., 2012; Ma et al., 2010; Morrison et al., 2011; Wang et al., 2013). Once activated, AMPK phosphorylates several downstream substrates, the effect of which is to stimulate ATP-producing pathways and inhibit ATP-consuming pathways, thus causing a rise in the ATP:AMP ratio within the cell (Carling, 2004; Moussa and Li, 2012; Paiva et al., 2011; Young, 2008).
AMPK contains α, β, and γ subunits (Morrison and Li, 2011; Wang and Li, 2009; Young, 2008) where α is the catalytic subunit that transfers a phosphate group from ATP to serine sites on target proteins (Oliveira et al., 2012; Young, 2008). The β and γ subunits perform structural and regulatory functions (Oliveira et al., 2012). There are two distinct α isoforms (Hardie, 2003). The α2 isoform is considered more enriched than the α1 in cardiomyocytes and more sensitive to changes in the intracellular AMP concentration (Li et al., 2006; Salt et al., 1998). AMPKα2 deficiency in murine models led to a phenotype of glucose intolerance and insulin resistance as well as elevated fatty acid levels (Paiva et al., 2011). Previous studies also showed that genetic deletion of AMPKα1 or AMPKα2 or overexpression of dominant-negative AMPKα reveals no effect on ventricular mass or function under unstressed conditions (Zhang et al., 2008). However, the specific role of AMPKα1 or AMPKα2 in mediating the contractile function of cardiomyocytes remains unclear.
In cardiac muscle, elevation of intracellular calcium concentration is subject to mediating contractile function of the heart (Wang et al., 2011). Direct phosphorylation of the troponin–tropomyosin, a key myofilament regulatory protein complex in the contractile apparatus, has shown to play an important role in modulation of myofilament properties and contractile function (Layland et al., 2005; Metzger and Westfall, 2004; Oliveira et al., 2012). cTnI is recognized as the “inhibitory” subunit of the troponin complex playing a critical regulatory role in cardiac muscle contraction and relaxation (Layland et al., 2004). Alternations in cTnI phosphorylation status were found in failing human hearts (Metzger and Westfall, 2004; van der Velden et al., 2003; Zakhary et al., 1999; Zhang et al., 2011). Phosphorylation at Ser23/24 of cTnI by PKA or PKG results in promising effects on cardiac contractility (Layland et al., 2005). Recent studies also showed that AMPK changes the phosphorylation status of cTnI at Ser149, suggesting a promising mechanism of AMPK mediation in contractility of cardiomyocytes (Nixon et al., 2012; Oliveira et al., 2012; Sancho Solis et al., 2011).
Although previous study demonstrated an important role of AMPK signaling in regulating contractile function (Oliveira et al., 2012), the contribution of AMPK isoforms to cardiac contractile functions is still unclear. To elucidate the specific role of AMPKα1 or AMPKα2 in mediating cardiomyocyte contractile function, we used the AMPKα2 KO mice and AMPK kinase dead (KD) transgenic mice, which have overexpression of dominant-negative α2 isoform in the heart (Mu et al., 2001; Russell et al., 2004). The AMPK KD demonstrated an abolished activity of α2 and partially decreased activity of α1 (Russell et al., 2004).
Several molecules have been identified as AMPK activity modulators such as A-769662, which shows its effect by reducing plasma glucose and triglycerides, reducing hepatic triglycerides and reducing the expression of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase and lipogenic enzyme fatty acid synthase (Cool et al., 2006; Kim et al., 2011). Goransson et al. further demonstrates that A-769662 is highly specific for AMPK which has a dual effect, causing both allosteric activation and inhibition of dephosphorylation (Goransson et al., 2007). Therefore, A-769662 is a promising molecule to study the consequence of AMPK activation on cardiomyocyte contractility.
Herein, we employed A-769662 to activate AMPK in cardiomyocytes and explored the role of AMPKα1, ATP, Ca2+ and cTnI in mediating the contractility of cardiomyocytes. We hypothesize that A-769662 facilitates the contractile function of cardiomyocytes by modulating the activation of AMPK. This effect may be due to the phosphorylation of cTnI.
Methods
Experimental animals
Male wild-type (WT) mice, male AMPKα2 knockout (KO) mice and male transgenic mice (C57BL/6) that express a kinase dead (KD) α2 isoform (K45R mutation) driven by the muscle creatine kinase promoter (Mu et al., 2001) were used in the experiments. All of these mice were of a C57BL/6 background and 4–6 months of age. All of the animal procedures conducted in this study were approved by the University at Buffalo (SUNY) Institutional Animal Care and Use Committee (IACUC).
Isolation of cardiomyocytes and cell mechanics
Mice were given 100 units of heparin i.p. (Sagent Pharmaceuticals, Schaumburg, IL) for anticoagulation before anesthetized with 100 mg/kg sodium pentobarbital i.p. (Sigma, St. Louis, MO). The heart was excised and fastened onto the cardiomyocyte perfusion apparatus (Radnoti, Monrovia, CA) and perfusion was initiated in the Langendorff mode. Hearts were perfused at 37 °C with a Ca2 +-free Krebs–Henseleit based buffer (pH 7.3) containing: 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 10 mM HEPES, 14.7 mM KCl, 1.7 mM MgSO4, 120.3 mM NaCl, 4.6 mM NaHCO3, 30 mM taurine, 10 mM glucose, and 10 mM 2,3-butanedione monoxime that was bubbled with 95% O2/5% CO2. After a few minutes of stabilization, the heart was then digested with the same perfusion buffer containing 0.067 mg/mL Liberase Blendzyme 4 (Roche, Indianapolis, IN). After digestion, the heart was removed and minced. Extracellular Ca2+ was added back to the cells to reach a final concentration of 1 mM. Cardiomyocytes were then subjected to pharmacological drug treatment with either vehicle (DMSO), 10 mM Compound C (Enzo Life Sciences, Farmingdale, NY) or A-769662 (SelleckBio) for 20 min at room temperature (20–25 °C).
Measurement of cardiomyocyte contractile function
The mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA) (Li et al., 2008; Ma et al., 2010; Wang et al., 2011). Cardiomyocytes were placed in a chamber and stimulated with a suprathreshold voltage at a frequency of 0.5 Hz. IonOptix SoftEdge software was used to capture changes in sarcomere length during shortening and re-lengthening. Cell shortening and re-lengthening were assessed using the following indices: peak shortening (PS), the amplitude myocytes shortened on electrical stimulation, which is indicative of peak ventricular contractility; time-to-90% relengthening (TR90), the duration of myocytes to reach 90% relengthening, an indicative of diastolic duration; and maximal velocities of shortening and re-lengthening.
Intracellular Ca2+ transient measurement
Intracellular Ca2+ was measured using a dual-excitation, single-emission photomultiplier system (IonOptix) (Li et al., 2008; Wang et al., 2011; Zhao et al., 2009). Cardiomyocytes were loaded with fura 2-AM (2 μM) and were exposed to light emitted by a 75 W halogen lamp through either a 340- or 380-nm filter while being stimulated to contract at a frequency of 0.5 Hz. Fluorescence emissions were then detected.
Immunoblotting
Isolated cardiomyocytes were lysed in a lysis buffer containing: 50 mM β-glycerol phosphate, 2 mM EGTA, 1 mM DTT, 10 mM NaF, 1 mM sodium orthovanadate, 20 mM HEPES (pH 7.4), 1% Triton X-100, 10% glycerol, and a protease inhibitor cocktail tablet. Protein levels of AMPK-α, p-AMPK, ACC, p-ACC, and p-cTnI were examined by standard western immunoblotting (Cui et al., 2013; Tong et al., 2013). Membranes were probed with anti-rabbit AMPK-α (1:1000, Cell Signaling), anti-rabbit phosphor-AMPK (Thr172, 1:1000, Cell Signaling), anti-rabbit ACC (1:1000, Cell Signaling), and anti-rabbit phosphor-ACC (Ser79, 1:1000, Cell Signaling), followed by incubation with horseradish peroxidase (HRP)-coupled anti-rabbit secondary antibody (Cell Signaling Technology Inc, Beverly, MA). For the detection of cTnI Ser149 phosphorylation, a polyclonal antibody was generated against the phosphopeptide LRRVRIS(phos)ADAMMQA and purified with affinity cross-absorption with the nonphosphorylated peptide (Sancho Solis et al., 2011). Blue X-ray film (Phenix, Candler, NC) was used for photon detection and image development. Films were scanned with the Bio-Rad GS-700 scanner in the core facility of the School of Medicine and Biomedical Sciences and the relative density of the bands on the film was determined by Image J software.
Immunoprecipitation
Immunoprecipitation was performed as previously described (Li et al., 2005; Morrison et al., 2011; Tong et al., 2013). Briefly, cardiomyocytes were homogenized and lysed in lysis buffer. The homogenate was incubated with the appropriate antibody precoupled to 2 μg AMPKα1 or AMPKα2 antibodies (Santa Cruz, CA) at 4 °C overnight. The beads were then washed 3 times with lysis buffer and loading samples were made in a total volume of 100 μL with lysis buffer, 4 × LDS-PAGE loading buffer (Invitrogen, Grand Island, NY) and 5% DTT. Immune-complexes were eluted at 95 °C on a heating block for 8 min, vortexed, and spun down (repeated twice). The eluted IP solution was then subjected to SDS-PAGE electrophoresis as described in the “Immunoblotting” method.
Intracellular ATP measurement
The isolated cardiomyocytes were subjected to pharmacological drug treatment. Cells were used to measure the concentration of intracellular ATP with the ATP Bioluminescent Somatic Cell Assay kit (Sigma) according to the manufacturer's instructions. In brief, isolated cardiomyocytes were lysed in somatic cell ATP releasing reagent. ATP standard was diluted to 1 μM/L, 0.1 μM/L, 0.01 μM/L, 0.001 μM/L and 0.0001 μM/L to make a standard curve. 0.1 mL of ATP Assay Mix Working Solution was added to the reaction wells and allowed to stand at room temperature for 3 min. The ATP standard and cell sample were transferred to the reaction well containing ATP Assay Mix Working Solution, and the amount of light emitted with a luminometer was immediately measured. Protein levels of cell samples were measured. The amount of ATP in the cell sample was calculated.
Statistical analysis
Data are presented as the means ± SEM. Differences between groups were assessed using analysis of variance (ANOVA), followed by Newman–Keuls' post hoc test. p < 0.05 was considered significant.
Results
Effect of A-769662 on AMPK signaling pathway in the cardiomyocytes
To explore the potential role of AMPK in the modulation of cardiomyocyte contractility, the freshly isolated cardiomyocytes from WT, AMPKα2 KO and AMPK KD mice were treated with vehicle (DMSO), AMPK inhibitor Compound C (Zou et al., 2004), AMPK activator A-769662 (Cool et al., 2006) and the combination of Compound C and AMPK activator A-769662 (100 μM) (Kim et al., 2011) for 20 min. Protein expression and phosphorylation of AMPK and its downstream signaling target acetyl-CoA carboxylase (ACC) were examined (Fig. 1). The results demonstrated that WT, AMPKα2 KO and AMPK KD group displayed an up-regulated level of phosphorylated AMPK and downstream ACC phosphorylation after treatment with A-769662 compare to individual vehicle groups (Fig. 1). After pretreating cardiomyocytes with Compound C for 5 min, A-769662 was added for a total treatment of 15 min, WT, AMPKα2 KO and AMPK KD group showed a decrease of p-ACC and p-AMPK levels compared with respective A-769662 treatment group (Fig. 1). We found a lower expression of both AMPKα and p-AMPK in AMPKα2 KO group compared with WT group, while knock out of AMPKα2 did not significantly affect the phosphorylation of ACC due to the possible compensation of α1 subunit (Fig. 1). The phosphorylation of AMPK induced by A-769662 in AMPK KD heart was increased as well (Fig. 1). Together, these data suggest that A-769662 triggers cardiac AMPK signaling in the cardiomyocytes and the phosphorylation of AMPK α1 subunit could be major contributor.
Fig. 1.

A-769662 treatment induces phosphorylation of AMPK and downstream ACC in isolated cardiomyocytes. (A) Immunoblots of phosphorylated (p-) AMPK and downstream ACC in isolated cardiomyocytes from WT, AMPKα2 KO and AMPK KD mice with or without compound C (10 μM), A-769662 (100 μM) or the combination of compound C and A-769662 treatment. (B) Bars represent the relative levels of phosphorylated AMPK. Phosphorylation of AMPK was normalized to the amount of total AMPK. (C) Bars represent the relative levels of phosphorylated ACC. Phosphorylation of ACC was normalized to the amount of total ACC. Values are expressed as means ± SEM, n = 4–5 per group. *p < 0.05 vs. the corresponding control group, respectively; †p < 0.05 vs. corresponding A-769662 group, ‡p < 0.05 vs. corresponding WT group, respectively.
Effect of AMPK activator on mechanical properties of intracellular Ca2+
We wonder whether AMPK activation could be involved in the regulation myocardial function. We investigated the effect of AMPK activator A-769662 on contractile function in myocytes isolated from mouse hearts. The contractility was measured in the isolated cardiomyocytes by IonOptix system (Ma et al., 2010; Wang et al., 2011). Data shown in Fig. 2 indicate that A-769662 significantly increased maximal velocity of shortening (+dL/dt) (Fig. 2C) and maximal velocity of re-lengthening (−dL/dt) in AMPKα2 KO group (Fig. 2D), peak height and peak shortening (PS) amplitude in both WT and AMPKα2 KO groups (Fig. 2B and E) compared with cardiomyocytes from respective vehicle groups, while A-769662 treatment didn't affect time-to-90% relengthening (TR90) (Fig. 2F). Interestingly, the cardiomyocytes from AMPK KD mice illustrated contractile dysfunction (Fig. 2B–F) compared with cardiomyocytes from WT and AMPKα2 KO mice. Fig. 2G shows the representative sarcomeric shortening traces of isolated cardiomyocytes obtained from WT, AMPKα2 KO or AMPK KD hearts with or without A-769662 treatment. These data indicated that A-769662 facilitates the contractility of cardiomyocytes and α1 catalytic subunit of AMPK appears to be critical factor contributing to maintain efficient cardiomyocyte contractile function.
Fig. 2.

Contractile properties of cardiomyocytes isolated from WT, AMPKα2 KO and AMPK KD mice with or without A-769662. A-769662 facilitates the contractile function of cardiomyocytes. Freshly isolated cardiomyocytes from WT, AMPKα2 KO and AMPK KD mice were incubated with or without A-769662 (100 μM) prior to mechanical assessment. (A) Resting sarcomere length; (B) peak height; (C) maximal velocity of shortening (+dL/dt); (D) maximal velocity of re-lengthening (−dL/dt); (E) peak shortening (normalized to the resting sarcomere length); (F) time-to-90% relengthening (TR90); and (G) representative sarcomeric shortening traces obtained from WT, WT + A-769662, AMPKα2 KO, AMPKα2 KO + A-769662, AMPK KD + A-769662 isolated cardiomyocytes. Means ± SEM, n = 50–120 cells from 2 to 4 mice per group,*p < 0.05 vs. vehicle group, respectively; †p < 0.05 vs. WT vehicle group.
To understand the potential mechanism involved in the role of A-769662 in elevating the contractility of cardiomyocytes and cardiomyocyte contractile defects of AMPK KD mice, we evaluated the intracellular Ca2+ transients using the fura-2 fluorescence technique (Wang et al., 2011). The results show that the fura-fluorescence intensity change in response to electrical stimuli, which indicates the rise of intracellular Ca2+ levels (Fig. 3B), resting intracellular Ca2+ levels (Fig. 3A), as well as the mean time constant of Ca2+ transient decay (Tau) (Fig. 3C) have no significant difference among WT, AMPKα2 KO and AMPK KD groups with and without the presence of A-769662. Fig. 3D displays the representative Ca2+ transient traces of isolated cardiomyocytes obtained from WT, AMPKα2 KO or AMPK KD hearts with or without A-769662 treatments. These data suggest that AMPK activation does not modulate intracellular Ca2+ levels in cardiomyocytes.
Fig. 3.

A-769662 significantly increased cardiomyocyte contractility without changing the amplitude of Ca2+ transient. (A) Histograms showing the resting intracellular calcium level; (B) histograms showing mean Ca2+ transient amplitude; (C) the mean time constant of Ca2+ transient decay (Tau); (D) representative Ca2+ transient traces obtained from WT, WT + A-769662, AMPKα2 KO, AMPKα2 KO + A-769662, AMPK KD and AMPK KD + A-769662 isolated cardiomyocytes. Mean ± SEM, n = 50–120 cells from 2 to 4 mice per group.
Immunoprecipitation confirms activation of AMPKα1 by A-769662 in cardiomyocytes
In order to confirm that A-769662 did indeed activate AMPK via AMPKα1 subunit in cardiomyocytes, AMPKα1 and AMPKα2 were immunoprecipitated with respective specific antibodies from isolated cardiomyocyte lysates. The results demonstrated that A-769662 treatment significantly increased activation of α1 catalytic subunit but not α2 catalytic subunit of AMPK versus control group (Fig. 4). These data confirm that activation of α1 catalytic subunit of AMPK could be a major contributor for modulation of cardiomyocyte contractility by A-769662.
Fig. 4.

A-769662 treatment triggers phosphorylation of α1 catalytic subunit of AMPK. (A) Extracted proteins of isolated cardiomyocytes from WT mice were immunoprecipitated by anti-AMPKα1 and anti-AMPKα2 antibodies, respectively and then separated by 7% SDS-PAGE. The transferred membrane was immunoblotted with either p-AMPK (Thr172) or AMPKα antibody that recognizes both AMPKα1 and AMPKα2. (B) Bars represent the relative levels of phosphorylated AMPK. Phosphorylation of AMPK was normalized to the amount of total AMPK; mean ± SEM, n = 3 per group, *p < 0.05 vs. vehicle group; †p < 0.05 vs. A-769662 group.
Effect of A-769662 on phosphorylation of troponin I in the cardiomyocytes
In order to examine whether activation of AMPKα1 catalytic subunit modulates contractile functions of cardiomyocytes via phosphorylation of troponin I (cTnI) which is involved in modulating cardiomyocyte contractility (Nixon et al., 2012; Oliveira et al., 2012), the isolated cardiomyocytes were treated with or without A-769662. The results showed that A-769662 significantly triggered phosphorylation of ACC and cTnI (Ser149) in WT and AMPKα2 KO cardiomyocytes, whereas this action of A-769662 in AMPK KD cardiomyocytes is dramatically attenuated as shown by phosphorylation of cTnI (Ser149) (Fig. 5).
Fig. 5.

A-769662 treatment induces phosphorylation of AMPK downstream ACC and cardiomyocyte troponin I (cTnI). (A) Immunoblots of phosphorylated (p-) ACC and cTnI (Ser149) of isolated cardiomyocytes from WT, AMPKα2 KO and AMPK KD mice with or without A-769662 (100 μM) treatment. (B) Bars represent the relative levels of phosphorylated cTnI (Ser149). Phosphorylation of cTnI (Ser149) was normalized to the amount of β-actin; mean ± SEM, n = 4 per group.
The change of energy substrates in cardiomyocytes
AMPK activation switched off ATP-consuming process and switched on ATP-generating process (Young, 2008). It's also found that ATP can enhance the strength of contraction in the heart (Song and Belardinelli, 1994). To study whether A-769662 treatment modulates the intracellular ATP levels, hence facilitates the contractility of cardiomyocytes, intracellular ATP levels of cardiomyocytes with Compound C, A-769662 or the combination of Compound C and A-769662 were measured using luminescence-based technique. We found that the difference of intracellular ATP level between each group was not significant (Fig. 6). Thus A-769662 mediated AMPK activation may not attribute to metabolic changes which would affect contraction.
Fig. 6.
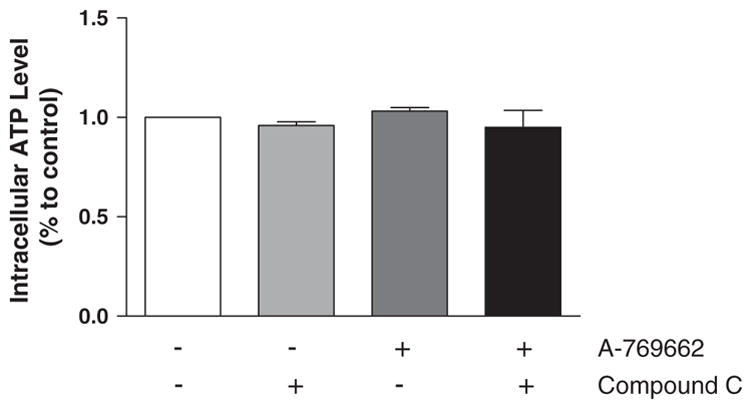
Intracellular ATP levels of cardiomyocytes. (A) The intracellular ATP levels in cardiomyocytes with Compound C (10 μM), A-769662 (100 μM) or the combination of Compound C and A-769662 treatment using luminescence-based measurement; values are expressed as means ± SEM, n = 3 per group.
Discussion
AMPK, known as a “cellular fuel gauge”, turns off the energy consuming processes and switches on energy producing processes during activation (Hardie and Hawley, 2001; Moussa and Li, 2012). Impaired cardiac function and susceptible cell death were found in transgenic mice which lacking functional AMPK (Russell et al., 2004). In this study, we explored the specific role of AMPKα1 and AMPKα2 in mediating the cardiomyocyte contractile function and its underlying mechanism.
A-769662 is a valuable molecule for the study of AMPK. It is highly specific for AMPK and is able to activate AMPK without changing the AMP:ATP ratio (Cool et al., 2006, Goransson et al., 2007). In order to confirm that AMPK activation is involved in the facilitation of cardiomyocyte contractility, we test the levels of phosphorylated AMPK and phosphorylated ACC with AMPK inhibitor compound C, AMPK activator A-769662 and combination of compound C and A-769662 in the isolated cardiomyocytes. Our data revealed that A-769662 triggers phosphorylation of AMPK in the isolated cardiomyocytes from WT, AMPKα2 KO and AMPK KD hearts. This is in consistent with the changes in phosphorylation of ACC, a downstream target of AMPK. We also observed that the effects of A-769662 on AMPK phosphorylation were smaller than that on ACC phosphorylation. It's possibly due to the susceptibility of ACC upon AMPK activation, thus only a small amount of AMPK phosphorylation could lead to maximal phosphorylation of ACC (Goransson et al., 2007). Knock out of AMPKα2 did not affect the phosphorylation of ACC due to the possible compensation of α1 subunit (Morrison and Li, 2011; Moussa and Li, 2012). However, AMPK KD mice displayed a lower p-ACC compared to WT mice and AMPKα2 KO mice. The reason may be the less activity of AMPKα1 and no AMPKα2 activity in AMPK KD mice compared to WT mice (Russell et al., 2004; Wang et al., 2013). Compound C is considered as an AMPK inhibitor, which could be used in intact cells (Zhou et al., 2001). Here we used a pharmacological way to ensure the activation of AMPK in isolated cardiomyocytes by a small molecule A-769662. Both p-ACC to ACC ratio and p-AMPK to AMPKα ratio were decreased after AMPK inhibitor Compound C treatment. Down-regulations of both p-ACC and p-AMPK were seen after treatment of cardiomyocytes with a mixture of Compound C and A-769662 as compared to respective A-769662 group, which further confirmed that A-769662 is able to activate AMPK.
Previous studies showed that AICAR, an AMPK activator, increased myocyte contractility without changing the amplitude of Ca2+ transient (Oliveira et al., 2012; Zhao et al., 2009). In our study, we used the similar way to illustrate the effect of A-769662 on cardiomyocyte contractile function. We further identified the role of AMPKα1 and AMPKα2 by studying the functional consequence of their deletions on the contractility. It's interesting to find out that activation of AMPK by A-769662 stimulated peak height and peak shortening (PS) amplitude in both WT and AMPKα2 KO groups compared with cardiomyocytes from respective control groups without affecting time-to-90% relengthening (TR90). A-769662 treatment group in WT and AMPKα2 KO mice also exhibited higher maximal velocity of shortening (+dL/dt). These results suggest that A-769662 regulates the contractility of cardiomyocytes via activation of AMPKα1 subunit. Using transgenic mice of which both AMPKα1 and AMPKα2 have been inactivated by overexpression of a dominant inhibitory catalytic subunit (AMPK KD) in muscle (Mu et al., 2001; Russell et al., 2004), we observed that cardiomyocytes from this group illustrated contractile dysfunction compared with cardiomyocytes from WT and AMPKα2 KO mice, which is consistent with our hypothesis that AMPKα1 does play an important role in mediating the contractility of cardiomyocytes after activation by A-769662. From immunoprecipitation result of which AMPKα1 and AMPKα2 were immunoprecipitated from WT cardiomyocyte lysates respectively; we confirmed that A-769662 does indeed activate AMPK via AMPKα1 subunit in cardiomyocytes.
Ca2+ stores in the sarcoplasmic reticulum (SR) play a major role in regulating cardiac contractility. The amount of Ca2+ accumulated in the SR is thought to control the contractile force (Andino et al., 2008). Here we hypothesize that phosphorylation of AMPK by A-769662 may contribute to an increase in Ca2+ uptake, therefore enhancing the contractility of cardiomyocytes. However, our results showed that A-769662 increased cardiomyocyte contractility without changing the intracellular Ca2+ level, which is consistent with the effect of AICAR (Oliveira et al., 2012). This could be explained as A-769662 promotes the contractility of cardiomyocytes by increasing myofilament Ca2+ sensitivity (Oliveira et al., 2012). Myofilament properties play an important role in the modulation of contractile function (Layland et al., 2004). Phosphorylation of cTnI is a key regulatory mechanism in cardiac muscle contraction and relaxation. Recently, phosphorylation of cTnI at Ser149 has been identified as a substrate for AMPK activation (Oliveira et al., 2012; Sancho Solis et al., 2011). The results in our study displayed that cTnI (Ser149) from WT cardiomyocytes has been up-regulated after A-769662 treatment compared to vehicle group, which supports our hypothesis that activation of AMPK by A-769662 could regulate the phosphorylation status of cTnI, leading to the result of increase contractility of cardiomyocytes.
ATP is the primary energy source for contractile proteins and many enzymes. AMPK phosphorylates several downstream substrates, the effect of which is to stimulate ATP-producing pathways and inhibit ATP-consuming pathways (Carling, 2004; Young, 2008). Thus, we reasoned that whether the cellular effect of A-769662 is mediated by modulating energy metabolism in cardiomyocytes. To extend our findings, the intracellular ATP levels in cardiomyocytes were measured using a luminescence-based measurement. Our results showed that A-769662 did not induce energy substrates (intracellular ATP level). We assume that the intracellular ATP is saturated in cardiomyocytes. Either inhibition of AMPK by compound C or activation of AMPK by A-769662 would not alter the intracellular ATP level significantly.
Limitations
Our results showed that the phosphorylation of AMPK and ACC was significant in AMPK KD mice upon A-769662 treatment. One possibility is due to the remaining activity of AMPKα1 even though there is less activity of AMPKα1 and no AMPKα2 activity in AMPK KD mice compared to WT mice. However, there are contradictive conclusions regarding whether A-769662 acts by binding to β1 subunit of AMPK (Goransson et al., 2007; Treebak et al., 2009). We could not rule out the possibility that A-769662 may contribute to the phosphorylation of AMPK and ACC through AMPKβ1 activation. Moreover, in order to make this study more convinced, specific AMPKα2 activation should be tested by using AMPKα1 KO model in future study.
Theoretically, an increase of Ca2+ sensitivity by phosphorylation of cTnI at Ser149 would accelerate shortening and slow relengthening. Previous studies revealed that activation of AMPK by AICAR phosphorylated cTnI at Ser149 without changing the phosphorylation status of Ser23/24 (Oliveira et al., 2012). On the contrary, PKA-mediated phosphorylation of TnI at Ser23/24 results in reduction in myofilament Ca2+ sensitivity, leading to acceleration of relaxation and decreased contraction (Layland et al., 2005). We can't exclude the possibility that A-769662 would activate through PKA signaling thus phosphorylate Ser23/24, leading to a decrease of Ca2+ sensitivity and faster re-lengthening. The possible cross-talk between cAMP and AMPK signaling will be of interest and currently is under investigation in our laboratory.
It has been demonstrated that incubation of cells with AICAR elevates ZMP, which binds domains on γ subunit of AMPK without affecting ATP (Corton et al., 1995; Henin et al., 1995). Study also showed that A-769662 inhibits Na+–K+-ATPase in L6 cells, which would decrease ATP consuming, thus they assumed A-769662 would lead to an increase of AMP:ATP ratio. However, our results showed that the intracellular ATP level didn't change significantly with or without A-769662 treatment in cardiomyocytes. Whether the positive contractile function mediated by A-769662 in cardiomyocytes is due to metabolic alteration is under question. To our knowledge, A-769662 may not be ideal to investigate metabolic changes in cardiomyocytes.
Conclusions
Our findings suggest that activation of AMPK signaling may be essential in mediating the contractile function of cardiomyocytes. These data favor the notion that AMPK activator A-769662 facilitates the contractility of cardiomyocytes via activating AMPKα1 subunit. Although our present study shows that activation of AMPK by A-769662 or AMPK deficiency did not change the intracellular Ca2+ level, we did found that cTnI is affected by the alternation of AMPK status, and, therefore helps regulate the contractility of cardiomyocytes. A better and profound understanding of the mechanisms and roles of AMPKα1 and cTnI phosphorylation in health and disease may allow the development of novel approaches for improving cardiac contractile function.
Acknowledgments
This study was supported by the American Heart Association SDG 0835169N and 12GRNT 11620029 (J.L.), American Diabetes Association Basic Sciences Grant 1-11-BS-92 (J.L.), National Natural Science Foundation of China (No. 31171121), the Major International (Regional) Joint Research Project 2008DFA31140 and 2010DFA32660 (P.Z.), and Guangdong Natural Science Fund 10251008002000002 and S2011010005836 (P.Z.), and Chinese Science and Technology Support Program 2011BAI11B22 (J.Z.).
Footnotes
Conflict of interest statements
All authors have no conflict of interest statements.
Contributor Information
Jian Zhuang, Email: zhuangjianzggd@yahoo.cn.
Ji Li, Email: jli23@buffalo.edu.
References
- Andino LM, Takeda M, Kasahara H, Jakymiw A, Byrne BJ, Lewin AS. AAV-mediated knockdown of phospholamban leads to improved contractility and calcium handling in cardiomyocytes. J Gene Med. 2008;10:132–42. doi: 10.1002/jgm.1131. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade — a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–65. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Costa R, Morrison A, Wang J, Manithody C, Li J, Rezaie AR. Activated protein C modulates cardiac metabolism and augments autophagy in the ischemic heart. J Thromb Haemost. 2012;10:1736–44. doi: 10.1111/j.1538-7836.2012.04833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Yu H, Wang J, Gao J, Li J. Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J Diabetes Res. 2013;2013:852754. doi: 10.1155/2013/852754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282:32549–60. doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–9. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Henin N, Vincent MF, Gruber HE, Van den Berghe G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995;9:541–6. doi: 10.1096/fasebj.9.7.7737463. [DOI] [PubMed] [Google Scholar]
- Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, et al. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM. Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol. 2004;556:835–47. doi: 10.1113/jphysiol.2004.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, III, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–9. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- Li J, Coven DL, Miller EJ, Hu X, Young ME, Carling D, et al. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006;291:H1927–34. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- Li Q, Ceylan-Isik AF, Li J, Ren J. Deficiency of insulin-like growth factor 1 reduces sensitivity to aging-associated cardiomyocyte dysfunction. Rejuvenation Res. 2008;11:725–33. doi: 10.1089/rej.2008.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–92. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–58. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- Morrison A, Li J. PPAR-gamma and AMPK — advantageous targets for myocardial ischemia/reperfusion therapy. Biochem Pharmacol. 2011;82:195–200. doi: 10.1016/j.bcp.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Morrison A, Yan X, Tong C, Li J. Acute rosiglitazone treatment is cardioprotective against ischemia–reperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am J Physiol Heart Circ Physiol. 2011;301:H895–902. doi: 10.1152/ajpheart.00137.2011. [DOI] [PubMed] [Google Scholar]
- Moussa A, Li J. AMPK in myocardial infarction and diabetes: the yin/yang effect. Acta Pharm Sin B. 2012;2:368–78. doi: 10.1016/j.apsb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–94. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, et al. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J Biol Chem. 2012;287:19136–47. doi: 10.1074/jbc.M111.323048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, et al. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circ Res. 2012;110:1192–201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- Paiva MA, Rutter-Locher Z, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, et al. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H2123–34. doi: 10.1152/ajpheart.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RR, III, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, et al. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–87. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho Solis R, Ge Y, Walker JW. A preferred AMPK phosphorylation site adjacent to the inhibitory loop of cardiac and skeletal troponin I. Protein Sci. 2011;20:894–907. doi: 10.1002/pro.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Belardinelli L. ATP promotes development of afterdepolarizations and triggered activity in cardiac myocytes. Am J Physiol. 1994;267:H2005–11. doi: 10.1152/ajpheart.1994.267.5.H2005. [DOI] [PubMed] [Google Scholar]
- Tong C, Morrison A, Mattison S, Qian S, Bryniarski M, Rankin B, et al. Impaired SIRT1 nucleocytoplasmic shuttling in the senescent heart during ischemic stress. FASEB J. 2013;27:4332–42. doi: 10.1096/fj.12-216473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treebak JT, Birk JB, Hansen BF, Olsen GS, Wojtaszewski JF. A-769662 activates AMPK beta1-containing complexes but induces glucose uptake through a PI3-kinase-dependent pathway in mouse skeletal muscle. Am J Physiol Cell Physiol. 2009;297:C1041–52. doi: 10.1152/ajpcell.00051.2009. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, et al. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- Wang J, Li J. Activated protein C: a potential cardioprotective factor against ischemic injury during ischemia/reperfusion. Am J Transl Res. 2009;1:381–92. [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yang L, Rezaie AR, Li J. Activated protein C protects against myocardial ischemic/ reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost. 2011;9:1308–17. doi: 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tong C, Yan X, Yeung E, Gandavadi S, Hare AA, et al. Limiting cardiac ischemic injury by pharmacologic augmentation of MIF-AMPK signal transduction. Circulation. 2013;128:225–36. doi: 10.1161/CIRCULATIONAHA.112.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation. 2008;117:832–40. doi: 10.1161/CIRCULATIONAHA.107.713115. [DOI] [PubMed] [Google Scholar]
- Zakhary DR, Moravec CS, Stewart RW, Bond M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–10. doi: 10.1161/01.cir.99.4.505. [DOI] [PubMed] [Google Scholar]
- Zhang P, Hu X, Xu X, Fassett J, Zhu G, Viollet B, et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–24. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guy MJ, Norman HS, Chen YC, Xu Q, Dong X, et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res. 2011;10:4054–65. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Wang J, He L, Ma H, Zhang X, Zhu X, et al. Deficiency in TLR4 signal transduction ameliorates cardiac injury and cardiomyocyte contractile dysfunction during ischemia. J Cell Mol Med. 2009;13:1513–25. doi: 10.1111/j.1582-4934.2009.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–51. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]