

Abstract
OBJECTIVE
Internal tandem duplication (ITD) mutations of the FLT3 receptor are associated with a high incidence of relapse in acute myeloid leukemia (AML). Expression of the CXCR4 receptor in FLT3-ITD–positive AML is correlated with poor outcome, and inhibition of CXCR4 was shown to sensitize AML blasts toward chemotherapy. The aim of this study was to evaluate the impact of FLT3-ITD on cell proliferation and CXCR4-dependent migration in human hematopoietic progenitor cells and to investigate their response to CXCR4 inhibition.
Materials And Methods
We used primary blasts from patients with FLT3-ITD or FLT3 wild-type AML. In addition, human CD34+ hematopoietic progenitor cells were transduced to >70% with retroviral vectors containing human FLT3-ITD.
Results
We found that FLT3-ITD transgene overexpressing human hematopoietic progenitor cells show strongly reduced migration toward stromal-derived factor–1 in vitro and display significantly reduced bone marrow homing in nonobese diabetic severe combined immunodeficient mice. Cocultivation of FLT3-ITD–positive AML blasts or hematopoietic progenitor cells on bone marrow stromal cells resulted in a strong proliferation advantage and increased early cobblestone area–forming cells compared to FLT3–wild-type AML blasts. Addition of the CXCR4 inhibitor AMD3100 to the coculture significantly reduced both cobblestone area–forming cells and proliferation of FLT3-ITD–positive cells, but did not affect FLT3–wild-type cells—highlighting the critical interaction between CXCR4 and FLT3-ITD.
Conclusion
CXCR4 inhibition to decrease cell proliferation and to control the leukemic burden may provide a novel therapeutic strategy in patients with advanced FLT3-ITD–positive AML.
Keywords: CXCR4, FLT3-ITD, AML, leukemia, migration, AMD3100
Acute myeloid leukemia (AML) is a heterogeneous clonal disorder that originates from leukemic stem cells with the ability to generate an excessive amount of malignant myeloid blasts. A concert of genetic aberrations underlies the multistep pathogenesis of AML. It appears that one class of mutations, such as the internal tandem duplication of the receptor tyrosine kinase FLT3 (FLT3-ITD) results in uncontrolled proliferation of hematopoietic progenitors and that the other class of mutations, such as RUNX1-ETO or RUNX1-EVI1, induces a block in differentiation leading to consecutive blast cell accumulation 1 and 2. The ITD mutation is located in the juxtamembrane domain of FLT3 and results in ligand-independent constitutive activation of the FLT3 receptor leading to activation of downstream signaling proteins, such as signal transducer and activator of transcription (STAT)5, STAT3, and extracellular signal-regulated kinase (ERK) [3]. ITD mutations of the FLT3 receptor occur in about 25% of patients with AML, and this mutation is associated with poor prognosis 4 and 5. The high relapse rate is due, in part, to the inefficient elimination of leukemic stem cells in the bone marrow niche, where these cells seem to be protected from chemotherapeutic therapies through interactions with the microenvironment. While integrins such as very late antigen–4 and the adhesion molecule CD44 have been demonstrated to be crucial for the persistence of leukemic progenitors in the bone marrow, the chemokine receptor CXCR4 and its ligand stromal cell–derived factor-1 (SDF-1) have been identified as key players for homing of AML cells to their niche and their in vivo growth 6, 7 and 8. CXCR4 is a G-protein–coupled receptor that induces migration upon binding of SDF-1, activates adhesion molecules and integrins and induces cell proliferation and cell survival [9]. Inhibition of CXCR4 has been shown to sensitize leukemic blasts to chemotherapy 10, 11, 12 and 13. Recent reports suggest a functional interaction between the FLT3 receptor and CXCR4. While CXCR4 expression in FLT3-ITD–positive CD34+ AML cells was correlated with poor outcome [14], FLT3-ITD overexpression in murine hematopoietic cell lines was suggested to result in enhanced SDF-1–mediated cell migration [15].
We show that FLT3-ITD transgene overexpression in human hematopoietic progenitor cells (HPC) confers reduced SDF-1–mediated migration in vitro, as well as reduced homing of human HPC to the bone marrow of nonobese diabetic severe combined immunodeficient (NOD/SCID) mice. Furthermore, we demonstrate that the growth advantage of FLT3-ITD–positive HPC or AML blasts in coculture with bone marrow stromal cells largely depends on the activation of CXCR4. Inhibition of CXCR4 selectively reduced cell proliferation and formation of cobblestone area–forming cells (CAFCs) of FLT3-ITD transgene–positive HPC and FLT3-ITD–positive human AML blasts.
MATERIALS AND METHODS
Source of healthy human CD34+ progenitor cells and primary AML blasts
CD34+ HPC from mobilized peripheral blood or from cord blood of healthy donors were used. After informed consent following the Institutional Review Board–approved protocols, CD34+ cells were selected by MACS MicroBeads (Miltenyi, Bergisch Gladbach, Germany). The purity of CD34+ after selection was >97%. Primary AML cells were harvested from bone marrow aspirates after informed consent. Vials of CD34+ cells and AML cells were stored in liquid nitrogen and thawed for each experiment.
Source of primary human bone marrow stromal cells (hBMSC)
Bone marrow aspirates were collected from healthy donors after informed consent and approval of the Institutional Review Board. After density centrifugation, human BMSC were isolated by plastic adherence and cultivation in Dulbecco’s modified Eagle’s medium/0.1% glucose and 10% fetal bovine serum (passaged maximally three times). After 1 week, cells were stained with peridinin-chlorophyll protein complex anti–human-CD45 immunoglobulin G (B&D, Heidelberg, Germany) to monitor possible contamination with hematopoietic cells. For further characterization of isolated hBMSC, osteogenic and adipogenic differentiation potential was confirmed by alkaline phosphatase activity and Oil Red staining, respectively.
Generation of γ-retroviral transfer vectors and virus vector production
FLT3-ITD complementary DNA was cloned from the human acute monocytic leukemia cell line MV4-11 [16]. FLT3 wild-type (FLT3-wt) complementary DNA was cloned from human HPC. To generate MFGS-FLT3-ITD-IRES-GFP and MFGS-FLT3-ITD-IRES-CFP, respectively, FLT3-ITD was cloned directionally into the XhoI-BamHI cloning sites of MFGS-IRES-GFP and MFGS-IRES-CFP vectors [17]. To generate MFGS-FLT3-wt-IRES-GFP, FLT3-wt was cloned directionally into the XhoI-BamHI cloning site of the MFGS-IRES-GFP vector. The MFGS-GFP, MFGS-CFP, and MFGS-IRES-GFP vectors were published elsewhere and served as controls [18]. To obtain various RD114-pseudotyped MFGS virus vectors from stable virus vector producer cell lines, FLYRD18 packaging cells were transduced with transiently produced virus vector, and for each vector a high-titer FLYRD18 producer clone was selected [17]. FLYRD18 cells with a FLT3-ITD–containing construct were cultured in medium supplemented with GTP-14564 at 2 μM. After ultracentrifugation, the virus vector pellet was suspended in fresh X-VIVO10/1% human serum albumin. The neat titers of the virus vector as determined on cord blood CD34+ HPC were 2–5 × 106.
Transduction of HPCs
Cultures were initiated in Retronectin (Takara Shuzo Ltd., Otsu, Japan) precoated six-well plates with 3 × 105 CD34+ HPC per well. HPC were cultured in X-VIVO10 (Bio Whittaker, Walkersville, MD, USA) supplemented with 1% human serum albumin, 50 ng/mL FLT3 ligand, 10 ng/mL stem cell factor, 10 ng/mL thrombopoietin, and 10 ng/mL interleukin-3. All cytokines were purchased from R&D Systems (Wiesbaden, Germany). HPC were transduced at 48 hours and 72 hours after culture initiation. Naïve and control-transduced HPC served as negative control. The transduction efficiency was controlled by flow cytometry and cell viability determined by trypan blue (Merck, Darmstadt, Germany) exclusion.
Analysis of STAT5a, STAT3, and ERK1/2 phosphorylation by flow cytometry
The 1 × 105 cells were fixed, permeabilized (BD PhosFlow Perm Buffer; B&D) and stained according to manufacturer’s instructions with Alexa Fluor 647–conjugated antibodies directed against phospho-STAT5a, phospho-STAT3, and phospho-ERK1/2 (B&D), respectively, and subsequently analyzed by flow cytometry.
CXCR4 expression by flow cytometry
Cell surface and total (surface plus intracellular) CXCR4 expression were analyzed in transduced CD34+ HPC by flow cytometry. Cells were stained with allophycocyanin-conjugated anti-human CD184 (anti-CXCR4; clone 12G5; B&D) or isotype control (Mouse IgG2a; B&D) after fixation and permeabilization (total CXCR4, Fix&Perm reagents; Caltag Laboratories, Hamburg, Germany) or without permeabilization (surface CXCR4).
In vitro migration assay
MFGS-FLT3-ITD-IRES-GFP or MFGS-FLT3-ITD-IRES-CFP transduced human CD34+ HPC were pre-incubated for 3 hours in RPMI without FBS. The 5 × 104 of transduced HPC were loaded in 59 μL growth medium on the membrane of a 5-μm ChemoTx microplate (96-well plate; Neuroprobe, Gaithersburg, MD, USA). The bottom chamber contained 29 μL growth medium with or without 100 ng/mL SDF-1α. After 120 minutes, migrated cells in the bottom chamber were collected and analyzed by flow cytometry for green fluorescent protein (GFP) expression and for the total number of migrated cells using fixed time settings. All runs were performed in triplicates.
Analyses of CXCR4 and suppressor of cytokine signaling–3 (SOCS3) messenger RNA expression by quantitative reverse transcription polymerase chain reaction (PCR)
Total RNA from 3- and 7-day cultured HPC was prepared with the RNeasy Mini Kit (Qiagen, Hilden, Germany) and reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen, Karlsruhe, Germany). Gene transcription was examined with FAM/TAMRA-labeled probes. Probes and primers for glyceraldehyde 3-phosphate dehydrogenase (forward primer: GAAGGTGAAGGTCGGAGTC; reverse primer: GAAGATGGTGATGGGATTTC; probe: CAAGCTTCCCGTTCTCAGCC) and CXCR4 (forward primer: CTCTATGCTTTCCTTGGAGCC; reverse primer: TGGACCCTCTGCTCACAG; probe TTAAAACCTCTGCCCAGCACGCACTCAC) were synthesized by Eurofins MWG (Ebersberg, Germany). SOCS3 TaqMan gene expression assay (Hs00269575_s1) was obtained from Applied Biosystems (Darmstadt, Germany). PCR reactions were carried out in a 7300 Real Time PCR System (Applied Biosystems). Gene transcription of CXCR4 and SOCS3 was normalized in relation to transcription of glyceraldehyde 3-phosphate dehydrogenase.
Analysis of hematopoietic cell growth during coculture on stromal cells
MFGS-FLT3-ITD-IRES-GFP and MFGS-CFP transduced human CD34+ HPC were mixed at equal numbers (5 × 104 cells each) and seeded in six-well plates onto hBMSC or irradiated M210B4 murine stromal cells. This approach was repeated for cells from six different cord blood units. For control studies, MFGS-FLT3-wt-IRES-GFP or MFGS-GFP transduced HPC were used. In addition, primary AML blasts from five donors with FLT3-wt and from six donors with FLT-ITD mutation were seeded separately in six-well plates onto irradiated M210B4 murine stromal cells. Transduced cells and blasts were cultured in MyeloCult H5100 medium (StemCell Technologies Inc., Köln, Germany) with hydrocortisone in the presence or absence of 1 μM, 10 μM, or 20 μM AMD3100 (Sigma-Aldrich, Hamburg, Germany). Half of the medium was changed after 7 days. Human hematopoietic cells were distinguished from the stromal feeder layer by forward scatter and side scatter gating as well as staining for the human CD45 hematopoietic cell marker.
CAFC assay
CAFC assay on M210B4 cells was performed as described previously [19]. Cocultures were maintained at 37°C and 5% CO2 with changing half of the medium weekly. Cobblestone-forming areas/units were counted by microscopic evaluation.
Analysis of hematopoietic cell proliferation using carboxyfluorescein succinimidyl ester (CFSE)
FLT3-ITD–positive AML blasts, FLT3-wt AML blasts, MFGS-FLT3-ITD-IRES-CFP–transduced and MFGS-CFP control–transduced CD34+ HPC were pulse-labeled with the cell tracker dye CFSE (Molecular Probes, Invitrogen GmbH, Karlsruhe, Germany) at a final concentration of 10 μM and subsequently cocultured on M210B4 cells [18]. Half of the CFSE-labeled cells were treated with 1 μM, 10 μM, or 20 μM of AMD3100 and cell proliferation analyzed by flow cytometry 7 days after initiation of coculture with stromal cells.
Homing of transduced HPC into NOD/SCID mice
NOD/SCID mice were housed in microisolator cages and provided with autoclaved food and acidified water. Eight- to 10-week-old mice were sublethally irradiated with 300 cGy prior to transplantation and MFGS FLT3-ITD-IRES-GFP and MFGS-CFP–transduced human CD34+ HPC, respectively were cotransplanted at equal numbers (2 × 106 cells each) via tail vein injection. Spleens and bone marrow from tibias and femurs were harvested 18 hours after transplantation. Red cells were lysed with ACK lysis buffer (Quality Biological Inc., Gaithersburg, MD, USA). Human cells were detected by flow cytometry using anti-human CD45+ allophycocyanin-conjugated antibody (Becton Dickinson, Heidelberg, Germany), and CD45-positive cells were further analyzed for cyan fluorescent protein (CFP) and GFP expression to distinguish FLT3-ITD expressing from control-transduced cells.
Statistical analysis
Statistical analyses were performed using a paired t-test and analysis of variance analysis with subsequent Tukey multiple comparison test. Statistical significance was established at *p < 0.05; **p < 0.01; and ***p < 0.001. All results are presented as mean values, with error bars corresponding to standard deviation (SD). The values for AML blasts are the mean ± SD of six FLT3-ITD–positive AML donors and five FLT3-wt AML donors. For transduced HPC, results are the average of at least three different donors.
RESULTS
FLT3-ITD transgene-positive cells demonstrate constitutive phosphorylation of STAT5a, ERK1/2, and STAT3
Mobilized human peripheral blood and cord blood progenitor cells were transduced to >70% using our high-titer γ-retroviral vector MFGS-FLT3-ITD-IRES-GFP. FLT3-ITD transgene expression was confirmed by flow cytometry and Western blot (not shown). We demonstrate that overexpressed FLT3-ITD transgene was functional and resulted in the expected activation of downstream signaling proteins, as was shown by constitutive activation of STAT5a, STAT3, and ERK1/2 (Fig. 1), while GFP-control–transduced or nontransduced human HPCs did not show any constitutive phosphorylation of STAT5a, STAT3, or ERK1/2.
Figure 1.

Hematopoietic progenitor cells (HPC) overexpressing the FLT3-internal tandem duplication mutation (FLT3-ITD) transgene demonstrate constitutive phosphorylation of signal transducer and activator of transcription (STAT)5a, extracellular signal-regulated kinase (ERK)1/2, and STAT3. Using γ-retroviral vector transduction, we achieved unprecedented transgene overexpression of human FLT3-ITD in >70% of human cord blood hematopoietic progenitors (A). The functionality of the FLT3-ITD–transgene was demonstrated by constitutive phosphorylation of STAT5a (B), STAT3 (C), and ERK1/2 (D) in the FLT3-ITD overexpressing cells. Representative histograms are shown. GFP, green fluorescent protein.
FLT3-ITD transgene-positive HPC and FLT3-ITD-positive human AML blasts demonstrate significantly reduced CXCR4 expression
Unlike published data [15], we found reduced surface and reduced total (surface plus intracellular) CXCR4 expression as measured by flow cytometry for FLT3-ITD transgene overexpressing HPC compared to naïve or control-transduced HPC. While 42% ± 8% of FLT3-ITD transgene-positive cells showed CXCR4 surface expression, the control-transduced HPC demonstrated CXCR4 surface staining in 72% ± 6%. Similarly, only 47% ± 7% of FLT3-ITD transgene-positive cells showed total CXCR4 expression compared to 77% ± 4% for control-transduced HPC (Fig. 2A). The level of CXCR4 expression as measured by the median fluorescence intensity (MFI) was also significantly reduced for FLT3-ITD–positive HPC. Gene expression analysis confirmed these data and demonstrated an 8.8-fold reduction of CXCR4 messenger RNA expression in FLT3-ITD overexpressing HPC compared to GFP-control–transduced HPC (*p < 0.05, Fig. 2B).
Figure 2.

Hematopoietic progenitor cells (HPC) overexpressing the FLT3-internal tandem duplication mutation (FLT3-ITD) transgene demonstrate significantly reduced CXCR4 expression and reduced in vitro and in vivo migration potential. Fewer FLT3-ITD transgene-positive HPCs (solid black line) show total CXCR4 expression compared to green fluorescent protein (GFP) control transduced cells (GFP Ctr, grey line; isotype control, dashed line) as analyzed by flow cytometry (A; 47% vs 77%). In addition, the level of CXCR4 expression as measured by the median fluorescence intensity was also significantly reduced for FLT3-ITD–positive HPC compared to control-transduced cells (87.7 vs 173.5). A representative histogram is displayed. Similarly, FLT3-ITD transgene-positive HPCs show significantly less CXCR4 expression (*<0.05%) as measured by quantitative reverse transcription polymerase chain reaction (RT-PCR) (B). Furthermore, compared to control-transduced cells, FLT3-ITD transgene-positive cells demonstrate reduced migration in vitro (C; ***p< 0.001) and reduced homing to the bone marrow of nonobese diabetic severe combined immunodeficient (NOD/SCID) mice (D; **p< 0.01). CFP = cyan fluorescent protein; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; SDF-1 = stromal cell-derived factor 1.
In addition, we found that significantly (*p < 0.05) fewer FLT3-ITD–positive AML blasts expressed surface CXCR4 compared to FLT3-wt cells (23% ± 8.6% vs 63.7% ± 20%, Fig. 3A, B). The total CXCR4 expression of FLT3-ITD AML blasts was not significantly different compared to FLT3-wt blasts.
Figure 3.

Internal tandem duplication mutation of the receptor tyrosine kinase FLT3 (FLT3-ITD)–positive acute myeloid leukemia (AML) blasts show significantly reduced CXCR4 surface expression. Compared to FLT3 wild-type (FLT3-wt) AML blasts (A; 63.7% ± 20%), fewer FLT3-ITD–positive AML blasts show CXCR4 surface expression (B; 23% ± 8.6%), whereas total CXCR4 expression levels show no significant difference (dotted line: isotype; dashed line: surface expression, solid line: total expression). Cocultivation on human bone marrow stromal cells (hBMSC) resulted in a significantly increased number of FLT3-ITD–positive AML blasts with surface CXCR4 expression (C) compared to initiation of culture. Addition of AMD3100 reduces proportion of surface CXCR4 expression in FLT3-ITD–positive AML blasts, but not in FLT3-wt AML blasts (D), as determined by median fluorescence intensity (MDI). Representative histograms are shown. *p < 0.05.
FLT3-ITD transgene-positive HPC demonstrate significantly reduced migration and bone marrow homing potential
Consistent with the reduced CXCR4 expression of FLT3-ITD–positive cells, we demonstrate a 12.8-fold reduced SDF-1–dependent migration potential (***p < 0.001) of FLT3-ITD transgene-positive cells compared to control-transduced cells ( Fig. 2C) in vitro. Specifically, FLT3-ITD–positive cells migrated in 1.6% ± 0.71% (spontaneous migration: 0.6% ± 0.45%), while GFP-control–transduced cells migrated in 20.6% ± 3.2% (spontaneous migration: 0.6% ± 0.49%). Results are from three individual experiments. Migration of FLT3-wt–transduced cells did not differ significantly from the GFP-control–transduced cells (data not shown).
Furthermore, FLT3-ITD overexpressing and control-transduced HPC were cotransplanted at equal numbers into eight NOD/SCID mice and homing into the bone marrow and spleen was analyzed by flow cytometry 18 hours posttransplantation. We found significantly (**p < 0.01) reduced bone marrow homing of FLT3-ITD–positive HPC (0.06% ± 0.04% for FTL3-ITD) compared to CFP-positive control cells (0.21% ± 0.09%; Fig. 2D). The numbers of FLT3-ITD–overexpressing HPC that homed to the spleens were not statistically different from CFP-positive control HPC.
SOCS3 is significantly upregulated in FLT3-ITD–transgene overexpressing HPC
SOCS3 has been shown to impair SDF-1–mediated migration by binding to CXCR4 [20]. It is known that STAT proteins upregulate SOCS3 [21]. To analyze whether SOCS3 is upregulated in our FLT3-ITD–positive hematopoietic cells, we determined SOCS3 messenger RNA levels by quantitative RT-PCR on days 3 and 7 after initiation of culture. We found significantly (***p < 0.001) upregulated SOCS3 in FLT3-ITD transgene-positive HPC compared to control-transduced cells (14.5-fold on day 3 and 3.7-fold upregulation on day 7; Fig. 4).
Figure 4.
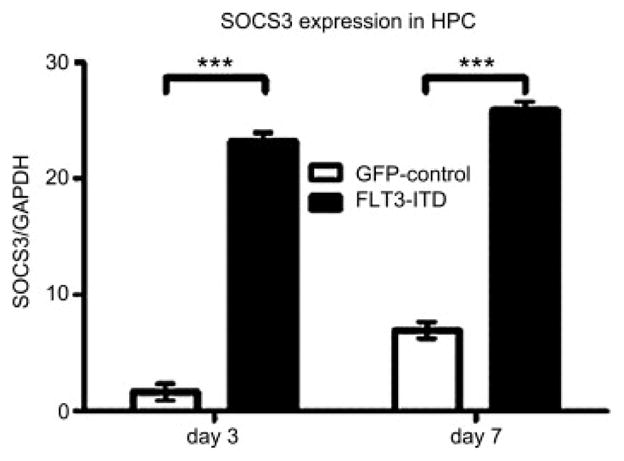
Hematopoietic progenitor cells (HPC) overexpressing the FLT3-internal tandem duplication mutation (FLT3-ITD) transgene demonstrate upregulated expression of suppressor of cytokine signaling–3 (SOCS3). Using quantitative reverse transcription polymerase chain reaction, we found SOCS3, a known negative regulator of CXCR4 signaling, to be significantly (***p< 0.001) upregulated in FLT3-ITD transgene-positive HPC on days 3 and 7 after initiation of culture. GAPDH = glyceraldehyde 3-phosphate dehydrogenase.
FLT3-ITD transgene-positive HPC and primary FLT3-ITD–positive AML blasts demonstrate enhanced cell growth
To mimic the bone marrow microenvironment, the primary human HPCs were cultured on a stromal cell layer. We found that the FLT3-ITD transgene-positive HPC from six different donors cocultured on stromal cells exhibit a strong and statistically significant growth advantage (***p < 0.001) during ex vivo coculture compared to control-transduced HPC, as seen in Figures 5A and B. Beside the growing percent of FLT3-ITD transgene-positive cells over time of culture, the MFI of FLT3-ITD transgene-positive cells as measured by MFI of the IRES-linked fluorescence protein increased up to 1.5-fold over time, whereas the MFI for MFGS-CFP–transduced control cells decreased twofold during the same observation period. Furthermore, surface CXCR4 expression of nonadherent FLT3-ITD blasts increased from 23% ± 9% at initiation of culture to 38% ± 5% at 10 days of coculture. Although FLT3-ITD–positive blasts show both a high and a low surface CXCR4-expressing population, the CXCR4 distribution becomes very homogenous during coculture (data not shown). Moreover, the migration potential of the cultured nonadherent FLT3-ITD–positive blasts increased to 15% ± 4% compared to migration at initiation of culture (8.6% ± 2%). There was no significant difference in cell growth for adherent or nonadherent HPC and, there was no significant difference in cell growth between FLT3-wt–transduced or GFP-control–transduced cells (data not shown). When FLT3-ITD–positive cells were grown in liquid culture or in transwells to provide growth factor supply from stromal cells without direct cell-to-cell contact, the growth advantage for FLT3-ITD transgene-positive cells was less pronounced and statistically not significant. Furthermore, addition of SDF-1 to the liquid culture did not significantly enhance cell growth. Similarly, we found a 1.4- to 1.8-fold growth advantage for six donors for nonadherent FLT-ITD–positive AML blasts on M210B4 compared to nonadherent FLT3-wt AML blasts from five donors (Fig. 5C, D). The adherent fractions of blast cells were not analyzed for cell growth. In coculture using transwells to prevent cell-to-cell contact, the cell growth of FLT3-ITD–positive blasts was not significantly different from FLT3-wt blasts.
Figure 5.

The internal tandem duplication mutation of the receptor tyrosine kinase FLT3 (FLT3-ITD) mediates enhanced cell growth, which is selectively reduced by CXCR4 inhibition. Cyan fluorescent protein (CFP) transgene control-transduced hematopoietic progenitor cells (HPC) (A), FLT3-ITD transgene-positive HPC (B), FLT3-wild-type (wt) acute myeloid leukemia (AML) blasts (C), and FLT3-ITD–positive blasts (D) were cocultivated on stromal cells. Compared to control-transduced HPC and FLT3-wt blasts, FLT3-ITD–positive cells demonstrated significantly enhanced cell growth (open bars). The FLT3-ITD–mediated cell growth was significantly reduced (***p < 0.001) by the CXCR4 antagonist AMD3100 (B and D, filled bars). However, CXCR4 inhibition had no effect on cell growth of FLT3-wt cells (A and C, filled bars).
The cell viability of FLT-ITD–positive cells, determined by trypan blue exclusion, as well as the rate of apoptotic and necrotic cells, determined by propidium iodide and Annexin-V analysis, did not differ significantly from that of FLT3-wt cells (data not shown), suggesting that differences in cell viability did not account for the enhanced growth advantage of FLT3-ITD–positive cells.
CXCR4 antagonist AMD3100 selectively reduces cell growth of the FLT3-ITD overexpressing HPC and of primary FLT3-ITD–positive AML
When HPC were cocultured on stromal cells and CXCR4 was blocked by addition of the CXCR4 antagonist AMD3100 to the medium, cell growth was significantly reduced for FLT3-ITD transgene-positive cells (***p < 0.001), as well as for primary FLT3-ITD–positive AML samples (***p < 0.001; Fig. 5B, D) compared to cocultures without AMD3100. The different AMD3100 concentrations used (1 μM, 10 μM, and 20 μM) resulted in comparable data, with 20 μM having the strongest effect. No significant difference in cell viability and apoptosis was found for FLT3-ITD–positive cells with or without AMD3100 (data not shown). In nonadherent AML blasts, AMD3100 reduces the proportion of surface CXCR4 in FLT3-ITD samples as determined by MFI, but not in FLT3-wt samples (Fig. 3D). Addition of AMD3100 to control-transduced or primary FLT3-wt AML samples had no effect on cell growth or viability, highlighting the impact of SDF-1/CXCR4 signaling on cell growth of ITD-positive cells (Fig. 5A, C).
Tracking cell divisions, FLT3-ITD transgene-positive HPC and FLT3-ITD–positive AML blasts show enhanced cell proliferation
Because we did not see significant differences in apoptosis between the cell populations, we investigated—using CFSE labeling—whether enhanced cell proliferation accounts for the growth advantage seen for FLT3-ITD–positive cells in coculture with M210B4. We found increased cell proliferation for all FLT3-ITD–containing cell populations studied, compared to cells with naïve FLT3 expression (Fig. 6). More specifically, we demonstrate an increased rate of cell division for adherent and nonadherent FLT3-ITD transgene-positive cells compared to GFP-control–transduced cells (Fig. 6A, B). In addition, nonadherent FLT3-ITD AML blasts demonstrated increased proliferation compared to FLT3-wt AML blasts (Fig. 6E, F). Using the CXCR4 antagonist AMD3100 to block SDF-1/CXCR4 signaling, we found that the cell proliferation of FLT3-ITD–positive cells was greatly reduced, compared to cocultures without AMD3100 (Fig. 6C, D, and F). In contrast, cell proliferation of the FLT3-wt–containing cells was not influenced by CXCR4 inhibition (Fig. 6E).
Figure 6.

CXCR4 inhibition selectively reduces cell proliferation of cells expressing the internal tandem duplication mutation of the receptor tyrosine kinase FLT3 (FLT3-ITD) as measured by flow cytometric analyses of carboxyfluorescein succinimidyl ester (CFSE)–labeled cells on stromal cells. Both, adherent (A) and nonadherent (B) FLT3-ITD transgene-positive hematopoietic progenitor cells (HPC) show a greater proliferation rate compared to control (green fluorescent protein [GFP] Ctr, gray line) transduced adherent and nonadherent hematopoietic progenitor cells (HPC). CXCR4 inhibition with AMD3100 (dotted histograms) reduced proliferation of both, adherent (C) and nonadherent (D) FLT3-ITD transgene-positive HPC, in contrast to cultures without AMD3100 (continuous line in C and D). Similarly, while CXCR4 inhibition (dotted line) has no effect on cell proliferation of FLT3-wild-type (wt) acute myeloid leukemia (AML) blasts (E), AMD3100 reduces the proliferation of FLT3-ITD–positive AML blasts as seen in (F) (C–F: continuous line, without AMD3100; dotted line, with AMD3100). Representative histograms are shown. CFP = cyan fluorescent protein; CFSE = carboxyfluorescein succinimidyl ester.
CXCR4 inhibition prevents CAFC formation in FLT3-ITD-transduced cord blood HPC and primary FLT3-ITD-positive AML cells
When FLT3-wt AML blasts or control-transduced HPC were cocultured on stromal cells, CAFC (>10 cells per colony) appeared at day 12 of ex vivo culture and increased in total number and in cells per colony during the observation period (Fig. 7A, C). In contrast, when FLT3-ITD AML blasts or FLT3-ITD transgene-positive HPC were cocultured on stromal cells, early cobblestone area cell formation was observed as soon as 3 days after initiation of culture (Fig. 7B, D).
Figure 7.

CXCR4 inhibition reduces the formation of cobblestone area forming cells (CAFC) derived from cells positive for the internal tandem duplication mutation of the receptor tyrosine kinase FLT3 (FLT3-ITD) CAFC derived from green fluorescent protein (GFP)-control transduced hematopoietic progenitor cells (HPC) from three FLT3 wild-type (wt) donors appeared at day 12 and increased in number and size over the observation period (A, open bars). For FLT3-ITD transgene-positive HPC from three donors, CAFC were observed as early as 3 days after initiation of coculture (B, open bars). Inhibition of stromal-derived factor–1 (SDF-1)/CXCR4 signaling by addition of AMD3100 significantly (***p < 0.001) suppressed the early and overall formation of CAFC from FLT3-ITD–positive HPC (B, filled bars). Inhibition of CXCR4, however, had no effect on the number of CAFC from GFP-control transduced cells with FLT3-wt expression (A, filled bars). Similarly, for FLT3-ITD–positive AML blasts from five donors, CAFC were observed as early as 7 days after initiation of coculture (D, open bars). Inhibition of SDF-1/CXCR4 signaling by addition of AMD3100 significantly (***p < 0.001) suppressed the early formation of CAFC from FLT3-ITD–positive acute myeloid leukemia (AML) blasts (D, filled bars). In contrast, CXCR4 inhibition had no effect on the formation of CAFC of FLT3-wt AML blasts (C).
Inhibition of SDF-1/CXCR4 signaling by addition of AMD3100 significantly (***p < 0.001) suppressed the early and overall formation of CAFC from FLT3-ITD–positive HPC and FLT3-ITD–positive AML blasts ( Fig. 7B, D). Inhibition of CXCR4, however, had no effect on the number of CAFC from FLT3-wt blasts (Fig. 7C) or GFP-control–transduced cells with naïve FLT3 expression (Fig. 7A).
Discussion
Several recent publications suggest an interaction between the mutated FLT3-ITD receptor and the chemokine receptor CXCR4. Based on these reports, we studied the influence of FLT3-ITD expression on cell proliferation and migration in human HPCs, as well as FLT3-ITD–positive AML blasts, and investigated their response to CXCR4 inhibition.
Using retroviral vector transduction, we achieved unprecedented transgene overexpression of human FLT3-ITD in >70% of cord blood hematopoietic progenitors (HPC, Fig. 1A). Compared to control-transduced HPC, FLT3-ITD–positive human HPC demonstrated strongly impaired homing to the bone marrow of NOD/SCID mice (Fig. 2D), confirming previous results by Fukuda and Pelus using FLT3-ITD–overexpressing murine hematopoietic cells [21]. Unlike their study that describes enhanced migration for FLT3-ITD–positive murine cells in vitro, we found that FLT3-ITD–positive human HPC displayed significantly reduced in vitro migration toward the CXCR4 ligand SDF-1 compared to FLT3-wt or control transduced human HPC (Fig. 2C). In line with the reduced migration potential, we found significantly fewer FLT3-ITD–positive cells that express CXCR4 (Figs. 2A, B, and 3B). Furthermore, we analyzed expression levels of the SOCS3, a known negative regulator of CXCR4 signaling 20 and 22, and confirmed upregulated expression of SOCS3 in FLT3-ITD transgene overexpressing HPC (Fig. 4) [23]. The high SOCS3 expression levels in FLT3-ITD–positive cells are likely a result of the constitutively activated STAT3 and STAT5, as the expression level of the STAT inhibitor SOCS3 is associated with constitutive STAT activation 24 and 25. SOCS3 upregulation, which is likely to inactivate CXCR4 function, corroborates the reduced migratory capacity of FLT3-ITD–positive cells seen in our in vitro and in vivo studies with human cells. However, further analyses to elucidate the detailed signaling interactions between FLT3-ITD and CXCR4 are needed.
For individually tested CD34+ HPC from six cord blood units, cocultivation of FLT3-ITD–overexpressing HPC on stromal cells resulted in an increased formation of early CAFC (Figure 7B) and in a significantly accelerated growth of FLT3-ITD–positive cells compared to naïve FLT3-wt or GFP-transduced HPC (Figs. 5A, B, and 6A, B). The growth advantage was in particular seen for cells with high FLT3-ITD transgene expression as measured by expression of the IRES-linked fluorescence protein GFP. Similarly, FLT3-ITD–positive AML blasts demonstrated significantly enhanced cell growth on hBMSC, as well as increased formation of early CAFC compared to FLT3-wt AML blasts (Figure 5, Figure 6 and Figure 7). The growth advantage was less pronounced and statistically not significant, when FLT3-ITD–positive cells were grown in liquid culture (with or without SDF-1) or in transwells on hBMSC (data not shown) to provide growth factor supply without direct HPC-hBMSC contact, highlighting the effect of the microenvironment on hematopoietic cell growth.
Addition of the CXCR4 chemokine inhibitor AMD3100 to the coculture significantly decreased the cobblestone formation and growth advantage of FLT3-ITD transgene-positive cord blood HPC. Similar results were obtained using FLT3-ITD–positive AML blasts (Figure 5, Figure 6 and Figure 7). Interestingly, we show that the reduced cell growth of FLT3-ITD–positive cells is due to diminished cell proliferation and not a result of increased cell death (Fig. 6). Thus, inhibition of the CXCR4/SDF-1 signaling in FLT3-ITD–positive cells abrogates the robust proliferation effect exerted by the FLT3-ITD mutation, stressing the synergistic effect of CXCR4 and FLT3-ITD in AML blast proliferation.
Unlike the studies by Tavor et al. 7 and 26, we did not observe any significant effect of AMD3100 on cell proliferation or survival of FLT3-wt AML blasts (Fig. 5A, C), nor did AMD3100 have an effect on CXCR4 or FLT3 expression in these cells. While in their studies, AMD3100 was applied to AML cells in liquid culture, we blocked CXCR4 in coculture experiments, suggesting that the direct cell-to-cell contact in our studies protected the AML cells from cell death. It is important to note that CXCR4 inhibition in coculture studies might also act on bone marrow stromal cells, which in turn may have an effect on the hematopoietic cell growth.
Rombouts et al. [14] demonstrated that CXCR4 signaling supports survival of CD34+ blasts and that CXCR4 expression is a strong negative prognostic factor in FLT3-ITD–positive AML. Our studies demonstrate that during the coculture, the initially low surface CXCR4 expression on FLT3-ITD–positive blasts increases and that, blocking of the SDF-1/CXCR4 signaling axis selectively reduces cell growth of FLT3-ITD hematopoietic cells. As CXCR4 signaling is a prerequisite for the strong proliferation of FLT3-ITD–positive AML blasts, it seems possible that blasts with the highest CXCR4 expression are selected over time and represent an aggressive phenotype.
Inhibition of CXCR4 has been shown to result in a homing and engraftment defect of AML blasts. In addition, we have recently demonstrated successful mobilization of leukemic blasts in a patient with advanced AML [27]. This maneuver allows dislodging leukemic cells from the protective microenvironment, making them accessible to chemotherapeutic drugs. In line with this report, other groups have presented evidence that AMD3100 overcomes the chemoresistance of leukemic cells mediated by the microenvironment and that CXCR4 inhibition adds synergistic effects to chemotherapy [10, 11, 12 and 13]. In addition to the sensitizing effects of AMD3100 for chemotherapy, the antiproliferative effect of AMD3100 on FLT3-ITD–positive blasts may provide a rationale to apply this strategy in patients with advanced and relapsed FLT3-ITD–positive AML over a longer period of time [28] to slow progression in situations when aggressive therapy, e.g. in elderly patients, is not warranted. Novel targeted therapies that selectively suppress leukemia progression and interfere with the leukemic niche may help to further improve outcomes of selected patients.
Acknowledgments
This work was supported by the ‘Deutsche Forschungsgemeinschaft’ (SFB 655 to CT, MB and SB); and by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH (Project Z01-AI-000988).
This work was supported by the ‘Deutsche Forschungsgemeinschaft’ (Bonn, Germany) (SFB 655 to CT, MB and SB).
Footnotes
Conflict of Interest Disclosure
No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Gilliland DG. FLT3-activating mutations in acute promyelocytic leukaemia: A rationale for risk-adapted therapy with FLT3 inhibitors. Best Pract Res Clin Haematol. 2003;16:409–417. doi: 10.1016/s1521-6926(03)00063-x. [DOI] [PubMed] [Google Scholar]
- 2.Lee BH, Tothova Z, Levine RL, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. 2007;12:367–380. doi: 10.1016/j.ccr.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–3914. [PubMed] [Google Scholar]
- 4.Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 5.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 6.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158–1165. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 7.Tavor S, Petit I, Porozov S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64:2817–2824. doi: 10.1158/0008-5472.can-03-3693. [DOI] [PubMed] [Google Scholar]
- 8.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 9.Broxmeyer HE, Kohli L, Kim CH, et al. Stromal cell-derived factor-1/CXCL12 directly enhances survival/antiapoptosis of myeloid progenitor cells through CXCR4 and G[alpha]i proteins and enhances engraftment of competitive, repopulating stem cells. J Leukoc Biol. 2003;73:630–638. doi: 10.1189/jlb.1002495. [DOI] [PubMed] [Google Scholar]
- 10.Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–6224. doi: 10.1182/blood-2008-05-158311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liesveld JL, Bechelli J, Rosell K, et al. Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk Res. 2007;31:1553–1563. doi: 10.1016/j.leukres.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Juarez J, Dela PA, Baraz R, et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia. 2007;21:1249–1257. doi: 10.1038/sj.leu.2404684. [DOI] [PubMed] [Google Scholar]
- 13.Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia [AML] following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–6214. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104:550–557. doi: 10.1182/blood-2004-02-0566. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha[CXCL12]/CXCR4 axis. Blood. 2005;105:3117–3126. doi: 10.1182/blood-2004-04-1440. [DOI] [PubMed] [Google Scholar]
- 16.Koch S, Jacobi A, Ryser M, Ehninger G, Thiede C. Abnormal localization and accumulation of FLT3-ITD, a mutant receptor tyrosine kinase involved in leukemogenesis. Cells Tissues Organs. 2008;188:225–235. doi: 10.1159/000118788. [DOI] [PubMed] [Google Scholar]
- 17.Brenner S, Whiting-Theobald NL, Linton GF, et al. Concentrated RD114-pseudotyped MFGS-gp91phox vector achieves high levels of functional correction of the chronic granulomatous disease oxidase defect in NOD/SCID/beta- microglobulin−/− repopulating mobilized human peripheral blood CD34+ cells. Blood. 2003;102:2789–2797. doi: 10.1182/blood-2002-05-1482. [DOI] [PubMed] [Google Scholar]
- 18.Brenner S, Ryser MF, Whiting-Theobald NL, Gentsch M, Linton GF, Malech HL. The late dividing population of gamma-retroviral vector transduced human mobilized peripheral blood progenitor cells contributes most to gene-marked cell engraftment in nonobese diabetic/severe combined immunodeficient mice. Stem Cells. 2007;25:1807–1813. doi: 10.1634/stemcells.2006-0581. [DOI] [PubMed] [Google Scholar]
- 19.Jo DY, Rafii S, Hamada T, Moore MA. Chemotaxis of primitive hematopoietic cells in response to stromal cell-derived factor-1. J Clin Invest. 2000;105:101–111. doi: 10.1172/JCI7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soriano SF, Hernanz-Falcon P, Rodriguez-Frade JM, et al. Functional inactivation of CXC chemokine receptor 4-mediated responses through SOCS3 up-regulation. J Exp Med. 2002;196:311–321. doi: 10.1084/jem.20012041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukuda S, Pelus LM. Internal tandem duplication of Flt3 modulates chemotaxis and survival of hematopoietic cells by SDF1alpha but negatively regulates marrow homing in vivo. Exp Hematol. 2006;34:1041–1051. doi: 10.1016/j.exphem.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 22.Pello OM, Moreno-Ortiz MC, Rodríguez-Frade JM, et al. SOCS up-regulation mobilizes autologous stem cells through CXCR4 blockade. Blood. 2006;108:3928–3937. doi: 10.1182/blood-2006-02-006353. [DOI] [PubMed] [Google Scholar]
- 23.Choudhary C, Brandts C, Schwable J, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110:370–374. doi: 10.1182/blood-2006-05-024018. [DOI] [PubMed] [Google Scholar]
- 24.Schuringa JJ, Wierenga AT, Kruijer W, et al. Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood. 2000;95:3765–3770. [PubMed] [Google Scholar]
- 25.Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–3914. [PubMed] [Google Scholar]
- 26.Tavor S, Eisenbach M, Jacob-Hirsch J, et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia. 2008;22:5151–5158. doi: 10.1038/leu.2008.238. [DOI] [PubMed] [Google Scholar]
- 27.Fierro FA, Brenner S, Oelschlaegel U, et al. Combining SDF-1/CXCR4 antagonism and chemotherapy in relapsed acute myeloid leukemia. Leukemia. 2008;23:393–396. doi: 10.1038/leu.2008.182. [DOI] [PubMed] [Google Scholar]
- 28.Hendrix CW, Collier AC, Lederman MM, et al. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37:1253–1262. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]