

SUMMARY
High field asymmetric waveform ion mobility spectrometry (FAIMS) is an atmospheric pressure ion mobility technique that separates gas-phase ions by their behavior in strong and weak electric fields. FAIMS is easily interfaced with electrospray ionization and has been implemented as an additional separation mode between liquid chromatography (LC) and mass spectrometry (MS) in proteomic studies. FAIMS separation is orthogonal to both LC and MS and is used as a means of on-line fractionation to improve detection of peptides in complex samples. FAIMS improves dynamic range and concomitantly the detection limits of ions by filtering out chemical noise. FAIMS can also be used to remove interfering ion species and to select peptide charge states optimal for identification by tandem MS. Here, we review recent developments in LC-FAIMS-MS and its application to MS-based proteomics.
Keywords: mass spectrometry, proteomics, FAIMS, DMS, ion mobility
THE NEED FOR SELECTIVITY OF GAS PHASE IONS
Identification of proteins from peptide fractionation by liquid chromatography coupled to a mass spectrometer (LC-MS) is a standard procedure in current biological research. In the typical shotgun or “bottom-up” approach [1], proteins are enzymatically digested (usually with trypsin to maintain a basic amino acid at the C-terminus and thus a charge) into peptides which are separated by high performance liquid chromatography (HPLC; usually reverse-phase). The chromatography column is interfaced with a mass spectrometer by electrospray ionization (ESI). Low flow ESI (< 1 μL/min) with pulled tip glass emitters is commonly referred to as nanospray or nanoESI (NSI). Peptide ions are separated by their mass-to charge ratio (m/z) in a precursor scan (MS1) and then individually selected for fragmentation (either in the same ion-trap or a in secondary quadrupole), and the fragments are again separated by m/z in a product ion scan (MS2). The most common means of fragmentation is collision-induced dissociation (CID), though electron transfer dissociation (ETD) is becoming common and is often used in tandem with CID. Fragment ion spectra are then matched to theoretical spectra [2] or a spectral library [3] with statistical validation [4] to determine peptide identity. These peptide spectrum matches (PSM) are used to infer the proteins present in the original sample. If the m/z is known for a peptide precursor ion and its corresponding fragment ions, these “transitions” can be monitored uniquely and their intensities used to quantify the peptide of interest in a selected reaction monitoring (SRM) experiment [5].
With the move toward a systems approach to biology in which the researcher attempts to understand the entirety of complex and dynamic biochemical networks, the burden has fallen on the mass spectrometrist to be able to identify and quantify thousands of proteins in complex biological samples [6]. The ability to select a peptide precursor ion for fragmentation and subsequent identification is limited by the mass analyzer’s duty cycle (how many mass spectra can be obtained per unit time), resolving power (the ability to distinguish amongst precursor ions of similar mass), and dynamic range (the greatest and least amount of analyte that produces a linear response from the detector). Peptides from a digest of a complex sample (e.g., cellular lysates) are typically identified using a data-dependent acquisition (DDA) approach: the most abundant ion in the precursor scan is selected for fragmentation, then the next most abundant, and so on. If the duty cycle of the mass analyzer is insufficient to sample all the precursor ions present in a scan, the least abundant ions will not be selected for fragmentation. Using a dynamic peptide ion exclusion list in real-time can enable detection of low-abundance ions by ignoring previously-analyzed precursors, but a complex sample will still overwhelm the instrumental duty cycle [7]. The resolving power of the mass analyzer (defined as M/ΔM, where M is the m/z of an ion and ΔM is the peak width at half of maximum) determines whether precursors with similar m/z can be distinguished from each other and individually selected for fragmentation. Even with high mass accuracy, greater sample complexity increases the likelihood of multiple precursors being simultaneously fragmented (resulting in difficult-to-interpret chimeric spectra [8]) or of a less abundant precursor being masked by a more abundant precursor. Finally, in order to be selected for fragmentation, a precursor ion must be distinguishable from the background noise.
Although mass analyzers continue to improve in resolving power, duty cycle, and dynamic range, effective sampling of every component in a complex protein digest is still beyond the reach of a single LC-MS experiment, so the issue of sample complexity is usually dealt with by some sort of fractionation prior to LC-MS analysis. Typical approaches include strong cation exchange[2], isoelectric focusing [9-11], or gel electrophoresis [12]. Sample pre-fractionation is routine, effective, and in some cases can be automated [13], but generally increases the amount of sample required, contributes to sample loss, and significantly increases variability between experiments. Fractionating peptide ions in the gas phase avoids sample handling. One approach to overcoming sample complexity by gas-phase fractionation in shotgun experiments is to select only precursors in a narrow range of m/z for fragmentation [14,15], and perform repeat injections of the same sample until the desired m/z range is covered. This approach to fractionation increases the likelihood of selecting a low-intensity precursor for fragmentation by relieving the constraints on the duty cycle of the mass analyzer, but does not solve the problem of interfering species or background signal.
A technique that solves fractionation reproducibility is high field asymmetric waveform ion mobility spectrometry (FAIMS). FAIMS is an atmospheric pressure ion mobility technique that separates gas-phase ions based on their characteristic differences in mobility in high and low electric fields. In this review, we will show how FAIMS is used to improve identification of peptides by LC-MS by providing additional fractionation without manual sample handling.
FAIMS: BASIC CONCEPTS AND TERMINOLOGY
FAIMS is alternatively known as differential mobility spectrometry (DMS). Some groups use the terms DMS or FAIMS to refer to devices with planar or curved electrode geometries, respectively, though many groups use the terms interchangeably. This review will use the term FAIMS to describe both. The theory and practice of FAIMS has been well-described. The reader is directed to a general review of FAIMS by Kolakowski and Mester [16], a series of theoretical and practical explorations of FAIMS by Nazarov and co-workers [17-22], a chapter by Eiceman and Karpas [23], and a book by Shvartsburg [24]. A brief treatment of the salient terminology and theory is provided here.
Gas-phase ions accelerated through a gas medium by an electric field (E) quickly reach a steady-state velocity, the magnitude of which is directly proportional to the ion’s charge and inversely proportional to the ion’s collision cross section (i.e. size). The velocity (cm/s) with respect to electric field strength (V/cm) of an ion is reported as its mobility K (cm2/V·s). At low field strengths K remains constant with varying E and allows comparison of ion behavior under different experimental conditions. As the magnitude of E increases, however, heating causes K vs. E to become non-linear. The non-linear responses are generalized as three types: Type A ions increase in mobility at high field (e.g. small molecules declustering from adducts, thus decreasing in collision cross section); Type C ions decrease in mobility (e.g. peptides unfolding, thus increasing in collision cross section); and Type B ions increase in mobility initially, but decrease in mobility as E increases due to high-energy collisions.
In traditional drift tube ion mobility spectrometry (IMS), a constant low field E is maintained in a chamber filled with drift gas. Gas phase ions separate based on size-to-charge, ions having greater K reaching the detector first. In FAIMS, gas-phase ions are carried by a flow of carrier gas between two electrodes in a direction orthogonal to the direction of E. E is generated as an asymmetric waveform that alternates between a high field voltage of one polarity and a low field voltage of the opposite polarity. The magnitude of the high field portion of the wave is reported as the dispersion voltage (DV). The duration of the low field portion of the wave is longer than the high field portion. In most of the devices described here, the low field portion of the wave is half the magnitude and twice the duration of the high field portion such that the area under the curve is equal for negative and positive polarity [25]. An ion in the field will alternate between travelling toward one electrode or the other as the field oscillates in polarity. If there is no difference in the ion’s mobility in the high and low field, there will be no net displacement toward either electrode and it will exit the FAIMS device and can be detected. If an ion has a higher mobility in the high or low field, it will experience a net displacement toward one electrode. If the ion reaches the electrode it will be neutralized and will not be detected. A small direct current compensation voltage (CV) added to the waveform will bias the net displacement and change which subset of ions will pass through the FAIMS device to the detector. In contrast to drift tube IMS in which the analyte ion stream is sampled in discrete packets and ions of all mobilities reach the detector, FAIMS is a continuous filtration technique that allows uninterrupted sampling of the ion stream, but only for the subset of the ion population allowed by the selected CV.
DEVELOPMENT OF ESI-FAIMS-MS
FAIMS was developed in the former Soviet Union in the early 1980’s and was shared with the international scientific community in the early 1990’s [26]. Both planar (p-FAIMS) and cylindrical (c-FAIMS) electrode geometries were described, and FAIMS devices were coupled both to electrometer detectors and to MS. Much of the early development of FAIMS-MS was performed by researchers in the lab of Roger Guevremont and employed FAIMS devices with cylindrical geometries [27]. A technical challenge with coupling ESI to FAIMS is allowing analyte ions to enter the FAIMS device while maintaining a controlled gas environment within. The solution presented by Purves et al. [28,29] was to pump an excess of carrier gas into the FAIMS device, thus maintaining a positive gas pressure within the device and creating a flow of “curtain gas” from the entrance aperture. This curtain gas aided in desolvation and blew away neutrals while charged ions were drawn into the device by a voltage bias. This approach to coupling ESI to FAIMS is universal among FAIMS devices of all geometries.
It was observed that the sensitivity of a c-FAIMS device (the percentage of analyte ions that passed through the device) increases as DV is increased [30]. With planar FAIMS electrodes, increasing field strength causes ions to travel further during each cycle of the waveform and collide with the electrodes, decreasing signal. In contrast, the inhomogeneous field created by the cylindrical electrodes actively focuses ions into a narrow region between the electrodes [30-33]. (See [101] for a very clear explanation of ion focusing in c-FAIMS). It is important to note that the effect of ion focusing is not uniform. Ions that exhibit high field dependence of mobility (that is, a large difference in mobility in high and low fields) will be focused more strongly and thus may have very high transmission efficiency through a c-FAIMS device, but ions with low field dependence will be weakly focused and may transmitted poorly or not at all [34].
The ion focusing of c-FAIMS limits which types of ions will pass through the device. In p-FAIMS, type A ions of a given polarity will have a net displacement toward one electrode and type C ions of the same polarity will have a net displacement toward the other. Type A ions can be brought into equilibrium with one polarity of CV, type C ions with CV’s of the opposite polarity. If the polarity of the FAIMS waveform is reversed, the FAIMS separation is identical as long as the polarity of the CV’s is also reversed accordingly. In c-FAIMS, however, a FAIMS waveform that focuses Type C ions of a given polarity will actively defocus Type A ions of the same polarity, causing them to expire on the electrodes. This defocusing cannot be rescued with CV. If the polarity of the waveform is reversed, type A ions will be focused but type C ions will be defocused (FIGURE 1 A). While this focusing phenomenon limits the type of ions that can be analyzed under a given set of FAIMS conditions, it is actually of some benefit to analysis of peptides. Analyses of poly-Gly peptides of various lengths by ESI-FAIMS-MS with c-FAIMS [29] showed that as peptides increase in length, they move from type A ion behavior (in which mobility increases in high field) to type C ion behavior (in which mobility decreases in high field). Peptides of interest in proteomics studies are typically ≥7 residues in length and exhibit type C behavior, so a cylindrical FAIMS tuned to transmit peptides will largely exclude short peptides and small molecules that would contribute to chemical noise. It should be kept in mind, however, that the extent of ion focusing (and thus transmission efficiency) decreases with field dependence, an ion-specific characteristic, so the efficiency of transmission through a c-FAIMS device is not uniform for peptides. It is also important to note that ion transmission through a c-FAIMS device is very low when the FAIMS waveform is absent [35]. Consequently, a c-FAIMS device cannot be operated in transparent mode to pass all ions and the hardware must be removed in order to perform non-FAIMS analyses.
Figure 1. Cylindrical FAIMS.
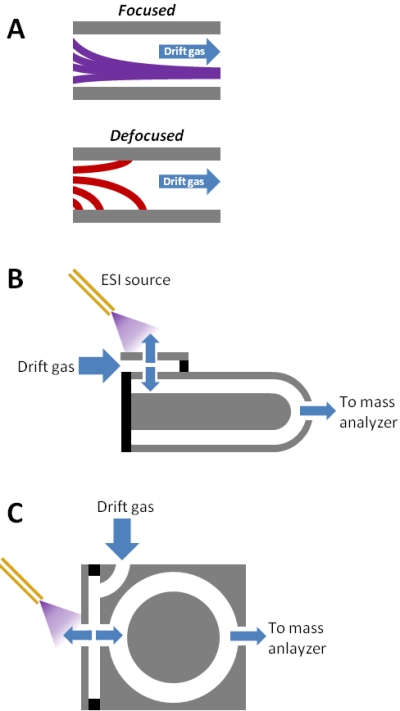
(A) Simulation of focusing and defocusing in a cylindrical FAIMS device like the one shown in (B). Ion trajectories are shown for the same ion originating at different radial distances from the center electrode. Ions will be focused or defocused depending on their properties and the instrumental parameters. If type C ions (e.g., peptides) are focused, type A ions will be defocused. The type C ions will defocus if the polarity of the FAIMS waveform is reversed. (B) Side cutaway view of the device from [36-40]. The electrodes are concentric cylinders that terminate in domes. Ions travel the length of the electrodes and encounter an additional focusing region in the domes. (C) Side cutaway view of the device from [43,45,46,49-54]. Ions travel the radius of the cylindrical electrodes rather than lengthwise as in (B).
(A) adapted from [18] , ©2010, with permission from Elsevier. (B) adapted with permission from [36], © 2000 American Chemical Society.
Ionalytics corporation was formed in 2001 and the c-FAIMS device developed by Guevremont et al. was commercialized and sold as the Selectra (FIGURE 1 B). Various ESI-FAIMS-MS experiments with tryptic digests of protein standards [36-38] demonstrated the potential of FAIMS to augment proteomics research. Signal-to-noise was improved by FAIMS, and in many cases the ion current for a peptide was greater with c-FAIMS than without, likely because the FAIMS device delivered ions into the MS more efficiently than was possible with the unaided source. It was also observed that singly charged peptide ions tended to be optimally transmitted at smaller CV’s while doubly and triply charged peptide ions were detected at CV’s of larger magnitude. Since multiply charged precursors are preferable for fragmentation by CID, it was proposed that it would be advantageous to implement FAIMS as a filtering step between LC and MS for proteomics.
CYLINDRICAL FAIMS FOR PROTEOMICS
To date, nearly all published examples of shotgun proteomics by LC-FAIMS-MS have employed c-FAIMS. The first example of LC-FAIMS-MS was reported by Venne et al. [39,40], who used the Ionalytics c-FAIMS device to couple a capillary LC (nanoLC) column to a Micromass (Waters) Q-TOF. The signal for an infusion of [Glu-1] fibrinopeptide B was 20-40% greater with FAIMS than without, and signal-to-noise was reduced 8-fold. A tryptic digest of an 8-protein standard mixture was separated on a 150 μm I.D. (inner diameter) × 10 cm column with a 60 minute gradient at 0.6 μL/min and analyzed by MS with and without FAIMS. A CV-stepping experiment was designed in which 3 separate CV’s were used in a single nanoLC-FAIMS-MS run: MS1 and subsequent data-dependent acquisition of MS2 scans were collected at one CV, then the next set of MS1 and MS2 scans were collected at a second CV, and then again at a third CV. In this fashion, FAIMS provided an additional dimension of separation and resulted in a 12% increase in the number of unique peptide ions identified compared to a nanoLC-MS analysis of the same sample without using FAIMS. The compatibility of nanoLC and FAIMS is significant since most modern shotgun proteomics platforms employ nanoLC due to the decreased sample requirements, improved separation efficiency [41], and increased ionization efficiency [42].
Thermo-Fisher Scientific acquired Ionalytics Corporation in 2005 and released a c-FAIMS device that interfaces with their instruments and software environment. The Thermo-Fisher Scientific c-FAIMS device retained the concentric cylinder design of the Ionalytics device, but rather than ions travelling lengthwise down the electrodes, they entered an aperture medial to the analytical region of the electrodes, traversed the interstitial space in a direction orthogonal to the axis of the electrodes, and exited through an aperture on the opposite side [43] (FIGURE 1 C). The inner and outer electrodes had diameters of 13 mm and 18 mm, respectively, and were 25 mm in length, corresponding to a residence time of ~150 ms (assuming 1.25 L/min carrier gas). The c-FAIMS device was compatible with standard high flow ESI probes (designed for flow rates in the μL/min to mL/min range) but not NSI probes, so an aftermarket NSI probe was required to couple a nanoLC column to FAIMS [44]. Canterbury et al. [45] used a prototype version of the Thermo-Fisher Scientific c-FAIMS device coupled to a LTQ linear ion trap to explore gas phase fractionation of peptides by FAIMS. Samples were separated on a 75 μm I.D. × 40 cm column with a 90 minute gradient at 0.35 μL/min. NanoLC-FAIMS-MS of a yeast lysate acquiring only precursor (MS1) scans was performed in order to ascertain the increase in peak capacity afforded by FAIMS. Stepping through 9 CV’s in a single nanoLC-FAIMS-MS experiment produced a 9-fold increase in peak capacity due to the additional separation dimension. The authors reported a loss of signal of at least one order of magnitude with nanoLC-FAIMS compared to no FAIMS, though the signal loss was offset by a 5-fold increase in dynamic range. To explain why such a significant signal loss was observed with the Thermo-Fisher Scientific c-FAIMS device when gains in signal had been reported with other c-FAIMS devices, the authors proposed that the problem was the direction that ions travel in the device. In the Ionalytics c-FAIMS device, ions were guided to the exit aperture by domes at the end of the electrodes that provide an additional region of ion focusing. In the Thermo-Fisher Scientific c-FAIMS device, ions were able to spread across the analyzer region by diffusion and electrostatic repulsion, but were not focused back to the exit aperture by a field; rather, ions were assumed to be guided out of the analytical region by the fluid dynamics of the carrier gas exiting the device. It is therefore conceivable that some ions were lost. Saba et al. [46] used the same FAIMS device coupled to a LTQ-Orbitrap and were able to significantly ameliorate the signal loss observed by Canterbury et al. [45], reporting only a two-fold decrease in the signal of highly abundant precursors in nanoLC-FAIMS-MS experiments, and an increase in the signal of low-abundance precursors. Both groups used the same FAIMS devices and NSI probe adaptor; however, Canterbury et al. [45] used 100% N2 as a carrier gas and did not regulate the temperature of the FAIMS electrodes, whereas Saba et al. used 1:1 He:N2 as the carrier gas and maintained the outer and inner FAIMS electrode temperatures at 90°C and 70°C, respectively. The addition of He to the carrier gas has been observed to significantly increase the transmission efficiency of peptides in FAIMS with planar and cylindrical electrode configurations [38,47,48]. The behavior of ions at high fields differs greatly in different gases [49]. This difference accentuates the field dependence of peptides, increasing CV’s and thus ion focusing (and thus transmission efficiency) as well as resolving power [47]. Elevated electrode temperatures improve transmission efficiency in c-FAIMS by increasing field strength with respect to gas density [43,50].
Saba et al. [46] used nanoLC-FAIMS-MS to analyze tryptic digests of U937 human monocytes activated with a tumor promoter. Samples were separated on a 150 μm I.D. × 10 cm column with a 53 minute gradient at 0.6 μL/min. Each nanoLC-FAIMS-MS technical replicate consisted of a total of 6 injections: a CV stepping experiment cycling through 5 CV’s and collecting only MS1 data, followed by 5 DDA experiments, each at a fixed CV. The MS1-only CV stepping experiment was used to obtain peak intensities used for label-free quantification and the DDA experiments were used to identify the peptides. A peak matching algorithm used accurate mass and retention time to match PSM’s to quantified precursors. Three sets of FAIMS analyses identified 46% more peptides from control and 55% more peptides from activated monocytes than three nanoLC-MS injections without FAIMS. Using FAIMS decreased the number of +1 ions observed while increasing the number of multiply charged ions observed, especially +2, validating earlier predictions to this effect. Bridon et al. used a similar technique and added ETD to improve detection of phosphopeptides [51]. The use of FAIMS increased the number of phosphopeptides identified by 60%. The population of phosphopeptides identified by nanoLC-MS without FAIMS was not subsumed by the nanoLC-FAIMS-MS analysis: 45% of the phosphopeptides identified without FAIMS (22% of all the phosphopeptides identified by the combined FAIMS and non-FAIMS experiments) were not identified by the FAIMS analysis. This fraction of peptides not observed by FAIMS likely represents a population of peptides that do not transmit optimally or at all through the FAIMS device at the selected CVs, suggesting that peptide discovery experiments benefit from some number of non-FAIMS replicates in addition to FAIMS analyses. Further investigation into the behavior of these peptide ions not observed with FAIMS is warranted.
Recently, we reported coupling the Thermo-Fisher Scientific c-FAIMS device to a LTQ Velos Orbitrap for nanoLC-FAIMS-MS analysis of tryptic digests [52]. A novel NSI-FAIMS interface was described, consisting of a high flow ESI probe that was modified to accommodate a capillary column. In addition to allowing the use of nanoLC with FAIMS, the modified probe also allowed the use of sheath gas, a feature not available with other adaptors. 100% N2 was used as the FAIMS carrier gas and the outer and inner FAIMS electrode temperatures were 90°C and 70°C, respectively. Samples were separated on a 75 μm I.D. × 15 cm column with a 60 minute gradient at 0.3 μL/min. Infusing human angiotensin-I, the signal with FAIMS was 4% of that without FAIMS, but adding sheath gas increased the signal to 17% of the signal without FAIMS. The signal-to-noise for the infused peptide was 5 times greater with FAIMS than without. DDA experiments with a SILAC-labeled haploid and diploid yeast tryptic digest were performed with and without FAIMS. For nanoLC-FAIMS-MS experiments, each injection was held at a fixed CV for the duration of the analysis. Multiple injections were performed across the range of CV’s at which peptides could be detected. The number of unique peptide sequences identified by multiple DDA experiments with FAIMS at different CV’s was greater than the number of peptides identified from an equal number of injections without FAIMS after as few as three injections, and the advantage of FAIMS increased with the number of injections at different CV’s (4% more unique peptide sequences after 3 injections, 14% more after 5 injections, 23% more after 10 injections, and 50% more after 35 injections).
In the experiments with the Thermo-Fisher Scientific c-FAIMS device described above, a pause of 0.1 to 0.3 seconds was programmed between acquisitions at different CV’s to accommodate the FAIMS residence time (the time required for an ion to traverse the length of the FAIMS device). This loss of acquisition time represents a significant loss in duty cycle, especially for modern rapid mass analyzers. Although CV stepping experiments increase the peak capacity of a LC-MS experiment, more MS2 can be acquired without CV stepping experiments, which may be why none of the groups employing this c-FAIMS device were able to improve peptide discovery when collecting MS2 in a CV stepping experiment.
The benefit of the enhanced selectivity and sensitivity provided by FAIMS has been demonstrated for quantitative proteomics by SRM. Xia et al. employed the Thermo-Fisher Scientific c-FAIMS device coupled to a TSQ Quantum triple quadrupole instrument to augment quantification of peptide standards spiked into rat plasma [53]. Sample was separated on a 2.1 mm I.D. × 50 mm column with a 3-minute gradient at 0.4 mL/min and ionized by heated ESI. 2:3 He:N2 was used as the FAIMS carrier gas, and the outer and inner FAIMS electrode temperatures were 90°C and 50°C, respectively. The optimal CV for the peptide of interest was determined experimentally by infusion and held constant for subsequent LC-FAIMS-MS analyses. The absolute peak area of the analyte of interest decreased by ~30% with FAIMS, but the signal-to-noise doubled, leading to overall improved precision in quantification, especially at low concentrations. Experimental reproducibility was also improved by FAIMS: after 120 injections, the coefficient of variance for peak area and signal-to-noise was 10% and 5.6%, respectively, compared to 12% and 65% without FAIMS. Klassen et al. performed a similar experiment with the Thermo-Fisher Scientific FAIMS device coupled to a TSQ Quantum Ultra. A peptide standard spiked in rat plasma was separated on a 2.1 mm I.D. × 30 mm column with a 6 min gradient at 0.2 mL/min [54]. 1:1 He:N2 was used as the FAIMS carrier gas, and the outer and inner FAIMS electrode temperatures were 90°C and 70°C, respectively. The peak area for the peptide of interest was on average 12% greater with FAIMS, though the peak area of the internal standard was 10% lower. There was an 18-fold improvement in signal-to-noise for a target peptide at the lower limit of quantification using FAIMS.
PLANAR FAIMS FOR PROTEOMICS
Most of the recent developments in FAIMS-MS technology have focused on planar FAIMS (p-FAIMS) devices that employ two flat parallel electrodes rather than concentric cylinders. There are a variety of advantages to p-FAIMS configurations over c-FAIMS: the simple electrode configuration of p-FAIMS is more amenable to miniaturization, allowing for decreased residence times and faster CV scanning speeds; p-FAIMS devices can be operated in transparent mode by setting the electrode potential to ground, allowing the user to switch between FAIMS and non-FAIMS analyses without altering any hardware; and p-FAIMS can achieve higher resolving power. The resolving power of p-FAIMS is greater than that of c-FAIMS because the parallel planar electrodes produce a uniform electric field with no focusing effect. Ions may be at equilibrium within a c-FAIMS device over a range of CV’s [30], and ions that would have been separated in a uniform field may be at equilibrium together, whereas a given ion will only be at equilibrium at a single CV in a p-FAIMS device [55], resulting in narrower FAIMS peaks (signal with respect to CV). The FAIMS peak capacity is further reduced in c-FAIMS because the width of the peak increases with the magnitude of the CV [30,33], whereas peak width is independent of CV magnitude in p-FAIMS [55]. Resolving power in p-FAIMS increases as the square root of residence time [56], whereas FAIMS peaks narrow slowly in c-FAIMS in a manner dependent on the extent of ion focusing [25,33]. Increased resolving power in p-FAIMS is not without cost: absent focusing to counteract diffusion, ion transmission in p-FAIMS decreases exponentially with residence time, necessitating a trade-off between selectivity and sensitivity. In addition to residence time, transmission efficiency in p-FAIMS depends on the mobility of the species: transmission efficiency decreases for high mobility ions because they exhibit higher diffusion and travel farther during each cycle of the FAIMS waveform, colliding with the electrodes sooner than low mobility ions. However, since there is no focusing or defocusing in p-FAIMS, transmission efficiency is determined only by absolute mobility and not field dependence, so ions that would be reduced in signal or lost entirely with c-FAIMS can be passed through a p-FAIMS device [34,35].
Levin et al. modified a commercial p-FAIMS device with a 1 mm gap (Sionex microDMX) to be compatible with NSI and MS [57,58]. Ions entered the device through an aperture in one of the two parallel plate electrodes, were carried lengthwise through the interstitial space, and exited through an aperture in the opposite electrode into the mass analyzer, a Waters single quadrupole detector (FIGURE 2A). The dimensions of the device decreased the residence time over an order of magnitude compared with the c-FAIMS devices described earlier, enabling the device to scan through 100 CVs in 1 second (when not constrained by the mass analyzer acquisition time). The effect of chemical modifiers in the carrier gas was studied, and it was observed that the presence of isopropanol in the carrier gas improved FAIMS separation of peptide standards. Addition of polar modifiers can improve FAIMS separation by forming clusters with gas phase ions, increasing the difference in mobility in high and low fields [20,22]. Transmission efficiency through the Sionex p-FAIMS device was not reported in these works.
Figure 2. Planar FAIMS.
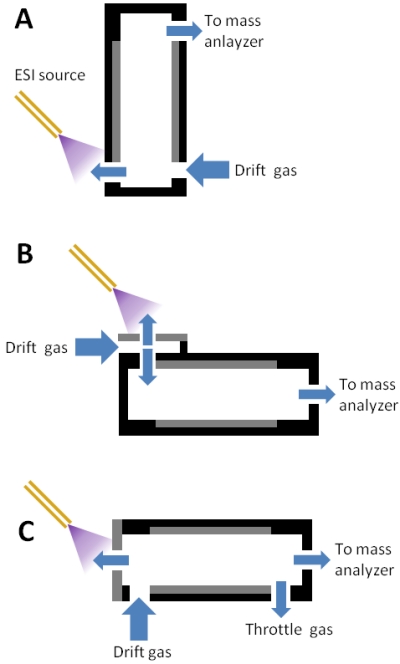
Side cutaway views of various ESI-FAIMS-MS interfaces employing planar electrodes. (A) The device from [57,58]. (B) The device from [55,59-63]. (C) The device from [18]. The throttle gas controls the flow rate of the carrier gas, allowing the user to vary between shorter residence time (increased sensitivity) and longer residence time (increased resolving power).
(A) adapted with permission from [58], © 2006 American Chemical Society. (B) adapted with permission from [55], © 2006 American Chemical Society. (C) adapted from [18], © 2010, with permission from Elsevier.
Shvartsburg et al. constructed a p-FAIMS device with dimensions similar to existing c-FAIMS devices for the purpose of directly comparing performance of c-FAIMS and p-FAIMS in ESI-FAIMS-MS experiments [55]. A curtain plate and curtain gas was employed to accommodate NSI. Ions entered the device through one of the parallel planar electrodes, travelled the length of the electrodes, and exited the gap between the two electrodes directly into the mass analyzer (FIGURE 2B). The ion transfer capillary of the mass analyzer was modified with a row of holes to accommodate the ribbon-shaped ion beam. Peptide standards were infused and MS data was continuously collected as the CV was scanned. Peak profiles of the signal with respect to CV for the infused peptides were used to calculate FAIMS resolving power and peak capacity. The resolving power of a p-FAIMS device with a 2 mm gap and 0.1 sec residence time was 3-4 times greater than with a comparable c-FAIMS device. Experiments with longer residence times ranging from 0.2 sec to 0.5 sec increased the resolving power another 3-fold to 10-fold [59,60], though signal loss was significant. Further experiments with extended residence times and increased concentration of He in the carrier gas demonstrated that high-resolution p-FAIMS could be used for separation of peptide sequence isomers [61], isomeric phosphopeptides with variable sequence modifications [62,63], and variably methylated histone tails[64].
In 2011, ABSciex released a commercial p-FAIMS device (termed SelexION technology) that interfaced with their ion trap and triple quadrupole mass analyzers. Residence time could be varied by adjusting carrier gas flow to facilitate choosing between optimized sensitivity or selectivity. The electrode dimensions corresponded to residence times as low as ~10 ms, enabling more rapid CV scanning than existing c-FAIMS designs. Chemical modifiers could be added to the carrier gas for enhanced separations. The SelexION device could be operated in transparent mode by turning off the dispersion voltage, allowing non-FAIMS analysis without requiring removal of hardware. Initial reports given at the 2012 American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics described using the ABSciex SelexION for infusion and LC-FAIMS-MS analysis of peptides and protein digests. Making FAIMS technology commercially available and readily integrated into commercial instrument platforms has already been seen to greatly increase use of the technology, and peer-reviewed reports of proteomic analyses with the ABSciex p-FAIMS device can be expected in the near future.
ULTRA FAIMS FOR PROTEOMICS
Rapid CV scanning capability is desirable for improved duty cycle in LC-FAIMS-MS. The duty cycles of c-FAIMS devices described to date are limited by long residence times that are the result of their size and the time required to achieve focusing. Shorter residence times have been demonstrated with p-FAIMS devices by decreasing the path length or increasing the carrier gas flow rate, but resolving power is lost as residence time decreases. Shvartsburg et al. [65] described a microfabricated p-FAIMS device manufactured by Owlstone Nanotech and demonstrated that resolving power can be partially maintained while drastically decreasing residence time by increasing the field strength of the asymmetric waveform. The microfabricated FAIMS device (termed ultra FAIMS) was constructed with interleaved plates creating multiple parallel gaps 35 μm in width and 300 μm in length (Figure 3). Compared to conventional p-FAIMS devices that typically obtain field strengths of ~20 kV/cm with DV’s of ~5 kV, the small dimensions of the ultra FAIMS (u-FAIMS) device facilitated field strengths up to 60 kV/cm with a DV of only 214 V. The maximum field strength in FAIMS is fundamentally limited by the voltage at which arcing occurs, but this maximum increases as gap width decreases, so u-FAIMS devices can employ much higher field strengths than can macro FAIMS devices and can achieve these elevated field strengths with significantly less power. Elevated field strengths decrease transmission efficiency due to increased diffusion, but these losses can be partially offset by decreasing residence time. As with macro p-FAIMS devices, decreased residence time leads to decreased resolving power, but in u-FAIMS this loss is partially offset by an increased FAIMS peak capacity that is a result of the elevated field strength.
Figure 3. Ultra FAIMS.
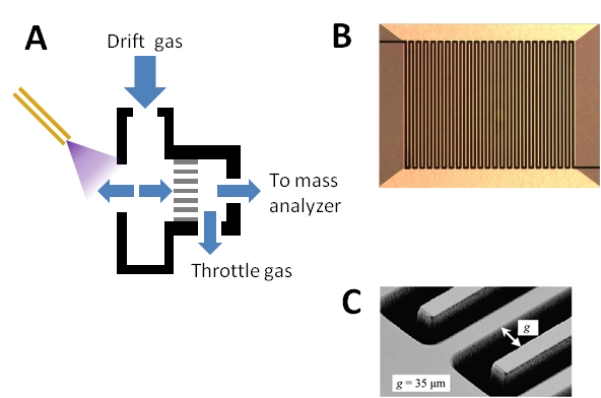
(A) Side cutaway view of the device from [65-68]. The throttle gas controls the flow rate of the carrier gas, allowing the user to vary between shorter residence time (increased sensitivity) and longer residence time (increased resolving power).Note the use of throttle gas (see Figure 2 (C)). (B) Head-on view of the ultra FAIMS device. Multiple channels are created by interleaving plates. (C) Electron micrograph of the device showing gap width.
(A) adapted with permission from [66], © 2009 American Chemical Society. (B) and (C) reprinted with permission from [65], © 2009 American Chemical Society.
Shvartsburg et al. adapted the Owlstone u-FAIMS device for ESI-FAIMS-MS [66] (FIGURE 3). The ESI-FAIMS interface employed curtain gas in a fashion similar to that described for other ESI-FAIMS interfaces. The flow rate of carrier gas could be varied to alter the residence time over the range of ~10 - 60 μs in order to choose between sensitivity and selectivity. Peptide standards were infused at 0.5 μL/min and analyzed by ESI-FAIMS-MS. Optimized for selectivity, the best resolving power achieved with the u-FAIMS was less than that observed with typical p-FAIMS devices or c-FAIMS devices. Optimized for sensitivity, the resolving power was too low to separate the peptide standards from each other, but multiply charged peptides ions as a class could be passed while excluding singly charged polymer contaminants. In this fashion u-FAIMS can be used as a gas-phase filter for sample clean-up. As a result of the extremely high field strengths employed in the u-FAIMS device, small ions (e.g. single amino acids) that would normally behave in a type A fashion (increasing in mobility at high field) instead behaved in a type C fashion (decreasing in mobility at high field) due to hard scattering resulting from high-energy collisions between ions and carrier gas molecules [24,49]. This effect fundamentally limits the separation power of u-FAIMS compared with macro p-FAIMS and c-FAIMS devices that separate type A and type C ions.
Brown et al. performed ESI-FAIMS-MS with the same modified Owlstone Nanotech u-FAIMS device coupled to a Thermo-Fisher Scientific LTQ ion trap as well as an Agilent TOF [67]. Peptide standards were infused at 100 μL/min with a sheath gas-enabled ESI source. Signal-to-noise with u-FAIMS was 10-fold greater than without FAIMS. Singly charged peptides were confined to a narrower region of CV’s (relative to peak capacity) than had been reported in experiments with the Thermo-Fisher Scientific c-FAIMS, and could be separated from multiply charged peptides. The u-FAIMS was used to improve detection of peptides separated by short LC gradients and identified from fragments generated by in-source CID (ISCID) [68]. A mixture of peptide standards was separated on a 2.1 mm × 5 cm column with a 3 minute isocratic elution at 0.2 mL/min, ionized by ESI with sheath gas, fragmented by ISCID, and detected by an Agilent TOF. Lutenizing hormone releasing hormone (LHRH) co-eluted with bradykinin, and the resulting ISCID created a chimeric spectrum containing fragments from singly and multiply charged species of both peptides. Isolating the doubly charged LHRH ion by u-FAIMS decreased the signal for the ion 10-fold but enabled exclusion of singly charged precursors and the doubly charged bradykinin peptide, producing a clean fragment spectrum for LHRH. A standard peptide was spiked into rat plasma at various concentrations spanning just over one order of magnitude and separated on a 2.1 mm × 7.5 cm column with a 10 minute gradient at 0.4 mL/min. The use of u-FAIMS facilitated isolation of the peptide from complex background, and the intensity of fragment ions generated by ISCID was linear with respect to peptide concentration. The authors of the study noted that the combination of FAIMS and ISCID provides quantitative capabilities similar to SRM on mass analyzers that are also capable of qualitative analyses (e.g., shotgun proteomics).
EXPERT COMMENTARY
FAIMS is well-suited to implementation as an additional mode of separation between LC and MS and helps to address some of the instrumental limitations of LC-MS proteomics: on-line sample fractionation by FAIMS is robust and highly reproducible, improving peptide discovery by decreasing sample complexity; FAIMS enables detection of co-eluting species with similar m/z by providing a dimension of separation that is orthogonal to both liquid chromatography and mass spectrometry; and FAIMS improves dynamic range and limits of detection by reducing chemical noise from singly charged peptides, solvated species, and chemical background.
Selection of FAIMS design is application-specific and requires finding a balance in the trade-offs among resolving power, sensitivity, and duty cycle. The greatest sensitivity for peptide analysis has been demonstrated with cylindrical electrode configurations. The focusing effect of curved electrodes offsets diffusion within the device, though the extent of signal focusing is species-dependent. The focusing effect also decreases resolving power. The wide CV peaks of c-FAIMS can actually be beneficial for protein discovery applications because more peptides can be observed at fewer CV’s, relieving the constraint of the poor duty cycle available with current c-FAIMS designs. Singly charged peptides are observed over a narrow range of small CV’s and can be largely excluded from multiply charged peptides by selecting larger CV’s, while multiply charged peptides are observed at larger CV’s with wide, overlapping CV peaks with c-FAIMS. If greater selectivity is required, p-FAIMS is preferable over c-FAIMS or u-FAIMS since the best FAIMS resolving power has been reported with planar electrodes designs. Though the highest resolving power is acquired at the cost of significant loss of sensitivity, superior resolving power can still be obtained with signal loss that is acceptable in light of gains in signal-to-noise and selectivity. To date, microfabricated u-FAIMS devices have demonstrated poorer resolving power than macro p-FAIMS and c-FAIMS devices. However, CV scanning rates that are orders of magnitude faster than is possible with macro devices make u-FAIMS ideal for applications that require rapid duty cycle. The sensitivity of current ultra FAIMS devices is comparable to larger p-FAIMS devices.
Adding FAIMS to an LC-MS workflow requires several considerations. FAIMS fractionates gas phase ions in such a way that those ions that are not passed on to the mass analyzer are lost. In order to analyze multiple FAIMS fractions or monitor multiple precursors, it is necessary to either perform multiple injections of the same sample at different fixed CV’s or to step through multiple CV’s in a single LC-MS experiment. The number of CV’s that is practical or beneficial in a CV stepping experiment is limited by the duty cycle of the mass analyzer, the speed at which the FAIMS device can switch between CV’s, and the width of the chromatographic peak. Currently, there is no tool for predicting CV values based on peptide sequence, so CV must be selected empirically based on experience with the sample of interest and the FAIMS system in use. Since the behavior of a peptide in FAIMS is based on intrinsic properties of the peptide, it should be fundamentally possible to predict optimum CV’s. Numerical values for the optimum CV for a given peptide are dependent on multiple instrumental parameters, including design, electrode dimensions, DV, waveform shape and frequency, electrode temperature, carrier gas composition, and carrier gas flow rate. Further experimentation is required to enable a more complete understanding of the underlying processes involved in peptide separation using FAIMS. Armed with this information, predictive algorithms should be easily obtained to provide accurate determination of parameters to be used for efficient and reproducible fractionation by FAIMS and conversion of these parameters between different designs.
FIVE YEAR VIEW
FAIMS as a technique for augmenting proteomics by MS has been clearly demonstrated, but has yet to be widely adopted by proteomics labs. Some of the obstacles to improved FAIMS performance are fundamental in nature, but many are engineering challenges. As design obstacles are overcome, it will be possible to evaluate various FAIMS configurations based on their fundamental performance potential rather than the limitations of their current implementation.
Early development of FAIMS for proteomics employed cylindrical electrode configurations, and until recently, only c-FAIMS devices were available integrated into commercial mass spectrometry platforms. Consequently, the majority of reports to date describing LC-FAIMS-MS for proteomics have employed c-FAIMS. However, current c-FAIMS designs suffer from poor duty cycle, which is incompatible with the increasingly narrow LC peaks and rapid spectral acquisition of typical proteomics experiments. Recent work has shown that reducing the volume (and thus the residence time) of a commercial c-FAIMS device by decreasing the gap width provided increased signal and resolving power without the need for He in the carrier gas [50]. If improved duty cycle can be achieved while retaining sensitivity, c-FAIMS will continue to be the preferred FAIMS configuration for shotgun proteomics.
The majority of recent developments in FAIMS have focused on planar electrode designs because p-FAIMS is better-suited for rapid CV scanning and exhibits superior resolving power compared to c-FAIMS. Theoretical calculations have shown that ion transmission efficiency with p-FAIMS should in some cases be superior to c-FAIMS, but this has yet to be demonstrated for analysis of peptides. To date, p-FAIMS devices have suffered from significant signal losses due to inefficient ion transfer from source to FAIMS and from FAIMS to mass analyzer. Future development of p-FAIMS devices should focus on eliminating unnecessary signal loss. It is likely that p-FAIMS will remain the configuration of choice for high-resolution applications such as studies of peptide isomers and conformers. The recent availability of a p-FAIMS device incorporated into a commercial proteomics platform (ABSciex SelexION) will enable wider application of p-FAIMS to proteomics research and allow evaluation of the technique against the more well-established Thermo Fisher Scientific c-FAIMS.
Microfabricated u-FAIMS devices are not likely to replace macro p-FAIMS devices in all applications. Miniaturization overcomes obstacles to duty cycle, but the physics of elevated field strengths fundamentally reduces signal transmission, resolving power, and the ability to separate type A and type C ions. Even so, duty cycles that are orders of magnitude greater than possible with macro FAIMS devices are very attractive, and u-FAIMS devices may be the configuration of choice for quantitative applications such as reaction monitoring experiments which require multiple data points to reconstruct the chromatographic peak.
Although FAIMS is a powerful and robust technique capable of significantly augmenting LC-MS for proteomics research, the large majority of reports to date have described only proof-of-concept experiments with standard peptides and model organisms. As the technique gains in visibility and is made commercially available by more vendors, it is likely that FAIMS will achieve greater acceptance in the field of proteomics and become part of the standard proteomics tool kit.
KEY ISSUES.
FAIMS is an atmospheric pressure ion mobility technique that fractionates gas-phase ions by their mobility in strong and weak electric fields. FAIMS is implemented between the ESI source and the mass spectrometer in proteomics experiments.
FAIMS can be used for isolating peptides of interest in quantitative studies, on-line sample fractionation in shotgun experiments, removing interfering species, and removing chemical noise.
Fractionation by FAIMS allows detection of peptides that would otherwise have been missed such as low-intensity precursors or co-eluting peptides with similar m/z.
Cylindrical electrode FAIMS offers the best sensitivity for peptide analysis but the poorest duty cycle. Planar electrode FAIMS can achieve greater duty cycle and resolving power than cylindrical FAIMS, but sensitivity decreases as selectivity increases. Microfabricated planar FAIMS devices have much faster duty cycles than macro devices but with lower resolving power and sensitivity than macro FAIMS devices.
Selection of carrier gas composition (e.g., N2 and He) and additives (e.g. isopropanol) affect the sensitivity and resolving power of FAIMS. Resolving power and sensitivity are improved by the presence of He in the carrier gas, but the cost of the He and the fact that it is a non-renewable resource are prohibitive.
Table 1. Summary of cylindrical FAIMS devices.
| FAIMS Device |
Electrode dimensions |
DV | Carrier Gas |
Ion Source | Mass Analyzer | Sample | Ref. |
|---|---|---|---|---|---|---|---|
| Homebuilt | ro/ri = 7/5 mm gap = 2 mm L not given |
−3.9 kV (19.5 kV/cm) |
N2 | Infused ESI |
PE Sciex API 300 triple quadrupole |
Pig hemoglobin tryptic digest |
[36] |
| Ionalytics Selectra |
ro/ri = 10/8 mm gap = 2 mm L = 30 mm |
−3.8 kV (19 kV/cm) |
N2 | Infused ESI |
MDS-Sciex QqTOF | Pig hemoglobin tryptic digest |
[37] |
| Ionalytics Selectra |
ro/ri = 10/8 mm gap = 2 mm L = 30 mm |
−4.0 kV (20 kV/cm) |
1:1 He:N2 |
Infused ESI |
PE Sciex API 300 triple quadrupole |
Protein standard tryptic digests |
[38] |
| Ionalytics Selectra |
ro/ri = 10/8 mm gap = 2 mm L = 30 mm |
−4.0 kV (20 kV/cm) |
1:1 He:N2 |
NanoLC NSI 0.6 μL/min |
Waters Q-TOF | Human monocyte tryptic digests |
[40] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm |
−5.0 kV (20kV/cm) |
N2 | NanoLC NSI 0.35 μL/min |
Thermo-Fisher Scientific LTQ linear ion trap |
Yeast tryptic digest |
[45] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm To/Ti = 90°/70° C |
−5.0 kV (20kV/cm) |
1:1 He:N2 |
NanoLC NSI 0.6 μL/min |
Thermo-Fisher Scientific LTQ- Orbitrap |
Human monocyte tryptic digest |
[46] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm To/Ti = 90°/70° C |
−5.0 kV (20kV/cm) |
1:1 He:N2 |
NanoLC NSI 0.6 μL/min |
Thermo-Fisher Scientific LTQ- Orbitrap XL |
Phosphopeptides enriched from Drosophila tryptic digest |
[51] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm To/Ti = 90°/70° C |
−5.0 kV (20kV/cm) |
N2 | NanoLC NSI 0.3 μL/min |
Thermo-Fisher Scientific LTQ Velos-Orbitrap |
Yeast tryptic digest |
[52] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm To/Ti = 90°/50° C |
−5.0 kV (20kV/cm) |
2:3 He:N2 |
LC HESI 0.4 mL/min |
Thermo-Fisher Scientific TSQ Quantum triple quadrupole |
Peptide standard spiked in rat plasma |
[53] |
| Thermo- Fisher Scientific |
ro/ri = 9/6.5 mm gap = 2.5 mm L = 25 mm To/Ti = 90°/70° C |
−5.0 kV (20kV/cm) |
1:1 He:N2 |
LC HESI 0.2 mL/min |
Thermo-Fisher Scientific TSQ Quantum Ultra triple quadrupole |
Peptide standard spiked in rat plasma |
[54] |
DV: dispersion voltage; ESI: electrospray ionization; L: length; LC: liquid chromatography; ro/ri: radius of outer/inner electrode; To/Ti: temperature of outer/inner electrode
Table 2. Summary of planar FAIMS and ultra FAIMS devices.
| FAIMS Device |
Electrode dimensions |
DV | Carrier gas | Ion Source | Mass Analyzer | Sample | Ref |
|---|---|---|---|---|---|---|---|
| Sionex SDP-1 microDMX |
L = ~40 mm W = 5 mm Gap = 1.0 mm |
0.3 - 1 kV (3-10 kV/cm) |
N2, Modifiers (e.g., IPA) |
Infused NSI 0.5 μL/min |
Micromass (Waters) ZQ single quadrupole |
Peptide standards | [57,58] |
| Homebuilt | L = 50 mm W = 20 mm gap = 2 mm |
3.9 kV (19.5 kV/cm) |
N2, 10-50% He |
Infused ESI 0.4 μL/min |
Agilent TOF with ion funnel |
Peptide standards | [55] |
| Homebuilt | L = 50 mm W = 20 mm gap = 1.88 mm |
4.0-5.4 kV (21-28.7 kV/cm) |
N2, 0-70% He |
Infused ESI 0.2 μL/min |
Thermo-Fisher Scientific LCQ ion trap, Thermo-Fisher Scientific LTQ linear ion trap |
Protein standard tryptic digest, peptide standards, phosphopeptides, sequence isomers |
[59-63] |
| Owlstone | L = 300 μm W = 115 mm (47 channels) gap = 35 μm |
214 V (61 kV/cm) |
N2 | LC ESI 0.4 mL/min |
Thermo-Fisher Scientific LTQ linear ion trap, Agilent 6230 TOF |
Protein standard tryptic digest, peptide standards |
[65-68] |
DV: dispersion voltage; ESI: electrospray ionization; IPA: isopropyl alcohol; L: length of electrode; LC: liquid chromatography; W: width of electrode
FINANCIAL DISCLOSURE & ACKNOWLEDGMENTS
The authors thank the National Science Foundation MRI (grant 0923536), the National Institute of General Medical Sciences (grant No. 2P50 GM076547/Center for Systems Biology) and the Luxembourg Centre for Systems Biomedicine and the University of Luxembourg for support.
REFERENCES
- 1.Wu CC, MacCoss MJ. Shotgun proteomics: tools for the analysis of complex biological systems. Curr Opin Mol Ther. 2002;4(3):242–250. [PubMed] [Google Scholar]
- 2.Link AJ, Eng J, Schieltz DM, et al. Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17(7):676–682. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 3.Lam H. Building and searching tandem mass spectral libraries for peptide identification. Mol Cell Proteomics. 2011;10(12):R111 008565. doi: 10.1074/mcp.R111.008565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deutsch EW, Mendoza L, Shteynberg D, et al. A Guided Tour of the Trans Proteomic Pipeline. Proteomics. 2010 doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lange V, Picotti P, Domon B, Aebersold R. Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bensimon A, Heck AJ, Aebersold R. Mass spectrometry-based proteomics and network biology. Annu Rev Biochem. 2012;81:379–405. doi: 10.1146/annurev-biochem-072909-100424. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 8.Houel S, Abernathy R, Renganathan K, Meyer-Arendt K, Ahn NG, Old WM. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J Proteome Res. 2010;9(8):4152–4160. doi: 10.1021/pr1003856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moritz RL, Ji H, Schutz F, et al. A proteome strategy for fractionating proteins and peptides using continuous free-flow electrophoresis coupled off-line to reversed-phase high-performance liquid chromatography. Anal Chem. 2004;76(16):4811–4824. doi: 10.1021/ac049717l. [DOI] [PubMed] [Google Scholar]
- 10.Moritz RL, Simpson RJ. Liquid-based free-flow electrophoresis-reversed-phase HPLC: a proteomic tool. Nat Methods. 2005;2(11):863–873. doi: 10.1038/nmeth1105-863. [DOI] [PubMed] [Google Scholar]
- 11.Malmstrom J, Lee H, Nesvizhskii AI, et al. Optimized peptide separation and identification for mass spectrometry based proteomics via free-flow electrophoresis. J Proteome Res. 2006;5(9):2241–2249. doi: 10.1021/pr0600632. [DOI] [PubMed] [Google Scholar]
- 12.Clauser KR, Hall SC, Smith DM, et al. Rapid mass spectrometric peptide sequencing and mass matching for characterization of human melanoma proteins isolated by two-dimensional PAGE. Proc Natl Acad Sci U S A. 1995;92(11):5072–5076. doi: 10.1073/pnas.92.11.5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 14.Yi EC, Marelli M, Lee H, et al. Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis. 2002;23(18):3205–3216. doi: 10.1002/1522-2683(200209)23:18<3205::AID-ELPS3205>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Scherl A, Shaffer SA, Taylor GK, Kulasekara HD, Miller SI, Goodlett DR. Genome-specific gas-phase fractionation strategy for improved shotgun proteomic profiling of proteotypic peptides. Anal Chem. 2008;80(4):1182–1191. doi: 10.1021/ac701680f. [DOI] [PubMed] [Google Scholar]
- 16.Kolakowski BM, Mester Z. Review of applications of high-field asymmetric waveform ion mobility spectrometry (FAIMS) and differential mobility spectrometry (DMS) Analyst. 2007;132(9):842–864. doi: 10.1039/b706039d. [DOI] [PubMed] [Google Scholar]
- 17.Krylov EV, Nazarov EG. Electric field dependence of the ion mobility. International Journal of Mass Spectrometry. 2009;285(3):149–156. [Google Scholar]
- 18.Schneider BB, Covey TR, Coy SL, Krylov EV, Nazarov EG. Planar differential mobility spectrometer as a pre-filter for atmospheric pressure ionization mass spectrometry. Int J Mass Spectrom. 2010;298(1-3):45–54. doi: 10.1016/j.ijms.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krylov EV, Coy SL, Vandermey J, Schneider BB, Covey TR, Nazarov EG. Selection and generation of waveforms for differential mobility spectrometry. Rev. Sci. Instrum. 2010;81(2):024101. doi: 10.1063/1.3284507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider BB, Covey TR, Coy SL, Krylov EV, Nazarov EG. Control of chemical effects in the separation process of a differential mobility mass spectrometer system. Eur J Mass Spectrom (Chichester, Eng) 2010;16(1):57–71. doi: 10.1255/ejms.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coy SL, Krylov EV, Schneider BB, et al. Detection of Radiation-Exposure Biomarkers by Differential Mobility Prefiltered Mass Spectrometry (DMS-MS) Int J Mass Spectrom. 2010;291(3):108–117. doi: 10.1016/j.ijms.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider BB, Covey TR, Coy SL, Krylov EV, Nazarov EG. Chemical effects in the separation process of a differential mobility/mass spectrometer system. Anal Chem. 2010;82(5):1867–1880. doi: 10.1021/ac902571u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eiceman GA, Kapras Z. Ion Mobility Spectrometry. CRC Press; Boca Raton, FL: 2005. * A book on ion mobility spectrometry including a chapter on FAIMS.
- 24.Shvartsburg AA. Differential Ion Mobility Spectrometry. CRC Press; Boca Raton, FL: 2009. * The first book devoted entirely to FAIMS
- 25.Shvartsburg AA, Tang K, Smith RD. Optimization of the design and operation of FAIMS analyzers. J Am Soc Mass Spectrom. 2005;16(1):2–12. doi: 10.1016/j.jasms.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK. A new method of separation of multi-atomic ions by mobility at atmospheric pressure using a high-frequency amplitude-asymmetric strong electric field. International Journal of Mass Spectrometry and Ion Processes. 1993;128(3):143–148. [Google Scholar]
- 27.Guevremont R. High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J Chromatogr A. 2004;1058(1-2):3–19. [PubMed] [Google Scholar]
- 28.Purves RW, Guevremont R, Day S, Pipich CW, Matyjaszczyk MS. Mass spectrometric characterization of a high-field asymmetric waveform ion mobility spectrometer. Review of Scientific Instruments. 1998;69(12):4094–4105. [Google Scholar]
- 29.Purves RW, Guevremont R. Electrospray ionization high-field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal Chem. 1999;71(13):2346–2357. doi: 10.1021/ac981380y. [DOI] [PubMed] [Google Scholar]
- 30.Guevremont R, Purves RW. Atmospheric pressure ion focusing in a high-field asymmetric waveform ion mobility spectrometer. Review of Scientific Instruments. 1999;70(2):1370. ** An explanation of ion focusing in cylindrical FAIMS.
- 31.Guevremont R, Purves RW, Barnett DA, Ding L. Ion trapping at atmospheric pressure (760 Torr) and room temperature with a high-field asymmetric waveform ion mobility spectrometer. Int J Mass Spectrom. 1999;193(1):45–56. [Google Scholar]
- 32.Guevremont R, Thekkadath G, Hilton CK. Compensation voltage (CV) peak shapes using a domed FAIMS with the inner electrode translated to various longitudinal positions. J Am Soc Mass Spectrom. 2005;16(6):948–956. doi: 10.1016/j.jasms.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Guevremont R, Purves R. Comparison of experimental and calculated peak shapes for three cylindrical geometry FAIMS prototypes of differing electrode diameters. J Am Soc Mass Spectrom. 2005;16(3):349–362. doi: 10.1016/j.jasms.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Krylov EV. Comparison of the planar and coaxial field asymmetrical waveform ion mobility spectrometer (FAIMS) International Journal of Mass Spectrometry. 2003;225:39–51. [Google Scholar]
- 35.Shvartsburg AA, Tang K, Smith RD. Modeling the resolution and sensitivity of FAIMS analyses. J Am Soc Mass Spectrom. 2004;15(10):1487–1498. doi: 10.1016/j.jasms.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 36.Guevremont R, Barnett DA, Purves RW, Vandermey J. Analysis of a tryptic digest of pig hemoglobin using ESI-FAIMS-MS. Anal Chem. 2000;72(19):4577–4584. doi: 10.1021/ac0000271. ** A novel means of joining ESI, a cylindrical FAIMS device, and MS is used to analyze a protein digest.
- 37.Barnett DA, Ding L, Ells B, Purves RW, Guevremont R. Tandem mass spectra of tryptic peptides at signal-to-background ratios approaching unity using electrospray ionization high-field asymmetric waveform ion mobility spectrometry/hybrid quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2002;16(7):676–680. doi: 10.1002/rcm.621. [DOI] [PubMed] [Google Scholar]
- 38.Barnett DA, Ells B, Guevremont R, Purves RW. Application of ESI-FAIMS-MS to the analysis of tryptic peptides. J Am Soc Mass Spectrom. 2002;13(11):1282–1291. doi: 10.1016/S1044-0305(02)00527-5. [DOI] [PubMed] [Google Scholar]
- 39.Venne K, Bonneil E, Eng K, Thibault P. Enhanced Sensitivity in Proteomics Analyses Using NanoLC–MS and FAIMS. PharmaGenomics. 2004;4:30–40. [Google Scholar]
- 40.Venne K, Bonneil E, Eng K, Thibault P. Improvement in peptide detection for proteomics analyses using NanoLC-MS and high-field asymmetry waveform ion mobility mass spectrometry. Anal Chem. 2005;77(7):2176–2186. doi: 10.1021/ac048410j. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. High-efficiency nanoscale liquid chromatography coupled on-line with mass spectrometry using nanoelectrospray ionization for proteomics. Anal Chem. 2002;74(16):4235–4249. doi: 10.1021/ac0202280. [DOI] [PubMed] [Google Scholar]
- 42.Gangl ET, Annan MM, Spooner N, Vouros P. Reduction of signal suppression effects in ESI-MS using a nanosplitting device. Anal Chem. 2001;73(23):5635–5644. doi: 10.1021/ac010501i. [DOI] [PubMed] [Google Scholar]
- 43.Barnett DA, Belford M, Dunyach JJ, Purves RW. Characterization of a temperature-controlled FAIMS system. J Am Soc Mass Spectrom. 2007;18(9):1653–1663. doi: 10.1016/j.jasms.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Hatsis P, Kapron JT. A review on the application of high-field asymmetric waveform ion mobility spectrometry (FAIMS) in drug discovery. Rapid Commun Mass Spectrom. 2008;22(5):735–738. doi: 10.1002/rcm.3416. [DOI] [PubMed] [Google Scholar]
- 45.Canterbury JD, Yi X, Hoopmann MR, MacCoss MJ. Assessing the dynamic range and peak capacity of nanoflow LC-FAIMS-MS on an ion trap mass spectrometer for proteomics. Anal Chem. 2008;80(18):6888–6897. doi: 10.1021/ac8004988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saba J, Bonneil E, Pomies C, Eng K, Thibault P. Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. J Proteome Res. 2009;8(7):3355–3366. doi: 10.1021/pr801106a. ** A commercially-available cylindrical FAIMS device is combined with nanoLC and a high mass accuracy mass analyzer to improve protein discovery from a complex protein digest.
- 47.Shvartsburg AA, Tang K, Smith RD. Understanding and designing field asymmetric waveform ion mobility spectrometry separations in gas mixtures. Anal Chem. 2004;76(24):7366–7374. doi: 10.1021/ac049299k. [DOI] [PubMed] [Google Scholar]
- 48.Shvartsburg AA, Danielson WF, Smith RD. High-resolution differential ion mobility separations using helium-rich gases. Anal Chem. 2010;82(6):2456–2462. doi: 10.1021/ac902852a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barnett DA, Ells B, Guevremont R, Purves RW, Viehland LA. Evaluation of carrier gases for use in high-field asymmetric waveform ion mobility spectrometry. J Am Soc Mass Spectrom. 2000;11(12):1125–1133. doi: 10.1016/S1044-0305(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 50.Barnett DA, Ouellette RJ. Elimination of the helium requirement in high-field asymmetric waveform ion mobility spectrometry (FAIMS): beneficial effects of decreasing the analyzer gap width on peptide analysis. Rapid Commun Mass Spectrom. 2011;25(14):1959–1971. doi: 10.1002/rcm.5078. [DOI] [PubMed] [Google Scholar]
- 51.Bridon G, Bonneil E, Muratore-Schroeder T, Caron-Lizotte O, Thibault P. Improvement of phosphoproteome analyses using FAIMS and decision tree fragmentation. application to the insulin signaling pathway in Drosophila melanogaster S2 cells. J Proteome Res. 2012;11(2):927–940. doi: 10.1021/pr200722s. [DOI] [PubMed] [Google Scholar]
- 52.Swearingen KE, Hoopmann MR, Johnson RS, Saleem RA, Aitchison JD, Moritz RL. Nanospray FAIMS Fractionation Provides Significant Increases in Proteome Coverage of Unfractionated Complex Protein Digests. Mol Cell Proteomics. 2012;11(4):M111 014985. doi: 10.1074/mcp.M111.014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia YQ, Wu ST, Jemal M. LC-FAIMS-MS/MS for quantification of a peptide in plasma and evaluation of FAIMS global selectivity from plasma components. Anal Chem. 2008;80(18):7137–7143. doi: 10.1021/ac8010846. [DOI] [PubMed] [Google Scholar]
- 54.Klaassen T, Szwandt S, Kapron JT, Roemer A. Validated quantitation method for a peptide in rat serum using liquid chromatography/high-field asymmetric waveform ion mobility spectrometry. Rapid Commun Mass Spectrom. 2009;23(15):2301–2306. doi: 10.1002/rcm.4147. [DOI] [PubMed] [Google Scholar]
- 55.Shvartsburg AA, Li F, Tang K, Smith RD. High-resolution field asymmetric waveform ion mobility spectrometry using new planar geometry analyzers. Anal Chem. 2006;78(11):3706–3714. doi: 10.1021/ac052020v. ** An ESI-FAIMS-MS interface using planar FAIMS provides superior resolving power.
- 56.Shvartsburg AA, Smith RD. Scaling of the resolving power and sensitivity for planar FAIMS and mobility-based discrimination in flow- and field-driven analyzers. J Am Soc Mass Spectrom. 2007;18(9):1672–1681. doi: 10.1016/j.jasms.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 57.Levin DS, Vouros P, Miller RA, Nazarov EG, Morris JC. Characterization of gas-phase molecular interactions on differential mobility ion behavior utilizing an electrospray ionization-differential mobility-mass spectrometer system. Anal Chem. 2006;78(1):96–106. doi: 10.1021/ac051217k. [DOI] [PubMed] [Google Scholar]
- 58.Levin DS, Miller RA, Nazarov EG, Vouros P. Rapid separation and quantitative analysis of peptides using a new nanoelectrospray- differential mobility spectrometer-mass spectrometer system. Anal Chem. 2006;78(15):5443–5452. doi: 10.1021/ac060003f. [DOI] [PubMed] [Google Scholar]
- 59.Shvartsburg AA, Tang K, Smith RD. Differential ion mobility separations of peptides with resolving power exceeding 50. Anal Chem. 2010;82(1):32–35. doi: 10.1021/ac902133n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shvartsburg AA, Smith RD. Ultrahigh-resolution differential ion mobility spectrometry using extended separation times. Anal Chem. 2011;83(1):23–29. doi: 10.1021/ac102689p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shvartsburg AA, Creese AJ, Smith RD, Cooper HJ. Separation of a set of Peptide sequence isomers using differential ion mobility spectrometry. Anal Chem. 2011;83(18):6918–6923. doi: 10.1021/ac201640d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shvartsburg AA, Singer D, Smith RD, Hoffmann R. Ion mobility separation of isomeric phosphopeptides from a protein with variant modification of adjacent residues. Anal Chem. 2011;83(13):5078–5085. doi: 10.1021/ac200985s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shvartsburg AA, Creese AJ, Smith RD, Cooper HJ. Separation of peptide isomers with variant modified sites by high-resolution differential ion mobility spectrometry. Anal Chem. 2010;82(19):8327–8334. doi: 10.1021/ac101878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shvartsburg AA, Zheng Y, Smith RD, Kelleher NL. Separation of Variant Methylated Histone Tails by Differential Ion Mobility. Anal Chem. 2012 doi: 10.1021/ac301541r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shvartsburg AA, Smith RD, Wilks A, Koehl A, Ruiz-Alonso D, Boyle B. Ultrafast differential ion mobility spectrometry at extreme electric fields in multichannel microchips. Anal Chem. 2009;81(15):6489–6495. doi: 10.1021/ac900892u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shvartsburg AA, Tang K, Smith RD, et al. Ultrafast differential ion mobility spectrometry at extreme electric fields coupled to mass spectrometry. Anal Chem. 2009;81(19):8048–8053. doi: 10.1021/ac901479e. ** An ESI-FAIMS-MS interface using a microfabricated FAIMS device provides rapid scanning.
- 67.Brown LJ, Toutoungi DE, Devenport NA, et al. Miniaturized ultra high field asymmetric waveform ion mobility spectrometry combined with mass spectrometry for peptide analysis. Anal Chem. 2010;82(23):9827–9834. doi: 10.1021/ac102125u. [DOI] [PubMed] [Google Scholar]
- 68.Brown LJ, Smith RW, Toutoungi DE, et al. Enhanced Analyte Detection Using In-Source Fragmentation of Field Asymmetric Waveform Ion Mobility Spectrometry-Selected Ions in Combination with Time-of-Flight Mass Spectrometry. Anal Chem. 2012;84(9):4095–4103. doi: 10.1021/ac300212r. [DOI] [PubMed] [Google Scholar]
Website
- 101. www.faims.com. ** A website providing a clear explanation of the fundamentals and operation of FAIMS, including a clear explanation of ion focusing in c-FAIMS.