

Abstract
Systemic autoinflammatory diseases are driven by abnormal activation of innate immunity1. Herein we describe a new syndrome caused by high penetrance heterozygous germline mutations in the NFκB regulatory protein TNFAIP3 (A20) in six unrelated families with early onset systemic inflammation. The syndrome resembles Behçet’s disease (BD), which is typically considered a polygenic disorder with onset in early adulthood2. A20 is a potent inhibitor of the NFκB signaling pathway3. TNFAIP3 mutant truncated proteins are likely to act by haploinsufficiency since they do not exert a dominant-negative effect in overexpression experiments. Patients’ cells show increased degradation of IκBα and nuclear translocation of NFκB p65, and increased expression of NFκB-mediated proinflammatory cytokines. A20 restricts NFκB signals via deubiquitinating (DUB) activity. In cells expressing the mutant A20 protein, there is defective removal of K63-linked ubiquitin from TRAF6, NEMO, and RIP1 after TNF stimulation. NFκB-dependent pro-inflammatory cytokines are potential therapeutic targets for these patients.
Monogenic autoinflammatory diseases are a heterogeneous group of diseases marked by unprovoked episodic or chronic inflammatory symptoms. Mutations in more than 20 causative genes have been reported, with many coding for proteins regulating signaling by pro-inflammatory cytokines such as interleukin-1, type 1 interferon and TNF 4-6. Therapies targeting pro-inflammatory cytokines, especially IL-1 and TNF, have been effective in many patients 7. However, a significant number of sporadic and familial cases remain genetically uncharacterized.
We studied a family of Caucasian ancestry (Family 1) with a dominant disorder manifesting early-onset systemic inflammation, arthralgia/arthritis, oral/genital ulcers and ocular inflammation (Fig. 1a, b, Supplementary Table 1). Whole exome sequencing (WES) on the affected mother and the two affected daughters identified 11 novel candidate variants. After Sanger sequencing of maternal unaffected family members, two novel variants in TNFAIP3 (tumor necrosis factor, alpha-induced protein 3; A20) and TNFRSF9 (tumor necrosis factor receptor superfamily, member 9) remained in consideration (Supplementary Table 2, Supplementary Fig. 1). Both genes function in the regulation of immune responses.
Figure 1. TNFAIP3 mutations cause a dominantly inherited systemic inflammatory disease.
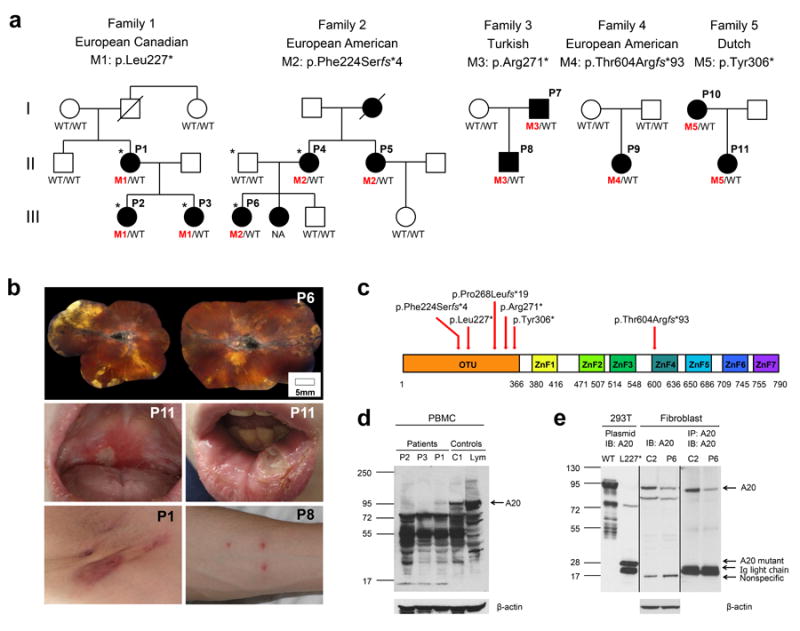
(a) Pedigrees of 5 families with heterozygous mutations in the TNFAIP3 gene. WT indicates wild-type TNFAIP3 alleles; M1, M2, M3, M4 and M5 indicate mutant alleles. The individuals selected for exome sequencing are marked with asterisk. NA: an affected sister of P6 had similar symptoms but was not available for this study.
(b) Clinical manifestations in patients: chorioretinal scarring and macular fibrosis secondary to retinal vasculitis causing a significant visual impairment (upper panel), recurrent oral ulcers (middle panel), axillary dermal abscesses and a pathergy-positive response (bottom panel).
(c) Schematic of TNFAIP3/A20 protein domains. Deubiquitinating (DUB) activity of A20 is mediated by the ovarian tumor (OTU) domain. ZFs mediate A20 ubiquitin ligase activity and binding to K63-linked ubiquitin chains. The locations of the mutations are indicated with red arrows. These mutations are predicted to affect A20 protein interactions, dimerization and its enzymatic activities.
(d-e) Reduced of TNFAIP3/A20 expression in patients’ PBMCs and fibroblasts. (d) Whole cell lysates were prepared from PBMCs isolated from three patients from Family 1, a healthy donor (C1), and purified control lymphocytes (Lym). Immunoblotting was performed against the N-terminus of A20. (e) Whole cell lysates from a healthy donor (C2) and patient P6 cultured fibroblast cells were either blotted with A20 N-terminal specific antibody (lane 3 and 4) or immunoprecipitated with one A20 N-terminal antibody and subsequently blotted with another A20 N-terminal antibody (lane 5 and 6). 293T lysates transfected with either FLAG-tagged wild-type (WT) A20 (lane 1) or A20 mutant L227* (lane 2) served as controls.
Independently, we performed WES in a family of Italian ancestry with an affected mother (P4) and daughter (P6). The family history was positive for a dominantly inherited inflammatory disease (Family 2, Fig. 1a). Patients P4, P6 and P5, who is the sister of P4 and aunt of P6, had in common oral/genital ulcers and polyarthritis. Patient P6 was initially diagnosed with an early-onset severe autoimmune condition resembling systemic lupus erythematosus, with CNS vasculitis and anterior uveitis (Fig. 1b, Supplementary Table 1). We identified 30 novel candidate variants that are shared between patients P4 and P6, including a novel variant in TNFAIP3. Sanger sequencing of patient P5 and the three unaffected family members confirmed that the novel variant p.Phe224Serfs*4 segregated with the inflammatory phenotype (Fig. 1a). Patient P6 did not have mutations in genes associated with early-onset autoimmune diseases8,9. We noted that Family 1 and Family 2 shared novel variants in a single candidate gene TNFAIP3 (Supplementary Fig. 2a).
Screening of an additional 150 patients with clinically similar disease identified three novel nonsense and frameshift variants in families of Turkish (Family 3), European -American (Family 4) and Dutch (Family 5) ancestry. In total, we identified 5 heterozygous truncating mutations in TNFAIP3 in five families (Fig. 1a, Supplementary Fig. 2b and Table 1). Targeted sequencing in 384 Turkish and 384 Japanese adult-onset Behçet’s cases from genome wide association studies identified one patient with a novel frameshift mutation in TNFAIP3, p.Pro268Leufs*19 (Supplementary Fig. 2c), which was subsequently identified in her two affected daughters. The identified mutations were considered pathogenic on the basis of proper family segregation and their absence from the Exome Aggregation Consortium (ExAC) database, dbSNP, and our exome database of more than 500 exomes. Previously, frameshift mutations in TNFAIP3 have been reported only in tumor tissues in the form of bi-allelic somatic mutations10. The pathogenic variants from our study were predicted to result in truncated proteins, suggesting haploinsufficiency or a dominant-negative effect of mutant TNFAIP3/A20 proteins.
Table 1.
Ancestry and sequence alterations in TNFAIP3 mutation positive families
| Family | Ancestry | Inheritance | Nucleotide alteration | Exon | cDNA alteration1 | Amino acid alteration | Domain | ExAC2 allele frequency |
|---|---|---|---|---|---|---|---|---|
| Family 1 | European Canadian | Dominant | chr6:138197178 T>A | 5 | c.680T>A | p.Leu227* | OTU | 0 / 122,972 |
| Family 2 | European American | Dominant | chr6:138197169 delT | 5 | c.671delT | p. Phe224Serfs*4 | OTU | 0 / 122,972 |
| Family 3 | Turkish | Dominant | chr6:138198218 C>T | 6 | c.811C>T | p.Arg271* | OTU | 0 / 122,972 |
| Family 4 | European American | De novo | chr6:138200391delG | 7 | c.1809delG | p.Thr604Argfs*93 | ZnF4 | 0 / 122,972 |
| Family 5 | Dutch | Dominant | chr6:138198325 C>G | 6 | c.918C>G | p.Tyr306* | OTU | 0 / 122,972 |
| Family 6 | Turkish | Dominant | chr6:138197297delG | 5 | c.799delG | p.Pro268Leufs*19 | OTU | 0 / 122,972 |
cDNA positions are according to the reference NM_006290.2.
ExAC: The Exome Aggregation Consortium includes 61,486 exomes.
TNFAIP3/A20 is a 790-residue protein that consists of an amino-terminal ovarian tumor domain (OTU) followed by 7 zinc finger domains (ZFs)11 (Fig. 1c). Five of the six disease-associated TNFAIP3 mutations are located in the OTU domain, which mediates the DUB activity, and they result in mutant truncated proteins of similar length. One disease-associated mutation, p.Thr604Argfs*93, resides in the ZnF4 domain, which recognizes K63-linked ubiquitin chains12,13 and is also essential for A20 E3 ligase activity12,13 and dimerization14. Expression of wild type A20 was reduced in patients’ PBMCs and fibroblasts, while mutant proteins were not detectable (Fig. 1d,e). We suspect that mutant proteins are not stable and undergo degradation.
We assessed the activity of each mutant A20 using an NFκB luciferase assay. Over-expressed mutant proteins failed to suppress TNF-induced NFκB activity in transfected human embryonic kidney (HEK) 293T cells (Fig. 2a) and a human T cell leukemia cell-line (Jurkat) (Supplementary Fig. 3). WT A20 suppresses NFkB activity, and co-transfection of a mutant truncated A20 construct with WT A20 did not reverse this outcome, suggesting that the mutant protein does not have a dominant-negative effect (Fig. 2a). IKK-mediated phosphorylation of IκBα resulting in IκB degradation, and subsequent nuclear translocation of the associated p50/65 NFκB heterodimer, is an essential step in the activation of the canonical NFκB pathway. To define the relationship of A20 mutants and NFκB induction in vivo, we studied activity of the canonical NFκB pathway in response to TNF stimulation. Stimulated patients’ cells showed increased phosphorylation of IKKα/β, increased phosphorylation of MAP kinases p38 and JNK, and increased degradation of IκBα compared to the controls (Fig. 2b, c and Supplementary Fig. 4a,b). Consistent with these findings, we observed increased nuclear translocation of p65 in patients’ PBMCs (Fig. 2d, and Supplementary Fig. 5a) and fibroblasts both at rest and after TNF stimulation (Fig. 2e, and Supplementary Fig. 5b,c). These data are strong evidence for enhanced signaling in the NFκB pathway.
Figure 2. Enhanced NFκB signaling in transiently transfected HEK293T cells and patients’ cells.
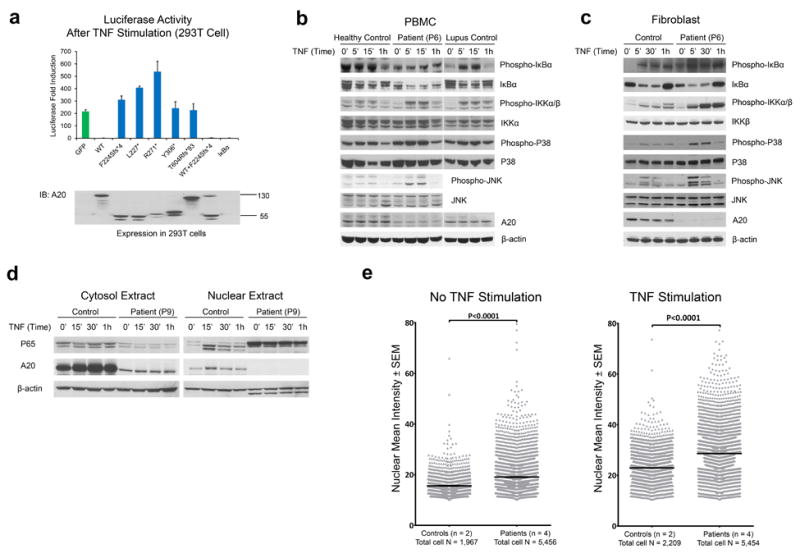
(a) The TNFAIP3/A20 mutants do not antagonize the inhibitory effects of wild type TNFAIP3/A20 on TNF-induced NFκB activation. HEK293T cells were transiently transfected with a NFκB reporter plasmid, Renilla luciferase control vector, and expression plasmids for either GFP-tagged wild-type or mutant A20. Plasmid expressing IκBα served as a positive control. Results are plotted as luc/ren to compensate for differences in transfection efficiency. One representative result of three independent experiments is shown. Values are expressed as mean of duplicates ± S.E.M. Protein expression of the transfected plasmids was confirmed by western blotting of cell lysates.
(b) Stimulated cells from A20-deficient patients show increased IκBα degradation and increased IKKα/β and MAPK activities. PBMCs from patient P6 were stimulated with TNF for the time period indicated in the graph. Whole cell lysates were immunoblotted against respective target proteins. Healthy individuals and an unrelated patient with SLE served as controls.
(c) Fibroblasts derived from A20-deficient patients show increased levels of phospho-IκBα, phospho-IKKα/β and MAPK activity. Whole cell lysates from TNF-stimulated fibroblasts were immunoblotted against respective target proteins.
(d) Cytosolic and nuclear fractions were prepared from patient P9 and immunoblotted with antibodies against p65, A20, and β-actin, respectively.
(e) Quantified nuclear NFκB p65 immunofluorescence staining in patients’ and controls’ fibroblasts under no-stimulation (left panel) and stimulation (right panel). Patient fibroblasts are significantly more activated than control fibroblasts (P<0.0001, Mann Whitney test). Data points represent mean ± S.E.M from analysis of quadruplicates of each individual. Data from patients (n = 4) and controls (n = 2) were pooled respectively.
The NFκB signaling cascade is under tight regulation by a number of post-translational modifications, including ubiquitination 15. A20 restricts cellular activation by cleaving K63-linked ubiquitin chains (Ub) from target substrates such as NEMO, RIP1, and TRAF612. Through its E3 ligase activity, A20 adds K48-linked Ub chains, targeting proteins for proteasome degradation 11,16. Upon TNF activation, TNFR forms a primary NF-kB activating signaling complex with TRADD, TRAF2 and RIP1. Consistent with reduced cellular levels of A20 in the cells of patients harboring TNFAIP3 truncation mutations, patient PBMCs displayed reduced recruitment of A20 to the TNFR complex compared to cells from healthy donors (Fig. 3a). To investigate whether the TNFAIP3/A20 mutants still retained their deubiquitinase function, WT and mutant A20 constructs were cotransfected into 293T cells with K63-linked ubiquitin and the A20 ubiquitination targets RIP1, NEMO/IKKγ and TRAF6 (Fig. 3b, c and d). Cells transfected with various disease-associated A20 mutants showed a marked defect in the deubiquitination of each of these target molecules, and cells co-transfected with wild-type and mutant A20 molecules, mimicking the situation in the patients, had intermediate levels of deubiquitination.
Figure 3. Impaired TNFR signaling and deubiquitinase function of mutant TNFAIP3/A20.
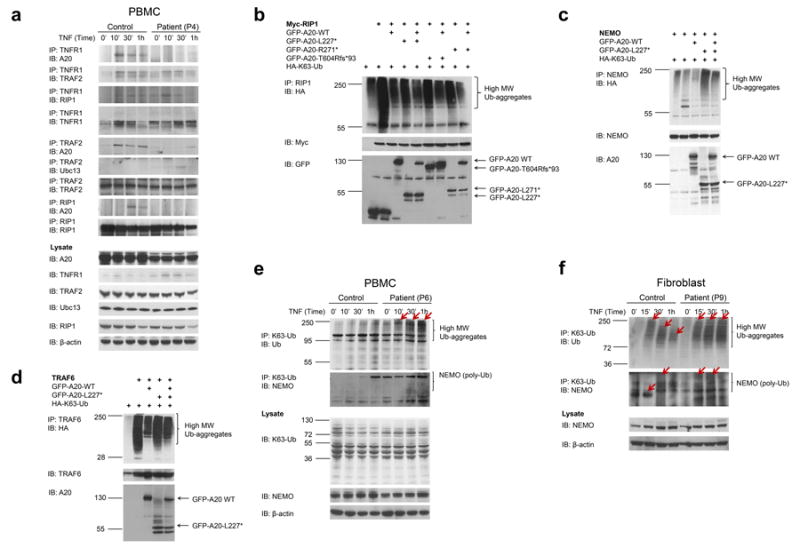
(a) Reduced recruitment of mutant A20 to the TNFR signaling complex. PBMCs from patient P4 and a healthy control were stimulated with TNF for the indicated times. Whole cell lysates were immunoprecipitated with antibodies against TNFR1, TRAF2, or RIP1, blotted with antibodies against N-terminal A20 and re-blotted with antibodies against TNFR1, TRAF2, RIP1 and Ubc13.
(b-d) The A20 mutants lose their ability to deubiquitinate K63-ubiquitin chains but do not antagonize the deubiquitinase function of WT A20. 293T cells were transiently transfected with expression plasmids for A20 target proteins RIP1 with Myc tag (b), NEMO (c) and TRAF6 (d), together with plasmids for HA-K63Ub and either GFP-tagged WT or mutant A20. Cells were harvested 48 hrs later and an equal amount of whole cell lysates was immunoprecipitated with antibodies against respective target proteins. High-molecular weight (MW) Ub-aggregates (top panel) were indicated by immunoblotting of precipitates with a HA-specific antibody. As controls for transfection efficiency, cell lysates were also blotted with antibodies against each target protein (middle panel) or antibodies against the N-terminus of A20 or GFP tag (bottom panel).
(e-f) TNF-stimulated patient PBMCs or fibroblasts showed increased levels and increased molecular weight of the K63-ubiquitinated NEMO protein as a result of insufficient A20 deubiquitinase activity. Primary cells from patients and healthy controls were stimulated for the indicated times. Whole cell lysates were subjected to immunoprecipitation with antibodies against K63-linked ubiquitin and subsequently blotted with antibodies against NEMO and Ub. Cell lysates were also blotted with antibodies against the target protein and β-actin as internal controls.
Consistent with the transfection data, TNFR1 signaling complexes from patients with A20 mutations accumulated sustained levels of K63-ubiquitinated NEMO and RIP1 and accumulated high-molecular weight (MW) Ub-aggregates (Fig. 3e and f, top panel, lane 5-8; Supplementary Fig. 6a,b, lane 5-8). In comparison, in healthy control PBMCs and fibroblasts, the expression of high MW Ub-aggregates gradually decreased over time following TNF stimulation (Fig. 3e, f, top panel, lane 1-4; Supplementary Fig. 6a,b, lane 1-4). Taken together, these results indicate that inefficient deubiquitination of A20 target proteins might explain a higher NFκB signaling activity in mutant cells.
Active NFκB subunits promote the transcription of genes encoding pro-inflammatory cytokines such as interleukin IL-1β, IL-6 and TNF, and facilitate the differentiation and activation of a variety of lymphocyte lineages. Multiple experiments showed evidence for increased expression of NFκB target genes in patients’ immune cells (Supplementary Fig. 7 and Supplementary Fig. 8). Levels of several proinflammatory cytokines were substantially increased in patients’ serum and in the supernatants of stimulated PBMCs relative to healthy controls (Fig. 4a, b). Intracellular cytokine staining also revealed increased polarization toward Th9 and Th17 CD4 T-effector cell lineages, which is consistent with the participation of IL-1β signaling in the differentiation of these lineages and may further contribute to tissue inflammation in A20-deficient patients (Fig. 4c). Finally, the frequency of CD14+ inflammatory monocytes was significantly higher in patients than in healthy controls (Fig. 4d).
Figure 4. Patients’ immune cells have a strong inflammatory signature.
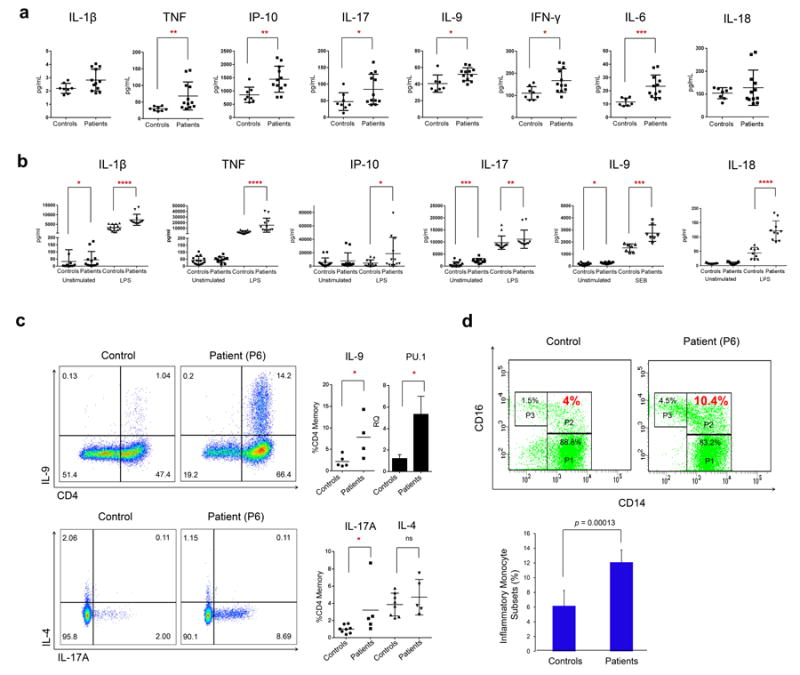
Cytokine profiles comparing A20-deficient patients with healthy controls. (a) Serum cytokine levels from 6 patients and 8 controls. (b) Cytokine levels from patients (n = 5) or controls (n = 6) PBMCs stimulated either with bacterial lipopolysaccharide (LPS) or Staphylococcal enterotoxin B (SEB). Each sample was assessed in duplicates or triplicates. Values are represented as mean ± S.E.M.
(c) Representative flow plots of CD3+ CD4+ CD45RO+ single live cells: Upper panels show IL-9 expression by intra-cellular cytokine (ICC) staining. The A20-deficient patients (n = 4) expressed significantly more IL-9 compared to paired controls (n = 5), and ex vivo PU.1 transcript levels were likewise significantly increased in patient PBMCs. Lower panels show representative flow plots of IL-17A and IL-4 expression in an A20-deficient patient compared to a paired control. A significant increase in intracellular IL-17A expression was observed in patients (n = 5) compared to controls (n = 8). Consistent with an absence of clinical allergic disease, no significant difference in intracellular IL-4 was seen. * P<0.05; Mann-Whitney test. Values are represented as mean ± S.E.M.
(d) Increased inflammatory monocyte subsets in patients. Top panel: Representative flow-cytometry analysis of monocyte subsets in one control (left) and one A20-deficient patient (right). Three monocyte subsets are identified as CD14highCD16neg (classical, P1), CD14highCD16pos (inflammatory, P2), CD14lowCD16pos (non-classical, P3). The population (%) of different subsets is shown in different gates. Bottom panel: Quantification of CD14highCD16pos (inflammatory) monocyte subsets in 4 A20-deficient patients and in 4 controls. Data are shown as mean ± SD (p = 0.00013, n = 4).
Recent studies in murine macrophages lacking A20 suggest that A20 functions as a negative regulator of the Nlrp3 inflammasome independently of its role in NFκB regulation 17,18. Consistent with these data, patients’ cells showed constitutive activation of the NLRP3 inflammasome (Fig. 5a), which resulted in activation of caspase-1 and increased secretion of active IL-1β and IL-18 (Fig. 4b). Patients’ PBMCs exhibited enhanced NLRP3 inflammasome-mediated caspase-1 activation and secreted IL-1β in response to LPS priming alone, and this effect was attenuated by use of a PLC inhibitor, an adenylate cyclase activator, or MCC950, all known NLRP3 Inflammasome inhibitors19,20 (Fig. 5b). Furthermore, initial experience in a patient with an agent targeting IL-1β has been positive (Fig. 5c).
Figure 5. Spontaneous NLRP3 inflammasome activity in PBMCs of patients with TNFAIP3 mutations.
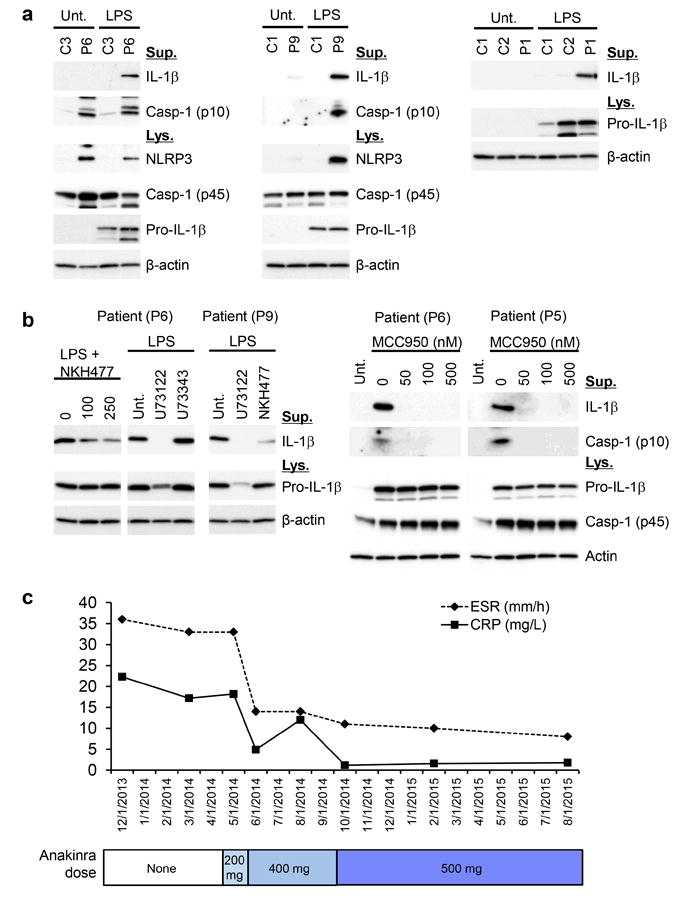
(a) Immunoblots of total cell lysates from LPS-stimulated patient cells and control show increased expression of pro IL-1β, NLRP3 and activated Caspase-1 (p10) and increased production of mature IL-1β. (b) IL-1β secretion in supernatants of LPS-stimulated patient cells is regulated by the activity of the NLRP3 inflammasome. Activation of adenylate cyclase (NKH477: adenylate cyclase activator), a PLC inhibitor (U73122), and a small molecule MCC950 attenuate the NLRP3 inflammasome activation. U73343: inactive analog of U73122. (c) Treatment with an IL-1 inhibitor, anakinra, normalized markers of systemic inflammation in Patient 6. Anakinra was initially given at a dose of 200mg daily (2014 May), later increased to 400mg daily (2014 Jun-2014 Sep), and then increased to 500 mg daily (2014 Oct-2015 Aug). The patient was also on prednisone 10mg and azathioprine 200 mg daily. Besides anakinra, she had no other medication changes from 2012-2014.
In summary, this is the first report of a human disease, Haploinsufficiency of A20 (HA20), caused by high-penetrance loss-of-function germline mutations in TNFAIP3/A20. The findings of multiple nonsense and frameshift variants associated with impaired regulatory function of TNFAIP3/A20 provide strong evidence that these are pathogenic mutations. Although patients with HA20 and Behçet’s disease share similar symptoms, the genetics are distinct. Most sporadic late-onset Behçet’s cases are not caused by highly penetrant germline mutations in A20. A20-deficient mice (A20-/-) display persistent NFκB activation, spontaneous multi organ inflammation and early lethality 21. Aging heterozygous mice (A20+/-) develop autoantibodies resembling human autoimmune conditions 15. Targeted deletion of A20 in specific hematopoietic cells leads to phenotypes reminiscent of autoimmunity 22-24. Although five patients in our study developed autoantibodies, an overt autoimmune disease was only diagnosed in Patient 6. Notably, none of our patients have developed lymphomas or other malignancies.
TNFAIP3/A20 variants have been linked to many human diseases 15. Somatic inactivating mutations in TNFAIP3 have been described in B cell lymphomas, suggesting that TNFAIP3/A20 acts as a tumor suppressor25. Common low-penetrance coding and non-coding variants in TNFAIP3 have been associated with multiple autoimmune diseases 26-36 and susceptibility to allergy and asthma37. The current study, however, is the first to delineate the consequences of germline inactivating TNFAIP3 mutations in human biology.
Next generation sequencing will likely identify other rare monogenic inflammatory diseases, possibly in the same pathway, which might further inform genetic studies of common polygenic immune diseases38-40. There are also potential therapeutic implications from studying patients with monogenic diseases, such as HA20, which may apply to the more common inflammatory conditions such as Behçet’s disease.
Online Methods
Human subjects
Seven patients were evaluated at the NIH Clinical Center, four were evaluated at University Medical Center Utrecht in Netherland and two at the Hacettepe University Children’s Hospital in Turkey. All patients enrolled in the study were evaluated under protocols approved by their respective institutional review boards. All the patients and family members provided written informed consent including consent to publish.
Whole exome sequencing
Whole exome sequencing and data analysis was performed as previously described41. Novel candidate variants were filtered by ExAC, 1000 genomes, dbSNP, NHLBI GO Exome Sequencing Project, Clinseq database, and an in-house database with over 500 exomes and selected based on autosomal dominant inheritance.
Targeted sequencing
Targeted sequencing of TNFAIP3 was performed on pooled DNA from 384 Turkish and 384 Japanese Behcet’s patients with adult-onset disease. Pooled DNA was prepared as previously described42. Ten amplicons covering the 9 exons of TNFAIP3 were generated using high fidelity polymerase AccuPrime™ Pfx SuperMix (Invitrogen). PCR products were pooled and prepared for library using Ovation® Ultralow Library Systems (NuGEN). Paired-end 300 bp reads were sequenced on a MiSeq instrument (Illumina). We called SNPs and INDELs, and calculated the frequencies from the pileup file by an in-house Perl script to identify SNPs and INDELs with frequency ≥1%.
Sanger sequencing
Sanger sequencing was used to confirm mutations identified by exome sequencing, targeted sequencing and for the candidate gene screening as previously described41.
Haplotype analysis
SNPs within the TNFAIP3 gene and STR markers across the chromosome 6 were genotyped for all family members from family 1. Haplotypes were reconstructed by PHASE and MERLIN.
Antibodies and expression plasmids
Antibodies specific for TNFAIP3 N-terminus (sc-166692), TRAF6 (sc-8409, sc-7221), Actin (sc-1615), Ubc13 (sc-376470, sc-292618), TNFR1 (sc-8436), RIP (sc-7881, sc-133102), NEMO (sc-56919, sc-8330), Ub (sc-271289), Caspase-1 (sc-515 and sc-622), GFP (sc-8334), HA-probe (sc-7392), c-MYC (sc-40) from Santa Cruz; TNFAIP3 N-terminal specific (#5630), K63-linkage Polyubiquitin (#5621), Phospho-IKKα/β (#2697), IKKα (#11930), IκBα (#4814, #9242), Phospho-IκBα (#2859), NFκB P65 (#8242), TNFR1 (#3736), RIP (#3493), Phospho-p38 MAPK (#4511), Phospho-p44/42 MAPK (Erk1/2) (#4370), Phospho-SAPK/JNK (#4668), p44/42 MAPK (Erk1/2) (#4695), SAPK/JNK (#9252), p38 MAPK (#8690), HRP-linked anti-rabbit IgG (#7074), HRP-linked anti-mouse IgG (#7076) from Cell Signaling; IL-1 β (AF-201-NA) from R&D Systems; NLRP3 (ALX-804-819-C100) from Enzo Life Sciences. pRK5-HA-Ubiquitin-K63 (Addgene plasmid # 17606) was a gift from Ted Dawson43. pEGFP-C1-TNFAIP3 (Addgene plasmid # 22141) was a gift from Yihong Ye 44. Myc-DDK-tagged-human RIPK1 (RC216024), Myc-DDK-tagged-human NFKBIA (RC200711), untagged human TRAF6 (sc109845) were from Origene. 3xFlag-TNFAIP3 (Ex-K6040-M120) was from Genecopoeia. pEF-NEMO was a gift from Dr. Chi Ma45. The GFP tagged or 3xFlag-TNFAIP3 mutant plasmids were constructed by PCR mutagenesis.
Cell cultures
HEK293T cells (human embryonic kidney cells expressing SV40 large T antigen, negative for mycoplasma, originally obtained from the American Type Culture Collection) and skin fibroblast cells derived from TNFAIP3-deficient patients or normal donors were grown in Dulbecco’s modified Eagle’s medium (Life Technologies) plus 10% fetal calf serum and 1x antibiotics (Life Technologies). Jurkat-T (human leukaemic T lymphoma cell line, negative for mycoplasma, originally obtained from the American Type Culture Collection) cells and human PBMCs were grown in RPMI1640 medium (Life Technologies), plus 10% fetal calf serum (Gemini Bio-Products) and antibiotics. For intracellular TNF staining, pan-T cells purified from PBMCs by negative selection (Miltenyi) were cultured in complete RPMI 1640 in the presence of plate-bound anti-CD3 and soluble anti-CD28 (both 1 μg/mL) for 5 days. For IL-9 staining, rhIL-4 (30 ng/mL), TGF-β (5 ng/mL), IL-1β (10 ng/ml), and IL-2 (10 U/ml), together with anti-IFNγ (10 μg/mL) were added to total PBMCs, and cells were cultured for 48 hours under the same conditions.
NFκB-dependent reporter assay and in vivo deubiquitination assay
Preformulated Cignal NFkB Pathway Reporter plasmids (Qiagen) (pGL4.32[luc2P/NF-κBRE/Hygro] luciferase reporter plus The pRLCMV-Renilla vector) were used to co-transfect in 293T cells with equal amount of expression plasmids for GFP-tag TNFAIP3 wild type or mutants using Polyjet transfection reagents (Signagen). After 24 hrs, cells were left untreated or stimulated with TNF (20 ng/ml). Luciferase activity (the firefly (luc) and renilla luciferase (ren) activity) was measured 5 hours afterwards by the dual-glo luciferase assay system (Promega). Results for NFkB activity are expressed as fold induction by normalizing firefly luciferase activity to Renilla luciferase activity.
For deubiquitination assay, 293T cells were transfected with 1 μg plasmids expressing target protein (RIP1, NEMO, or TRAF6), together with 0.5 μg HA-K63Ub, 0.5 ug GFP-tagged TNFAIP3 wild type or mutants. Empty vectors were used to equalize the plasmid transfection amount. Cells were harvested 48 hrs after transfection. Cell lysates were prepared and immunoprecipitation was conducted as previously described46.
NFκB reporter assay in Jurkat T cell line
5×106 3T8 cells (Jurkat T-cell line) were transiently transfected with 20 μg of expression plasmids encoding either wildtype TNFAIP3-GFP or mutant TNFAIP3-GFP fusion genes by electroporation. Sixteen hours post transfection, 20 ng/ml of human TNF was added to transfected 3T8 cells. Cells were collected 16 hours after the addition of TNF and surface stained with anti-Thy1-PECy7 and live/dead-violet for FACS analysis. Samples were acquired with a FACSVerse (Becton Dickinson Biosciences) and data were analyzed with FlowJo. The mean fluorescence intensity (MFI) of Thy1, the indicator of NFκB activity, was gated on the GFP+ live singlet populations.
PBMC preparation and stimulation, immunoprecipitation and immunoblotting
Heparinated peripheral blood mononuclear cells (PBMCs) were separated by Ficoll (Ficoll-Paque PLUS; GE Healthcare) gradient centrifugation. Recombinant human TNF (Peprotech) was used to stimulate PBMC (10 ng/ml) or fibroblast cells (20 ng/ml) from 5 min to 1 hr. Whole cell lysis were prepared using ice-cold 1xcell lysis buffer (Cell Signaling) supplemented with complete protease inhibitors. Nuclear extraction kit (Cayman) was used to prepare cytosolic and nuclear fractions. Immunoprecipitation and immunoblotting were conducted as described previously45,47 with specific antibodies. ImageJ was used to analyze the immunoblotting images.
Inflammasome inhibition
PBMCs (2×106 cells/well) were plated in 12-well plates and then primed with 1 μg ml-1 LPS in RPMI 1640 (Invitrogen) containing 10% FBS for 3 hrs. For the inhibition of inflammasome activation, LPS primed-PBMCs were treated with U73122 (5 μM), U73343 (5 μM), NKH477 (100 or 250 μM), or MCC950 (with three different doses: 50, 100, and 500 nM) in RPMI 1640. After 1 hr of treatment, supernatants and cell lysates were collected for immunoblot analysis.
Immunocytochemistry
Fibroblasts grown overnight in 96-well plates or on poly-L-lysine coated coverslips and treated with TNF were fixed in 4% paraformaldehyde and stained for NFκB p65. Primary (Cell Signaling 8242) and secondary antibodies (A21206 Molecular Probes) were incubated overnight at 4 °C and for 30 minutes at room temperature, respectively. Coverslips were mounted with Vectashield containing DAPI (H-1200, Vector Laboratories), imaged with a Zeiss AxioImager D2 using a 20x objective. 96-well plates were imaged with an Incucyte microscope (Essen Bioscience) using a 20x objective and a SYTO 59 (Molecular Probes) nuclear stain. Nuclear patterns were converted to regions of interest in ImageJ and used to measure nuclear p65 intensities. Data were analyzed in GraphPad Prism 6 and shown as mean ±S.E.M.
Immune cell cytokine production and serum cytokine detection
PBMCs (2×106 /ml) were stimulated with bacterial lipopolysaccharide (LPS) (Sigma), flagellin (Sigma), and Staphylococcal enterotoxin B (SEB) (Sigma) for 24 hour at 37°C. The concentrations of cytokines in the supernatants of stimulated and non-stimulated PBMCs and serum were determined using Bio-Plex Pro Human Cytokine 27-plex and 21-plex immunoassay kits (Bio-Rad Hercules). The Bio-plex pro human cytokine standard group I and group II were used as standards for the assays. The differences in the cytokine concentrations were statistically analyzed using the Mann-Whitney test, and plotted with GraphPad Prism software package.
Primary human monocyte isolation, differentiation and stimulation
Monocytes were purified from PBMCs by negative selection (Monocyte Isolation Kit II; Miltenyi Biotec). Monocytes were suspended in monocyte attachment medium (PromoCell) and seeded at a density of 150,000/cm2 for 2 hours. Monocytes were co-stained with APC-Cy7-conjugated anti-CD11b, PE-Cy7-conjugated CD11c, Fitc conjugated anti-CD14, APC-conjugated CD16 or relative isotype control and then were analyzed using a FACS Canton II (BD Biosciences).
Primary human monocytes were differentiated into M1 macrophages using 20 ng/mL human Granulocyte-macrophage colony-stimulating factor (hGM-CSF, Sigma) or into M2 macrophages using 100 ng/mL human macrophage colony-stimulating factor (hM-CSF, Sigma) for 10 days. hGM-CSF differentiated M1 macrophages were stimulated with or without LPS (Sigma) 100 ng/mL for 6 hours.
Real-time PCR
RNA was isolated from M1 macrophages using the RNeasy kit (Qiagen) and isolated from unstimulated PBMC using TRIzol. Reverse transcription was performed using the Taqman kit (Applied Biosystems). Real-time PCR reactions were performed using the iQ Syber Green kit (Biorad) or TaqMan gene expression master mix and TaqMan gene expression assays probes. 18S RNA served as control. The reactions were run on an Applied Biosystems 7500 Real-Time PCR System (Life Technologies).
Intracellular cytokine staining in T cells
Intracellular cytokine staining for IL-17A, TNF, IL-4, and IL-9 following either PMA and ionomycin stimulation (IL-17A, IL-4, and IL-9) or SEB stimulation (TNF) was performed as previously described.48 All events were collected on an LSRFortessa (BD Biosciences) and analyzed with FlowJo (Treestar).
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, the National Heart Lung and Blood Institute, the National Institute of Allergy and Infectious Diseases and the NIH Clinical Center. Dr. S. Ozen received royalties for consulting and speaking from Novartis and SOBI. Dr. H.L. Leavis received royalties for consulting from Baxter. Dr. E. Demirkaya was recipient of the Research Fellowship Program for International Researchers, which is supported by The Scientific and Technological Research Council of Turkey (TUBITAK; B.14.2.TBT.0.06.01-219-84). We thank Dr. Vishva Dixit for helpful discussions. We also thank all the patients and their families, and the healthy children and adult controls, for their enthusiastic support during this research study.
Footnotes
Author Contributions
Q.Z., D.L.K. and I.A. designed the study. Q.Z., H.Y.W., M.S., Y.H.P., Y.Z., D.Y., M.T., W.L.T., J.J.L., X.M.Y., C.O., C.C., D.T.C., S.C.C., E.P.H., and Z.Y. performed experiments. Q.Z., H.Y.W., K. Z., R.M.S., M.B., J.D.M., M.G., J.J.C., analyzed and interpreted the data. D.M.S., E.D., A.K.O., D.L.S., P.H., S.A.H, A.J., B.K.B., H.L.L., A.R., C.S., E.D.B., A.G., S.O., R.M.L., and D.L.K. enrolled the patients and collected and interpreted clinical information. K.Z., J.C.M. and I.W. provided technical support and comments. Q.Z., R.M.S., D.L.K. and I.A. wrote the manuscript. D.L.K. and I.A. directed and supervised the research. All authors contributed to the review and approval of the manuscript.
Competing financial interests
The authors declare no competing financial interests
URLs.
The Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org/; NHLBI GO Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/; 1000 genomes, http://www.1000genomes.org/; dbSNP,http://www.ncbi.nlm.nih.gov/projects/SNP/; Picard, http://broadinstitute.github.io/picard/; Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/; ANNOVAR, http://www.openbioinformatics.org/annovar.
Accession codes.
Whole-exome sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession number SRP062234.
Referenced accessions.
NCBI Reference Sequence
References
- 1.van Kempen TS, Wenink MH, Leijten EF, Radstake TR, Boes M. Perception of self: distinguishing autoimmunity from autoinflammation. Nature reviews Rheumatology. 2015 doi: 10.1038/nrrheum.2015.60. [DOI] [PubMed] [Google Scholar]
- 2.Sakane T, Takeno M, Suzuki N, Inaba G. Behcet’s disease. The New England journal of medicine. 1999;341:1284–91. doi: 10.1056/NEJM199910213411707. [DOI] [PubMed] [Google Scholar]
- 3.Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends in immunology. 2014;35:22–31. doi: 10.1016/j.it.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Russo RA, Brogan PA. Monogenic autoinflammatory diseases. Rheumatology. 2014;53:1927–39. doi: 10.1093/rheumatology/keu170. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, Aksentijevich I. New players driving inflammation in monogenic autoinflammatory diseases. Nature reviews. Rheumatology. 2015;11:11– 20. doi: 10.1038/nrrheum.2014.158. [DOI] [PubMed] [Google Scholar]
- 6.Moghaddas F, Masters SL. Monogenic autoinflammatory diseases: Cytokinopathies. Cytokine. 2015 doi: 10.1016/j.cyto.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annual review of medicine. 2014;65:223–44. doi: 10.1146/annurev-med-061512-150641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuehn HS, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345:1623–7. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flanagan SE, et al. Activating germline mutations in STAT3 cause early- onset multi-organ autoimmune disease. Nature genetics. 2014;46:812–4. doi: 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malynn BA, Ma A. A20 takes on tumors: tumor suppression by an ubiquitin-editing enzyme. The Journal of experimental medicine. 2009;206:977–80. doi: 10.1084/jem.20090765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nature reviews. Cancer. 2010;10:332–41. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- 12.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 13.Bosanac I, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Molecular cell. 2010;40:548–57. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Lu TT, et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity. 2013;38:896–905. doi: 10.1016/j.immuni.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nature reviews. Immunology. 2012;12:774–85. doi: 10.1038/nri3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. The Journal of biological chemistry. 2009;284:8217–21. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vande Walle L, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature. 2014;512:69–73. doi: 10.1038/nature13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duong BH, et al. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015;42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee GS, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–7. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coll RC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine. 2015;21:248–55. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin F, Dixit VM. A20 edits ubiquitin and autoimmune paradigms. Nature genetics. 2011;43:822–3. doi: 10.1038/ng.916. [DOI] [PubMed] [Google Scholar]
- 23.Matmati M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nature genetics. 2011;43:908–12. doi: 10.1038/ng.874. [DOI] [PubMed] [Google Scholar]
- 24.Tavares RM, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–91. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato M, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–6. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 26.Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nature genetics. 2008;40:1059–61. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nature genetics. 2007;39:1477–82. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomson W, et al. Rheumatoid arthritis association at 6q23. Nature genetics. 2007;39:1431–3. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodolce JP, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. Journal of immunology. 2010;184:7001–9. doi: 10.4049/jimmunol.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musone SL, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nature genetics. 2008;40:1062–4. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adrianto I, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nature genetics. 2011;43:253–8. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nair RP, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nature genetics. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fung EY, et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes and immunity. 2009;10:188–91. doi: 10.1038/gene.2008.99. [DOI] [PubMed] [Google Scholar]
- 34.Trynka G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–83. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 35.Musone SL, et al. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes and immunity. 2011;12:176–82. doi: 10.1038/gene.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nititham J, et al. Meta-analysis of the TNFAIP3 region in psoriasis reveals a risk haplotype that is distinct from other autoimmune diseases. Genes and immunity. 2015;16:120–6. doi: 10.1038/gene.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuijs MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349:1106–10. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 38.Rieux-Laucat F, Casanova JL. Immunology. Autoimmunity by haploinsufficiency. Science. 2014;345:1560–1. doi: 10.1126/science.1260791. [DOI] [PubMed] [Google Scholar]
- 39.Boisson B, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nature immunology. 2012;13:1178–86. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boisson B, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. The Journal of experimental medicine. 2015 doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 41.Zhou Q, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. The New England journal of medicine. 2014;370:911–20. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirino Y, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behcet disease. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8134–9. doi: 10.1073/pnas.1306352110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim KL, et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:2002–9. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, et al. Localization of A20 to a lysosome-associated compartment and its role in NFkappaB signaling. Biochimica et biophysica acta. 2008;1783:1140–9. doi: 10.1016/j.bbamcr.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma CA, et al. Dendritic cells from humans with hypomorphic mutations in IKBKG/NEMO have impaired mitogen-activated protein kinase activity. Human mutation. 2011;32:318–24. doi: 10.1002/humu.21439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang HY, et al. Cbl promotes ubiquitination of the T cell receptor zeta through an adaptor function of Zap-70. The Journal of biological chemistry. 2001;276:26004–11. doi: 10.1074/jbc.M010738200. [DOI] [PubMed] [Google Scholar]
- 47.Wang HY, et al. Antibody deficiency associated with an inherited autosomal dominant mutation in TWEAK. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5127–32. doi: 10.1073/pnas.1221211110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133:1400–9. 1409 e1–5. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.