

Abstract
Acute kidney injury (AKI) is commonly seen amongst critically ill and hospitalized patients. Individuals with certain co-morbid diseases have an increased risk of developing AKI. Thus, recognizing the co-morbidities that predispose patients to AKI is important in AKI prevention and treatment. Some of the most common co-morbid disease processes that increase the risk of AKI are diabetes, cancer, cardiac surgery and human immunodeficiency virus (HIV) acquired immune deficiency syndrome (AIDS). This review article identifies the increased risk of acquiring AKI with given co-morbid diseases. Furthermore, the pathophysiological mechanisms underlying AKI in relation to co-morbid diseases are discussed to understand how the risk of acquiring AKI is increased. This paper reviews the effects of various co-morbid diseases including: Diabetes, cancer, cardiovascular disease and HIV AIDS, which all exhibit a significant increased risk of developing AKI. Amongst these co-morbid diseases, inflammation, the use of nephrotoxic agents, and hypoperfusion to the kidneys have been shown to be major pathological processes that predisposes individuals to AKI. The pathogenesis of kidney injury is complex, however, effective treatment of the co-morbid disease processes may reduce its risk. Therefore, improved management of co-morbid diseases may prevent some of the underlying pathology that contributes to the increased risk of developing AKI.
Keywords: Acute kidney injury, Kidney disease, Human immunodeficiency virus, Co-morbidities, Diabetes, Cancer, Cardiac surgery, Acquired immune deficiency syndrome, Risk factors, Immune response, Cardiovascular disease
Core tip: In order to prevent, diagnose, and prophylactically treat patients, healthcare providers must identify co-morbidities that significantly increase the likelihood of acute kidney injury (AKI). Any treatments that compromise cardiac output, renal perfusion pressure, and glomerular hemodynamics risk ischemic injury to the kidney. The innate and adaptive immune responses, which are activated by renal epithelial cell necrosis contribute to the progression of AKI. These factors have been shown to be enhanced in diabetes, cancer, cardiac surgery and human immunodeficiency virus acquired immune deficiency syndrome patients.
INTRODUCTION
According to the Acute Kidney Injury Network (AKIN), AKI is an abrupt loss in kidney function within 48 h, as defined by an increase in serum creatinine of 26.4 μmol/L (0.3 mg/dL) or more; a percentage increase in serum creatinine of more than 50% from baseline; or a reduction in urine output, oliguria (< 0.5 mL/kg hourly for > 6 h)[1,2]. AKI can be characterized by severe changes in kidney function. The severity of these changes are time sensitive, thus, early treatment may minimize the complications associated with AKI[3]. AKI is most often secondary to extrarenal events in critically ill patients, specifically those that are hospitalized an are suffering from progressive degenerative diseases[4]. AKI has been shown to occur in 1% of patients admitted to the hospital and it has been shown that up to 7% of patients develop AKI during hospital stays[1,5,6]. The incidence of AKI in intensive care units (ICU) has been shown to range from 20% to 50%[7]. On average 5% of patients in the ICU with severe AKI require renal replacement therapy (RRT)[8].
Patients are at an increased risk of death from postoperative AKI. According to Hobson et al[9] the risk-adjusted 90-d postoperative mortality was 6.5% for patients with AKI (ranging from mild to severe) in comparison to 4.4% in patients without AKI. Some of these surgical procedures include thoracoabdominal aortic surgery[10], bone marrow transplantation[11] and cardiac surgery[12]. AKI, as a result of ischemia, is also a frequent clinical event. In the hospital setting, ischemic-AKI occurs in 50% of patients with AKI[13]. Ischemic-AKI occurs for a variety of reasons such as the use of vasoconstrictive drugs or radiocontrast agents and/or hypotension associated with sepsis or blood loss after surgery or trauma[2]. Individuals who survive AKI have an increased risk of short and long-term complications. Some of these complications include a 10-fold greater risk of chronic kidney disease, a 3-fold greater risk of end stage renal disease and double the risk of death[14,15].
Biomarkers have become a novel concept for the early diagnosis of AKI. A combination of two urinary cell-cycle arrest biomarkers, insulin-like growth factor-binding protein 7 (IGFBP-7) and tissue inhibitor of metalloproteinases-2 (TIMP-2) have been used to predict the risk of moderate and severe AKI (defined by stages 2 and 3 respectively according to the KDIGO classification of AKI)[16]. These biomarkers have been said to perform better than existing markers such as NGAL, KIM-1, interleukin (IL)-18, L-FABP and Cystatin C[17,18]. In AKI, these biomarkers localize in the site of injury where they are involved in the process of the G1 cell-cycle arrest, which acts to prevent cells from continuous division when DNA is damaged[19]. Two independent multicenter cohort studies conducted by Kashani et al[17] and Bihorac et al[18] allowed for the development of the FDA approved NEPHROCHECK® Test system. The test system is comprised of assays for TIMP-2 and IGFBP-7, which is to be used in conjunction with clinical evaluations. This system is used as a clinical aid in the risk assessment for moderate to severe AKI within 12 h of patient assessment[17,18]. As such, these new advancements allow for the early detection of AKI.
Several epidemiological studies have proposed a wide array of risk factors for AKI. These include acute clinical conditions, diagnostic, or therapeutic procedures, and chronic disease states. However, they do not highlight the relationship of co-morbid diseases with the pathophysiology of AKI in a systematic manner. As such, this paper seeks to identify important co-morbidities and illustrate mechanisms by which these co-morbidities increase the incidence of AKI. Identifying co-morbidities that significantly increase the likelihood of AKI will allow healthcare providers to prevent, diagnose, and prophylactically treat patients, thereby reducing the long-term complications associated with AKI.
Pathogenesis and co-morbid disease processes in AKI
Renal blood flow is highly regulated to ensure oxygen delivery for normal renal function[20]. Cardiac output, renal perfusion pressure, and glomerular hemodynamic factors are major determinants of renal blood flow autoregulation. If these factors are compromised, ischemic and toxic injury to the kidney can occur[20,21]. The afferent arteriole plays an important role in autoregulation to maintain glomerular filtration rate (GFR). There are two mechanisms by which the afferent arteriole regulates GFR: (1) the myogenic reflex occurs when renal perfusion pressure rises causing the smooth muscle of the afferent arteriole to constrict; and (2) tubuloglomerular feedback (TGF) is sensitive to sodium delivery to the macula densa causing vasoconstriction of the afferent arteriole[22]. Further, cyclooxygenase inhibitors such as aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) that may be taken by patients with co-morbid diseases can cause severe intrarenal vasoconstriction, serious decline in GFR, and worsen AKI[23-25].
The core pathology of AKI can be broken down into degenerative processes that target the tubular epithelium, vasculature and activate the immune response leading to a decline in kidney function. AKI associated with ischemia reperfusion injury, sepsis or toxins causes a rapid loss of proximal tubular cell cytoskeletal integrity and cell polarity[26]. As a result, there is a shedding of the proximal tubule brush border and loss of polarity with the mislocalization of adhesion molecules and other membrane proteins such as Na+/K+-ATPase and β-integrins[26]. ATP depletion can cause ER stress, which causes protein misfolding, including epithelial junction proteins, leading to loss of cell polarity and failure of sodium readsorption. This results in an aberrant TGF response (Figure 1)[2,26] caused by loss of the ability of the proximal tubular cells to reabsorb filtered sodium, thus, increasing sodium delivery to the distal nephron. With an increase in the delivery of sodium to the macula densa, the TGF mechanism of autoregulation senses hyperfiltration, causing afferent arteriole constriction[20]. However, the perfusion of the kidney may already be compromised by prerenal causes leading to an exaggerated TGF response, resulting in a sudden and substantial drop in GFR[20].
Figure 1.
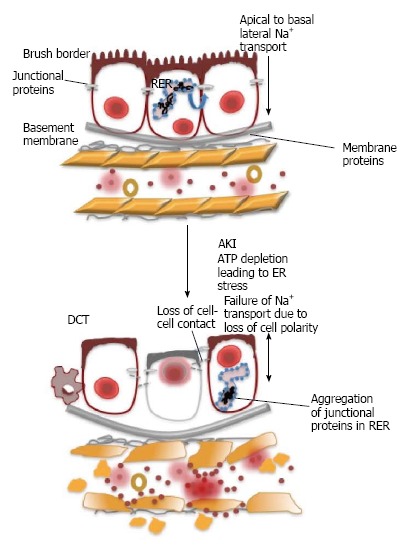
Epithelial cell damage. Ischemia reperfusion injury, sepsis or nephrotoxins are some the main causes of damage to epithelial cells resulting in AKI. The damage induces changes to the cytoskeleton, adhesion molecules and membrane proteins. ATP depletion results in the disruption of tight junctions causing back-leak of the filtrate as the actin cytoskeleton structure is altered. Endoplasmic reticulum stress caused by ATP depletion causes the aggregation of junctional proteins inducing an increase in the permeability of the endothelium. The loss of cell polarity due to AKI results in the failure of Na+ reabsorption allowing high concentrations of Na+ to reach the distal tubule stimulating an aberrant TGF response. AKI: Acute kidney injury; TGF: Tubuloglomerular feedback; ATP: Adenosine triphosphate; ER: Endoplasmic reticulum.
AKI and inflammation
Both the innate and adaptive immune responses, activated by tubular epithelial cell necrosis, are key contributors to the progression of AKI[1,21]. Activation of the inflammatory process triggers the expression of cytokines and chemokines like tumor necrosis factor (TNF) and IL-6 through toll-like receptors that detect materials released in response to injury and interact with their ligand receptors to activate a proinflammatory response to the site of injury. Upregulation of chemokines and adhesion molecules in the endothelium results in the infiltration of inflammatory cells such as neutrophils, lymphocytes, and macrophages from blood vessels to the interstitium of the kidney[1,21,27].
AKI and endoplasmic reticulum stress
AKI caused by ischemia, nephrotoxic drugs, or contrast agents has been associated with endoplasmic reticulum (ER) stress[27]. The ER has a pivotal role in the maintenance of protein homeostasis where it controls the concentration, conformation, folding and transport of synthesized proteins[27]. Disruptions such as hypoxia, glucose depletion, and oxidative stress can prevent the correct functioning of the ER where an accumulation of misfolded proteins in the ER lumen initiates the unfolded protein response (UPR)[27,28]. The UPR serves as an adaptive response attempting to re-establish normal ER functioning through the activation of calcium-dependent molecular chaperones such as glucose-regulated protein-78[1,27]. The UPR pathway can also induce the transcription of pro-apoptotic genes that cause cell death. Oxidative stress and inflammation are compounded by ER stress via the UPR, which contribute to glomerular and tubular damage in patients with AKI[27].
AKI and endothelium and vasculature damage
When the endothelium is damaged, the arteriole responds to a local high concentration of vasoconstrictive agents with a greater magnitude, as the injured endothelial cells produce a decreased amount of vasodilatory substance. There is an increase in the permeability of the endothelium post-injury, consequently resulting in a loss of fluid into the interstitium, thereby compromising blood flow[27]. Chronic hypoxia alongside the downregulation of angiogenic factors can cause a decline in the number of blood vessels and consequently lead to increased fibrosis that works in a positive feedback mechanism to reinforce its progression and ultimately cause epithelial cell injury and apoptosis[27,28]. Smaller constrictive vessels respond more intensely to vasoconstrictive agents (e.g., angiotensin II, thromboxane A2, prostaglandins etc.), but have a decreased response to vasodilators (acetylcholine, bradykinin, NO)[27]. These effects can be a consequence of alterations in the endothelium due to injury or enhanced leukocyte-endothelial adhesion. The latter effects can cause the obstruction of the small vessels and activate the inflammatory response, which becomes a vicious cycle of coagulation that prevents the delivery of vital nutrients and oxygen to the epithelial cells[27].
Co-morbidities and AKI
Damage to the kidneys, as a result of AKI, may be enhanced with the presence of co-morbidities and thereby complicate the treatment procedure. One study defined the incidence, risk factors and outcomes of AKI in a patient population from the Scottish Hip Fracture Audit database[29]. These patients who sought treatment for femur fracture and developed AKI showed an increase in inpatient morbidity, mortality (within 30 and 120 d) and length of hospital stay with multiple co-morbidities[29]. This study highlights the co-morbidities associated with the development of AKI including, diabetes mellitus, vascular disease, hypertension and pre-morbid chronic renal disease. The data presented in this study suggests that most cases of AKI occur post-surgery and the causes of AKI are multi-factorial comprising of pre, intra- and post-operative factors[29].
Diabetes-associated AKI
Globally, in 2014, it is estimated by the World Health Organization (WHO) that 387 million people suffer from Diabetes mellitus (DM), where 90% of the cases are of Type II diabetes[30]. The risk of AKI has been shown to be increased in patients with DM, with an adjusted odds ratio of 1.99, compared to non-DM controls with the same GFR[31]. It was determined that individuals who require dialysis, which is indicative of the severity of AKI, were an older patient group with DM and included individuals who had other complications such as hypertension and proteinuria[31]. A reason proposed for the higher risk of AKI in patients with DM is the frequent occurrence of complications associated with DM. Some of these complications include, cardiovascular disease; heart failure; exposure to medications such as diuretics and others that serve as nephrotoxic agents[32].
A greater susceptibility to ischemic insults of the diabetic kidney has been shown in experimental rodent models and in diabetic patients[33]. One study examined the influence of 30-min renal ischemia in rats with streptozotocin-induced DM. This study showed a complete recovery of the renal function in non-DM rats while DM animals showed a permanent loss of renal function[34]. DM rats, 8 wk after ischemia was induced, became completely anuric with tubular atrophy, and had extensive inflammation and tubulointerstitial fibrosis, which became evident within 4-wk post-surgery[34]. Another study led by the same investigators showed treatment of these rats with insulin prior to the ischemic event reduced ischemic injury[35].
The mechanism by which diabetes increases the severity of AKI has not yet been well established, but a great deal of research supports the connection between obesity, inflammation, and insulin resistance[36]. Inflammatory cytokines such as TNF-α and IL-6 are produced by adipocytes and have been shown to cause insulin resistance[36,37]. In rodent models of diabetes and diabetic humans, the increased upregulation of inflammatory cytokines in the kidney and urine have been shown[38]. These changes have been shown to result in long-term renal complications such as proteinuria and renal hypertrophy[38]. To experimentally determine the mechanistic role of TNF-α in facilitating the heightened risk of ischemic injury in Type II diabetic mice, one study used a neutralizing TNF-α antibody or nonimmune globulin control[39]. The mice were pre-treated with TNF-α antibody or nonimmune globulin injections 20 min before bilateral renal ischemia[39]. This study showed that the treatment with the TNF-α antibody was renal-protective against ischemic injury. Thus, the study concluded that diabetes increases the susceptibility to ischemic AKI due to an elevated TNF-α-mediated inflammatory response[39].
Although a majority of the scientific community agrees that diabetes increases the severity of AKI, some controversy surrounding DM and susceptibility to AKI exists. A study conducted by Venot et al[40] has shown no role of DM in increasing the risk of AKI or RRT. Instead, DM has been shown to only worsen the renal prognosis at discharge, determined by patients need for RRT, levels of serum creatinine and the recovery of renal function[40]. Additionally, the data from another study has shown that the history of DM is based on unclear self-reports of patients or records, and thereby does not reflect the current glucose control. Thus, using diabetes as a marker for a heightened risk of AKI at baseline clinical assessment in patients undergoing cardiac surgery may not be a useful tool in predicting renal injury outcomes[41]. Moreover, patients without a formal diagnosis of DM can suffer from chronic hyperglycaemia (CHG) due to pathological glycemic control or early stages of DM[41,42]. This study highlights that hyperglycaemia is also associated with cardiac dysfunction, susceptibility to infections and endothelial dysfunction, which pose as risk factors of perioperative morbidity and mortality after coronary artery bypass grafting (CABG) surgery[41]. The results of this study suggest that the measurement of Hemoglobin A1c (HbA1c) of ≥ 6.0%, which is an established tool used in the evaluation of diabetic control and CHG in patients with DM, is associated with a higher incidence of AKI after CABG[41]. Thus, a patient’s blood glucose levels should be evaluated for CHG, independent of DM, as it could be a strong determinant of AKI.
Cancer-associated AKI
AKI is an important complication of cancer and cancer-therapy where cancer patients are susceptible to a number of kidney lesions that can cause complications in the efficacy of treatment[43]. Factors such as the type and severity of malignancy (a solid tumour or hematologic process), associated complications such as co-morbidities and illnesses, and types of cancer management and therapy cause variability in when AKI is acquired[43]. One study conducted on Danish cancer patients reported the highest rates of AKI were in patients with kidney cancer at 44%, myeloma at 33% and liver cancer at 31.8%[44]. The rate of AKI in critically ill cancer patients was shown to be between 12% and 49%, with 9% to 32% of these patients requiring RRT[5,45,46], which is higher when compared to patient populations of an illness of similar severity[45,47,48]. Thus, AKI management in cancer patients is essential for patient survival and recovery.
AKI in cancer patients can be divided into prerenal, intrarenal or postrenal causes. Prerenal AKI is most commonly seen in cancer patients due to hypotension as a result of intravascular volume depletion caused by sepsis, vomiting, or diarrhea[43]. Hypercalcemia due to parathyroid hormone release, which increases bone resorption and renal tubular resorption of calcium, is seen in 10% to 30% of malignancies[49,50]. This can lead to a prerenal state of AKI due to vasoconstriction as well as volume depletion from natriuresis and diuresis[49,50]. Additionally, prerenal causes can result from the use of medications such as diuretics, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers or nonsteroidal anti-inflammatory agents for tumours, and/or other medical conditions such as hypertension or congestive heart failure[43]. Intrarenal causes of AKI in cancer patients consist of primary glomerular disease, acute tubular necrosis attributable to toxins or ischemia, infiltrative processes due to immune system activation, and microangiopathic processes[43]. Postrenal AKI is a result of kidney obstruction that is common in malignancies in the bladder, prostate, uterus, and cervix[43].
Nephrotoxicity by means of cancer therapy is one of the leading causes of AKI in cancer patients[51]. In a multivariate model, the OR for developing AKI from chemotherapy was 1.61, 4.55 for intravenous contrast and 1.52 for antibiotics[51]. Renal injury can be induced in a variety of ways by nephrotoxic drugs. In general, intrarenal vasoconstriction, direct tubular toxicity and intratubular obstruction are damaging results of nephrotoxic agents[51]. High levels of toxins are delivered and reabsorbed by the kidneys, which lead to increased intracellular concentrations of nephrotoxins in the tubular cell and medullary interstitium[51]. Further, the kidney is a site for drug metabolism and clearance[52]. Thus, the kidney can breakdown compounds that may be relatively harmless into toxic metabolites, or impairment of renal function can cause chemotherapeutic agents to concentrate in the kidneys without being cleared[52,53]. Delayed drug metabolism and excretion, due to increased concentrations of nephrotoxins, can result in increased systemic toxicity requiring an adjustment of treatment dosage[53]. As such, the nephrotoxic potential of anti-cancer agents can be significantly increased if there is pre-existing kidney damage and or a presence of concomitant co-morbidities such as heart failure and sepsis[53].
Cardiac surgery-associated AKI
Cardiac surgery-associated (CSA) - AKI is an important clinical problem that stems from a complex multifactorial pathogenic process. The incidence of CSA-AKI is 25%[54]. Mortality associated with the development of AKI can be as high as 60%, with an average of 15%-30% depending on the measurement and defining criteria of AKI[55]. Factors that increase the risk of CSA-AKI can be divided into preoperative and intraoperative (associated with and followed by postoperative CSA-AKI) categories.
The preoperative period is a critical point wherein renal injury can occur due to fluctuations in hemodynamics, exposure to nephrotoxic agents and the activation of the inflammatory response[55]. Injury, as a result of the aforementioned, can be substantiated when a patient undergoes surgery that decreases renal perfusion and reduces renal functional reserve. Patients undergoing conventional coronary bypass (CCB) often present with renal injuries that can range from minor to severe[55]. The pre-existing renal injury condition can be further amplified with the use of drugs such as diuretics, NSAIDs or angiotensin receptor blockers that can impair the autoregulation of renal blood flow[56]. Additionally, incidents of preoperative hypotension may lead to endothelial injury that can impair the production of vasodilatory substances such as NO causing vasoconstriction as a result of catecholamines and angiotensin II to further exacerbate injury[57,58].
The intraoperative period is when patients are exposed to anaesthesia and undergo CCB, these significantly impair hemodynamics and activate the innate and adaptive immune response[55]. Hemodynamic changes can be controlled and regulated given that a patient’s medical history is thoroughly assessed and the kidney is perfused accordingly during surgery. However, if not controlled, hemodynamic changes can lead to regional renal ischemia and can induce or extend renal injury[55]. Additionally, the activation of inflammatory mediators can initiate in the preoperative period and extend into the intraoperative period. An elevation of TNF-α levels have been observed in patients with pre-existing congestive heart failure, which further amplifies the inflammatory response during CCB in intraoperative period[59,60]. Neutrophils and the vascular endothelium are activated, inducing the upregulation of adhesion molecules such as platelets[60]. These events activate the upregulation of cytotoxic free-radicals[61], proteases[62], cytokines[63] and chemokines (IL-6, IL-8 and TNF-α)[63,64].
Postoperative events that impair renal function are similar to causative factors of AKI that are frequently found in intensive care setting such as the use of vasoactive agents, hemodynamic instability, exposure to nephrotoxic medications, volume depletion, and sepsis. Postoperative cardiac performance may be compromised with ventricular dysfunction causing reduced blood flow to the kidney and subsequently resulting in AKI[55].
Human immunodeficiency virus-associated AKI
Human immunodeficiency virus (HIV) infection that may progress to acquired immune deficiency syndrome (AIDS) creates an immunosuppressed state allowing for life-threatening opportunistic infections and cancers to thrive[65]. In contrast to AKI as a result of pre-renal and post-renal causes, HIV-associated AKI is most often due to HIV-mediated viral or immunological disease and or nephrotoxicity from treatments[66]. Risk factors for AKI in HIV infection include low CD4+ levels, AIDS, hepatitis C and liver disease[67]. Additionally, medications used to treat HIV such as anti-retroviral therapy (ART) or highly active antiretroviral therapy (HAART) may also increase the risk of developing AKI due to their nephrotoxic properties[66]. The OR of HIV patients acquiring AKI in pre-HAART has shown to be 2.9 and substantially increased to 6.0 in post-HAART[66]. ART causes severe immunosuppression where the CD4+ count becomes dangerously low at < 200 cells/mm3; normal values ranging from 500 cells/mm3 to 1200 cells/mm3[68]. The decreased CD4+ count is an independent predictor of experiencing AKI and is a vital predictor of HIV related morbidity and mortality[69]. Furthermore, co-viral-infections have been shown to increase the incidence of AKI. Hepatitis C virus co-infection occurs in 15%-30% of HIV-infected patients in the United States, where 30% of AKI events are a result of underlying liver damage[69].
Although no reliable data exists on the incidence and causes of AKI especially amongst HIV+ patients, South Africa, where 5.6 million of the 34 million people infected with HIV reside[67], faces problems of herbal intoxication, sepsis due to opportunistic infections, or severe gastroenteritis with dehydration[68,70]. AKI has been shown to be a critical cause of mortality particularly amongst indigenous black communities where herbal remedies are prescribed by traditional healers as curative measures for problems such as AIDS-related abdominal pain, diarrhea or to eliminate HIV from the system[70-72]. One of the most common nephrotoxic plants is the Impila (Callilepis laureola), found in regions of South Africa, Democratic Republic of Congo, Zimbabwe, and Zambia[73,74]. Nephrotoxicity from herbal remedies can arise from direct causes such as renal injury due to acute tubular necrosis and acute interstitial nephritis or indirectly as a result of intravascular hemolysis and dehydration due to diarrhea[68]. Therefore, HIV plays a major role in AKI from direct infection processes and treatment regimens.
CONCLUSION
AKI is an important clinical event that manifests in critically ill patients. AKI is associated with a multitude of risk factors that disrupt the homeostatic processes of the kidneys. Its complexity stems from pre-existing co-morbidities of patients that vary in severity, thereby making an overarching systematic treatment and management protocol difficult to deliver to patients suffering from AKI. A great deal of light has been shed upon the mechanistic basis by which AKI develops and progresses with the assessment of risk factors, however research efforts and emphasis should be placed on developing treatment interventions that can reverse or attenuate renal injury. To do this, therapeutic strategies need to be devised on a case-by-case basis where the identification of important co-morbid diseases such as DM, cancer, cardiac surgery and HIV takes place.
Footnotes
Conflict-of-interest statement: The authors declare no conflicts of interest regarding this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 13, 2015
First decision: November 7, 2015
Article in press: January 29, 2016
P- Reviewer: Eirini G, Trimarchi H S- Editor: Qiu S L- Editor: A E- Editor: Lu YJ
References
- 1.Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp Nephrol. 2009;112:e1–e9. doi: 10.1159/000210573. [DOI] [PubMed] [Google Scholar]
- 2.Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7:189–200. doi: 10.1038/nrneph.2011.16. [DOI] [PubMed] [Google Scholar]
- 3.Schrier RW. Early intervention in acute kidney injury. Nat Rev Nephrol. 2010;6:56–59. doi: 10.1038/nrneph.2009.170. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380:756–766. doi: 10.1016/S0140-6736(11)61454-2. [DOI] [PubMed] [Google Scholar]
- 5.Lameire N, Van Biesen W, Vanholder R. The changing epidemiology of acute renal failure. Nat Clin Pract Nephrol. 2006;2:364–377. doi: 10.1038/ncpneph0218. [DOI] [PubMed] [Google Scholar]
- 6.Waikar SS, Liu KD, Chertow GM. The incidence and prognostic significance of acute kidney injury. Curr Opin Nephrol Hypertens. 2007;16:227–236. doi: 10.1097/MNH.0b013e3280dd8c35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Case J, Khan S, Khalid R, Khan A. Epidemiology of acute kidney injury in the intensive care unit. Crit Care Res Pract. 2013;2013:479730. doi: 10.1155/2013/479730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiffl H, Lang SM, Fischer R. Long-term outcomes of survivors of ICU acute kidney injury requiring renal replacement therapy: a 10-year prospective cohort study. Clin Kidney J. 2012;5:297–302. doi: 10.1093/ckj/sfs070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobson C, Ozrazgat-Baslanti T, Kuxhausen A, Thottakkara P, Efron PA, Moore FA, Moldawer LL, Segal MS, Bihorac A. Cost and Mortality Associated With Postoperative Acute Kidney Injury. Ann Surg. 2015;261:1207–1214. doi: 10.1097/SLA.0000000000000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kashyap VS, Cambria RP, Davison JK, L’Italien GJ. Renal failure after thoracoabdominal aortic surgery. J Vasc Surg. 1997;26:949–955; discussion 955-957. doi: 10.1016/s0741-5214(97)70006-5. [DOI] [PubMed] [Google Scholar]
- 11.Parikh CR, McSweeney P, Schrier RW. Acute renal failure independently predicts mortality after myeloablative allogeneic hematopoietic cell transplant. Kidney Int. 2005;67:1999–2005. doi: 10.1111/j.1523-1755.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 12.Chertow GM, Levy EM, Hammermeister KE, Grover F, Daley J. Independent association between acute renal failure and mortality following cardiac surgery. Am J Med. 1998;104:343–348. doi: 10.1016/s0002-9343(98)00058-8. [DOI] [PubMed] [Google Scholar]
- 13.Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int. 2008;74:901–909. doi: 10.1038/ki.2008.314. [DOI] [PubMed] [Google Scholar]
- 14.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis. 2009;53:961–973. doi: 10.1053/j.ajkd.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81:442–448. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gocze I, Koch M, Renner P, Zeman F, Graf BM, Dahlke MH, Nerlich M, Schlitt HJ, Kellum JA, Bein T. Urinary biomarkers TIMP-2 and IGFBP7 early predict acute kidney injury after major surgery. PLoS One. 2015;10:e0120863. doi: 10.1371/journal.pone.0120863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, Bihorac A, Birkhahn R, Cely CM, Chawla LS, et al. Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care. 2013;17:R25. doi: 10.1186/cc12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bihorac A, Chawla LS, Shaw AD, Al-Khafaji A, Davison DL, Demuth GE, Fitzgerald R, Gong MN, Graham DD, Gunnerson K, et al. Validation of cell-cycle arrest biomarkers for acute kidney injury using clinical adjudication. Am J Respir Crit Care Med. 2014;189:932–939. doi: 10.1164/rccm.201401-0077OC. [DOI] [PubMed] [Google Scholar]
- 19.Yang QH, Liu DW, Long Y, Liu HZ, Chai WZ, Wang XT. Acute renal failure during sepsis: potential role of cell cycle regulation. J Infect. 2009;58:459–464. doi: 10.1016/j.jinf.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Edelstein CL, Ling H, Schrier RW. The nature of renal cell injury. Kidney Int. 1997;51:1341–1351. doi: 10.1038/ki.1997.183. [DOI] [PubMed] [Google Scholar]
- 21.Martin RK. Acute kidney injury: advances in definition, pathophysiology, and diagnosis. AACN Adv Crit Care. 2010;21:350–356. doi: 10.1097/NCI.0b013e3181f9574b. [DOI] [PubMed] [Google Scholar]
- 22.Johnson RJ, Feehally J, Floege J. Comprehensive clinical nephrology: Expert consult-online. Elsevier Health Sciences. 2014:22–29. [Google Scholar]
- 23.Lattanzio MR, Kopyt NP. Acute kidney injury: new concepts in definition, diagnosis, pathophysiology, and treatment. J Am Osteopath Assoc. 2009;109:13–19. [PubMed] [Google Scholar]
- 24.Himmelfarb J, Joannidis M, Molitoris B, Schietz M, Okusa MD, Warnock D, Laghi F, Goldstein SL, Prielipp R, Parikh CR, et al. Evaluation and initial management of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:962–967. doi: 10.2215/CJN.04971107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- 26.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 29.Bennet SJ, Berry OM, Goddard J, Keating JF. Acute renal dysfunction following hip fracture. Injury. 2010;41:335–338. doi: 10.1016/j.injury.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Hu FB. The global implications of diabetes and cancer. Lancet. 2014;383:1947–1948. doi: 10.1016/S0140-6736(14)60886-2. [DOI] [PubMed] [Google Scholar]
- 31.Hsu CY, Ordoñez JD, Chertow GM, Fan D, McCulloch CE, Go AS. The risk of acute renal failure in patients with chronic kidney disease. Kidney Int. 2008;74:101–107. doi: 10.1038/ki.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James MT, Grams ME, Woodward M, Elley CR, Green JA, Wheeler DC, de Jong P, Gansevoort RT, Levey AS, Warnock DG, et al. A Meta-analysis of the Association of Estimated GFR, Albuminuria, Diabetes Mellitus, and Hypertension With Acute Kidney Injury. Am J Kidney Dis. 2015;66:602–612. doi: 10.1053/j.ajkd.2015.02.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leblanc M, Kellum JA, Gibney RT, Lieberthal W, Tumlin J, Mehta R. Risk factors for acute renal failure: inherent and modifiable risks. Curr Opin Crit Care. 2005;11:533–536. doi: 10.1097/01.ccx.0000183666.54717.3d. [DOI] [PubMed] [Google Scholar]
- 34.Melin J, Hellberg O, Akyürek LM, Källskog O, Larsson E, Fellström BC. Ischemia causes rapidly progressive nephropathy in the diabetic rat. Kidney Int. 1997;52:985–991. doi: 10.1038/ki.1997.420. [DOI] [PubMed] [Google Scholar]
- 35.Melin J, Hellberg O, Larsson E, Zezina L, Fellström BC. Protective effect of insulin on ischemic renal injury in diabetes mellitus. Kidney Int. 2002;61:1383–1392. doi: 10.1046/j.1523-1755.2002.00284.x. [DOI] [PubMed] [Google Scholar]
- 36.Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab. 2006;4:417–419. doi: 10.1016/j.cmet.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 37.Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology. 1992;130:43–52. doi: 10.1210/endo.130.1.1727716. [DOI] [PubMed] [Google Scholar]
- 38.Navarro JF, Milena FJ, Mora C, León C, García J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. 2006;26:562–570. doi: 10.1159/000098004. [DOI] [PubMed] [Google Scholar]
- 39.Gao G, Zhang B, Ramesh G, Betterly D, Tadagavadi RK, Wang W, Reeves WB. TNF-α mediates increased susceptibility to ischemic AKI in diabetes. Am J Physiol Renal Physiol. 2013;304:F515–F521. doi: 10.1152/ajprenal.00533.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venot M, Weis L, Clec’h C, Darmon M, Allaouchiche B, Goldgran-Tolédano D, Garrouste-Orgeas M, Adrie C, Timsit JF, Azoulay E. Acute Kidney Injury in Severe Sepsis and Septic Shock in Patients with and without Diabetes Mellitus: A Multicenter Study. PLoS One. 2015;10:e0127411. doi: 10.1371/journal.pone.0127411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oezkur M, Wagner M, Weismann D, Krannich JH, Schimmer C, Riegler C, Rücker V, Leyh R, Heuschmann PU. Chronic hyperglycemia is associated with acute kidney injury in patients undergoing CABG surgery--a cohort study. BMC Cardiovasc Disord. 2015;15:41. doi: 10.1186/s12872-015-0028-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rydén L, Grant PJ, Anker SD, Berne C, Cosentino F, Danchin N, Deaton C, Escaned J, Hammes HP, Huikuri H, et al. ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD - summary. Diab Vasc Dis Res. 2014;11:133–173. doi: 10.1177/1479164114525548. [DOI] [PubMed] [Google Scholar]
- 43.Campbell GA, Hu D, Okusa MD. Acute kidney injury in the cancer patient. Adv Chronic Kidney Dis. 2014;21:64–71. doi: 10.1053/j.ackd.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Christiansen CF, Johansen MB, Langeberg WJ, Fryzek JP, Sørensen HT. Incidence of acute kidney injury in cancer patients: a Danish population-based cohort study. Eur J Intern Med. 2011;22:399–406. doi: 10.1016/j.ejim.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 45.Darmon M, Ciroldi M, Thiery G, Schlemmer B, Azoulay E. Clinical review: specific aspects of acute renal failure in cancer patients. Crit Care. 2006;10:211. doi: 10.1186/cc4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lameire N, Van Biesen W, Vanholder R. Acute renal problems in the critically ill cancer patient. Curr Opin Crit Care. 2008;14:635–646. doi: 10.1097/MCC.0b013e32830ef70b. [DOI] [PubMed] [Google Scholar]
- 47.Benoit DD, Hoste EA, Depuydt PO, Offner FC, Lameire NH, Vandewoude KH, Dhondt AW, Noens LA, Decruyenaere JM. Outcome in critically ill medical patients treated with renal replacement therapy for acute renal failure: comparison between patients with and those without haematological malignancies. Nephrol Dial Transplant. 2005;20:552–558. doi: 10.1093/ndt/gfh637. [DOI] [PubMed] [Google Scholar]
- 48.Darmon M, Thiery G, Ciroldi M, de Miranda S, Galicier L, Raffoux E, Le Gall JR, Schlemmer B, Azoulay E. Intensive care in patients with newly diagnosed malignancies and a need for cancer chemotherapy. Crit Care Med. 2005;33:2488–2493. doi: 10.1097/01.ccm.0000181728.13354.0a. [DOI] [PubMed] [Google Scholar]
- 49.Stewart AF. Clinical practice. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379. doi: 10.1056/NEJMcp042806. [DOI] [PubMed] [Google Scholar]
- 50.Seccareccia D. Cancer-related hypercalcemia. Can Fam Physician. 2010;56:244–246, 244-246. [PMC free article] [PubMed] [Google Scholar]
- 51.Lameire N. Nephrotoxicity of recent anti-cancer agents. Clin Kidney J. 2014;7:11–22. doi: 10.1093/ckj/sft135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cummings BS, Schnellmann RG. Pathophysiology of nephrotoxic cell injury. Diseases of the kidney and urinary tract. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 1071–1091. [Google Scholar]
- 53.Perazella MA. Onco-nephrology: renal toxicities of chemotherapeutic agents. Clin J Am Soc Nephrol. 2012;7:1713–1721. doi: 10.2215/CJN.02780312. [DOI] [PubMed] [Google Scholar]
- 54.Schopka S, Diez C, Camboni D, Floerchinger B, Schmid C, Hilker M. Impact of cardiopulmonary bypass on acute kidney injury following coronary artery bypass grafting: a matched pair analysis. J Cardiothorac Surg. 2014;9:20. doi: 10.1186/1749-8090-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosner MH, Okusa MD. Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol. 2006;1:19–32. doi: 10.2215/CJN.00240605. [DOI] [PubMed] [Google Scholar]
- 56.Okusa MD. The inflammatory cascade in acute ischemic renal failure. Nephron. 2002;90:133–138. doi: 10.1159/000049032. [DOI] [PubMed] [Google Scholar]
- 57.Goligorsky MS, Noiri E, Tsukahara H, Budzikowski AS, Li H. A pivotal role of nitric oxide in endothelial cell dysfunction. Acta Physiol Scand. 2000;168:33–40. doi: 10.1046/j.1365-201x.2000.00636.x. [DOI] [PubMed] [Google Scholar]
- 58.Caramelo C, Espinosa G, Manzarbeitia F, Cernadas MR, Pérez Tejerizo G, Tan D, Mosquera JR, Digiuni E, Montón M, Millás I, et al. Role of endothelium-related mechanisms in the pathophysiology of renal ischemia/reperfusion in normal rabbits. Circ Res. 1996;79:1031–1038. doi: 10.1161/01.res.79.5.1031. [DOI] [PubMed] [Google Scholar]
- 59.Hornick P, Taylor KM. Immune and inflammatory responses after cardiopulmonary bypass. Cardiopulmonary Bypass Principles and Practice. 2000:5: 303–319. [Google Scholar]
- 60.Asimakopoulos G, Taylor KM. Effects of cardiopulmonary bypass on leukocyte and endothelial adhesion molecules. Ann Thorac Surg. 1998;66:2135–2144. doi: 10.1016/s0003-4975(98)00727-9. [DOI] [PubMed] [Google Scholar]
- 61.Haga Y, Hatori N, Yoshizu H, Okuda E, Uriuda Y, Tanaka S. Granulocyte superoxide anion and elastase release during cardiopulmonary bypass. Artif Organs. 1993;17:837–842. doi: 10.1111/j.1525-1594.1993.tb00391.x. [DOI] [PubMed] [Google Scholar]
- 62.Faymonville ME, Pincemail J, Duchateau J, Paulus JM, Adam A, Deby-Dupont G, Deby C, Albert A, Larbuisson R, Limet R. Myeloperoxidase and elastase as markers of leukocyte activation during cardiopulmonary bypass in humans. J Thorac Cardiovasc Surg. 1991;102:309–317. [PubMed] [Google Scholar]
- 63.Frering B, Philip I, Dehoux M, Rolland C, Langlois JM, Desmonts JM. Circulating cytokines in patients undergoing normothermic cardiopulmonary bypass. J Thorac Cardiovasc Surg. 1994;108:636–641. [PubMed] [Google Scholar]
- 64.Paparella D, Yau TM, Young E. Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg. 2002;21:232–244. doi: 10.1016/s1010-7940(01)01099-5. [DOI] [PubMed] [Google Scholar]
- 65.Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Izzedine H, Baumelou A, Deray G. Acute renal failure in HIV patients. Nephrol Dial Transplant. 2007;22:2757–2762. doi: 10.1093/ndt/gfm404. [DOI] [PubMed] [Google Scholar]
- 67.Vachiat AI, Musenge E, Wadee S, Naicker S. Renal failure in HIV-positive patients-a South African experience. Clin Kidney J. 2013;6:584–589. doi: 10.1093/ckj/sft128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naicker S, Aboud O, Gharbi MB. Epidemiology of acute kidney injury in africa Proceedings of the Seminars in nephrology. Elsevier. 2008;28:348–353. doi: 10.1016/j.semnephrol.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 69.Franceschini N, Napravnik S, Eron JJ, Szczech LA, Finn WF. Incidence and etiology of acute renal failure among ambulatory HIV-infected patients. Kidney Int. 2005;67:1526–1531. doi: 10.1111/j.1523-1755.2005.00232.x. [DOI] [PubMed] [Google Scholar]
- 70.Cohen C, Karstaedt A, Frean J, Thomas J, Govender N, Prentice E, Dini L, Galpin J, Crewe-Brown H. Increased prevalence of severe malaria in HIV-infected adults in South Africa. Clin Infect Dis. 2005;41:1631–1637. doi: 10.1086/498023. [DOI] [PubMed] [Google Scholar]
- 71.Arendse CG, Wearne N, Okpechi IG, Swanepoel CR. The acute, the chronic and the news of HIV-related renal disease in Africa. Kidney Int. 2010;78:239–245. doi: 10.1038/ki.2010.155. [DOI] [PubMed] [Google Scholar]
- 72.Luyckx VA, Steenkamp V, Stewart MJ. Acute renal failure associated with the use of traditional folk remedies in South Africa. Ren Fail. 2005;27:35–43. [PubMed] [Google Scholar]
- 73.De Broe ME, Porter GA, Bennett WM, Verpooten GA. Clinical nephrotoxins. USA: Springer; 2008. [Google Scholar]
- 74.Lowenthal MN, Jones IG, Mohelsky V. Acute renal failure in Zambian women using traditional herbal remedies. J Trop Med Hyg. 1974;77:190–192. [PubMed] [Google Scholar]