

Abstract
Francisella tularensis, the causative agent of a fatal human disease known as tularemia, has been used in the bioweapon programs of several countries in the past, and now it is considered a potential bioterror agent. Extreme infectivity and virulence of F. tularensis is due to its ability to evade immune detection and to suppress the host's innate immune responses. However, Francisella-encoded factors and mechanisms responsible for causing immune suppression are not completely understood. Macrophages and neutrophils generate reactive oxygen species (ROS)/reactive nitrogen species as a defense mechanism for the clearance of phagocytosed microorganisms. ROS serve a dual role; at high concentrations they act as microbicidal effector molecules that destroy intracellular pathogens, and at low concentrations they serve as secondary signaling messengers that regulate the expression of various inflammatory mediators. We hypothesized that the antioxidant defenses of F. tularensis maintain redox homeostasis in infected macrophages to prevent activation of redox-sensitive signaling components that ultimately result in suppression of pro-inflammatory cytokine production and macrophage microbicidal activity. We demonstrate that antioxidant enzymes of F. tularensis prevent the activation of redox-sensitive MAPK signaling components, NF-κB signaling, and the production of pro-inflammatory cytokines by inhibiting the accumulation of ROS in infected macrophages. We also report that F. tularensis inhibits ROS-dependent autophagy to promote its intramacrophage survival. Collectively, this study reveals novel pathogenic mechanisms adopted by F. tularensis to modulate macrophage innate immune functions to create an environment permissive for its intracellular survival and growth.
Keywords: bacterial pathogenesis, cytokine induction, immunosuppression, p38 MAPK, redox signaling
Introduction
Francisella tularensis is a Gram-negative intracellular pathogen and the causative agent of a fatal human disease known as tularemia. F. tularensis is classified into four subspecies as follows: F. tularensis subspecies tularensis; F. tularensis subspecies holarctica; F. tularensis subspecies mediasiatica, and F. tularensis subspecies novicida. All classifications are based on virulence, genetics, and metabolic characteristics. F. tularensis subspecies tularensis (type A) is the most virulent of all four Francisella subspecies. About 70% of tularemia cases in North America are a result of type A Francisella with an infectivity dose of less than 10 colony-forming units (cfu) in humans (1). The live vaccine strain (LVS)3 is a derivative of Russian S15 strain of F. tularensis subspecies holarctica (type B). F. tularensis LVS is not approved for mass vaccinations in the United States due to adverse reactions in vaccinated individuals (2). F. tularensis LVS is relatively avirulent in humans and therefore commonly used as a surrogate for the more virulent SchuS4 strain to study tularemia pathogenesis. F. tularensis subspecies mediasiatica and novicida are rarely associated with human tularemia and have been isolated in Asia and in North America and Australia, respectively (3). In the past, F. tularensis was used in bioweapon programs, and now it is considered a potential bioterror agent (4). The extreme infectivity and virulence of F. tularensis in great part is due to its ability to evade immune detection and to suppress the host's innate immune response. However, Francisella-encoded factors and mechanisms responsible for causing immune suppression are not completely understood.
Francisella has the ability to survive in various cell types, including lung epithelial cells (5), fibroblasts (6), and phagocytic cells consisting of dendritic cells (7), neutrophils (8), and macrophages (9). Macrophages are considered to be the primary targets and are a widely studied cell type in Francisella research. F. tularensis has a unique intramacrophage cycle that involves entry (10), inhibition of phagosome-lysosome fusion (11–13), phagosomal escape, and cytosolic replication (14, 15). It has been shown that a fraction of cytosolic Francisella translocates into autophagic vacuoles after several hours of replication; however, most bacteria remain within the cytosol and proliferate without inducing autophagy (16). Macrophages and neutrophils generate reactive oxygen/nitrogen species (ROS/RNS) during oxidative burst as a defense mechanism for the clearance of phagocytosed microorganisms. The superoxide radicals (O2˙̄) are produced by the enzymatic reduction of molecular oxygen by NADPH oxidase, xanthine oxidase or non-enzymatically by mitochondrial electron transport chain. O2˙̄ radicals are highly reactive, unstable, have an ultra-short half-life, and thus rapidly dismutate to hydrogen peroxide (H2O2) either enzymatically by superoxide dismutases or non-enzymatically by spontaneous dismutation (17, 18). O2˙̄ radicals are highly reactive and can donate an extra electron to nitric oxide (NO) to produce highly toxic peroxynitrite (ONOO−) (19–21). Unlike O2˙̄, H2O2 is stable and can cross the lipid bilayer. Moreover, H2O2 can form highly reactive hydroxyl (OH−) radicals in the presence of iron (Fe2+) by Fenton chemistry. ROS serve a dual role; at high concentrations they act as microbicidal effectors that destroy intracellular pathogens by biologically targeting DNA, RNA, lipids, and proteins, and at low concentrations, they serve as secondary signaling messengers that regulate the expression of various inflammatory mediators (22, 23). The principal mediator of ROS-dependent signaling is H2O2 that acts by reversibly oxidizing the active site cysteines (Cys-Xaa5-Arg motif) in many redox-sensitive signaling components (24, 25).
F. tularensis possesses an elaborate antioxidant defense system to resist ROS and RNS generated by phagocytic cells. The antioxidant enzymes of F. tularensis are strategically localized to protect it from macrophage-derived oxidative insult. The iron-containing superoxide dismutase (SodB) is constitutively expressed and secreted (26, 27); and the copper-zinc-containing SodC is located in the periplasmic space. Previous studies from our laboratory with mutants of F. tularensis LVS in genes encoding for the antioxidant enzymes sodB, sodC, sodBC, and those of Lindgren et al. (28)., with catalase (katG), have shown that these antioxidant enzymes are required for intramacrophage survival and virulence in mice (26, 29, 30). We have also demonstrated that the sodBC mutant of F. tularensis LVS, which carries a point mutation in the sodB gene and a clean in-frame deletion of the sodC gene, exhibits enhanced sensitivity toward ROS and RNS, attenuated survival in macrophages, and virulence in mice (30). In addition to these primary antioxidant enzymes, a moxR family ATPase encoded by the FTL_0200 gene and two additional genes present on the moxR locus of F. tularensis LVS are required for resistance against oxidative and pH stresses (31). Additionally, the ohr gene of F. novicida and F. tularensis LVS is required for resistance to organic hydroperoxides (32). Recently, we have identified additional antioxidant defense mechanisms by screening a transposon insertion mutant library of F. tularensis LVS in the presence of H2O2 and reported that a membrane fusion protein EmrA1 encoded by the FTL_0687 gene contributes to oxidative stress resistance by affecting the secretion of SodB and KatG (33). In addition to these antioxidant defense mechanisms, F. tularensis has also evolved several strategies to limit NADPH-oxidase (Phox)-dependent ROS production in phagocytic cells to resist oxidative stress (34–36).
F. tularensis dampens or subverts the functions of cells involved in innate immune defenses. Recent studies have shown that F. tularensis infection leads to rapid bacterial replication in the host. Despite an exponential increase in bacterial numbers, activation of macrophages, dendritic cells and neutrophils is markedly suppressed (37–39). In addition, F. tularensis exerts profound suppressive effects on the production of pro-inflammatory cytokines by restricting signal transduction pathways (40, 41). Our previous work has demonstrated that KatG of F. tularensis LVS suppresses pro-inflammatory cytokine production and macrophage activation by interfering with PTEN-AKT redox-sensitive signaling pathways (29). In this study, we further investigated unique mechanisms of subversion of innate immune functions by antioxidant enzymes of F. tularensis. We hypothesized that antioxidant defenses of F. tularensis maintain redox homeostasis in infected macrophages to prevent activation of redox-sensitive signaling components that ultimately result in suppression of pro-inflammatory cytokine production and macrophage microbicidal activity. We demonstrate that antioxidant enzymes of F. tularensis prevent the activation of redox-sensitive MAPK signaling components, NF-κB signaling, and the production of pro-inflammatory cytokines by inhibiting the accumulation of ROS in infected macrophages. We also report that F. tularensis inhibits ROS-dependent autophagy to promote its intramacrophage survival. Collectively, this study reveals novel pathogenic mechanisms adopted by F. tularensis to modulate macrophage innate immune functions to create an environment permissive for its intracellular survival and growth.
Experimental Procedures
Bacterial Strains
F. tularensis LVS used in this study was obtained from BEI Resources, Manassas, VA. A sodBC double gene mutant previously reported from our laboratory (30), which carries a point mutation in sodB gene and deleted sodC gene, was used in this study. The transcomplemented strain of sodBC mutant (sodBC+psodC) was generated by providing a copy of the sodC gene in trans in the sodBC mutant. Francisella novicida was obtained from Microbiology Core at Albany Medical College. GFP expressing F. tularensis LVS and the sodBC mutants was prepared by transforming the GFP expression plasmid pKK214:GFP (42). All bacterial cultures were grown on Mueller-Hinton (MH)-chocolate agar plates (BD Biosciences) at 37 °C with 5% CO2 or Mueller-Hinton broth (BD Biosciences) with supplementations consisting of isovitalex and ferric pyrophosphate (BD Biosciences) at 37 °C with constant shaking at 175 rpm. Bacteria grown in Mueller-Hinton broth at mid-log phase were stored at −80 °C, and aliquots were thawed for use in experiments.
Mice
Wild type C57BL/6 and phox−/− mice obtained from The Jackson Laboratory were used for in vivo experiments and for isolation of bone marrow-derived macrophages (BMDMs) to be used in in vitro experiments. Mice were maintained in a specific pathogen-free environment in the animal facility of New York Medical College, Valhalla, NY. All experiments were performed on 6–8-week-old mice of either sex following the protocols approved by IACUC of New York Medical College. Mice were deeply anesthetized by intraperitoneal injection of ketamine/xylazine mixture and inoculated intranasally with 2 × 103 cfu of sodBC mutant or F. tularensis LVS in a 20-μl volume of PBS. Mice were sacrificed on days 1, 5, and 7 post-infection, and homogenates of lungs were prepared for quantitation of bacterial burden and pro-inflammatory cytokines following our previously published protocols (43, 44).
Primary Macrophages and Cell Lines
Raw264.7 macrophages were cultured in DMEM with 4.5 g/ml glucose (Corning Glass), 10% heat-inactivated fetal bovine serum (Sigma), 1% l-glutamine (Sigma), 1% sodium pyruvate (Sigma), and 1% penicillin/streptomycin (Invitrogen). BMDMs were isolated from wild type C57BL/6 and phox−/− mice using standard procedures.
Primary human monocytes were purchased from the University of Nebraska (Lincoln, NE) and cultured following our previously published protocol (29). Briefly, the cells were suspended in DMEM supplemented with l-glutamine, 10% human serum, and colony-stimulating factor-1 (R&D Systems). The medium was changed on days 2 and 4 of the culture, and human monocyte-derived macrophages (HMDMs) were infected on day 7 either with sodBC mutant or F. tularensis LVS unopsonized or opsonized with anti-F. tularensis LPS antibodies as described earlier (45). The production of ROS was inhibited by treating HMDMs with 250 μm apocynin (45). Cell culture supernatants from infected HMDMs were collected 24 h post-infection for quantification of pro-inflammatory cytokines TNF-α and IL-6.
Determination of Pro-inflammatory Cytokine Levels
Raw264.7 cells, BMDMs, and HMDMs were infected with F. tularensis LVS, the sodBC mutant, or its transcomplement (sodBC+psodC) at a multiplicity of infection (m.o.i.) of 100 and incubated for 24 h at 37 °C in 5% CO2. Cells that were co-infected with a combination of bacteria were inoculated with 50 m.o.i. of each strain to achieve an m.o.i. of 100. Cell culture supernatants were collected 24 h post-infection to determine levels of TNF-α and IL-6 using mouse or human cytometric bead array flex sets and analyzed via FCAP software (BD Biosciences).
Determination of Intracellular ROS in Macrophages Infected with F. tularensis LVS or the sodBC Mutant
Raw264.7 macrophages were seeded in 24-well plates and were either left uninfected or infected with WT F. tularensis LVS, the sodBC mutant, or the transcomplemented strain at an m.o.i. of 100. The plates were centrifuged at 1000 rpm for 10 min at 4 °C to synchronize the infection. Uninfected macrophages treated with 100 μm H2O2 or 100 μm paraquat were used as positive controls. After 30 min of infection, cells were harvested with a cell scraper and treated with 10 μm dihydroethidium (DHE) or 10 μm 2′,7′-dichlorofluorescin diacetate (DCFDA) to quantitate O2˙̄ radicals and total intracellular ROS, respectively. The cells were washed three times with sterile PBS and then analyzed by flow cytometry using the standard protocol. DHE and DCFDA were detected at an excitation of 518 and 495 nm, respectively.
Western Blotting Analysis
Macrophages were infected with F. tularensis LVS, the sodBC mutant, or the transcomplemented strain for 10, 20, and 30 min. Infection was synchronized by centrifugation of plates at 1000 rpm for 10 min at 4 °C. Macrophages were lysed using lysis buffer consisting of 50 mm Tris-HCl, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS. Pierce BCA protein assay kit (Thermo Scientific) was used to determine protein concentrations, and 30 μg of protein was separated on 10% SDS-polyacrylamide gel. Proteins were then transferred on a PVDF membrane and blocked with 5% nonfat milk in PBS containing 1% Tween for 1 h at room temperature. Primary antibodies against phosphorylated p38 and total p38, JNK, ERK, and ASK1 were diluted in PBS containing 1% Tween and incubated overnight at 4 °C. Western blots were developed by horseradish peroxidase-conjugated secondary antibodies and detected by incubating membranes with Pierce ECL Western blotting substrate (Thermo Scientific). Membranes were exposed to x-ray film, and the bands were detected and quantitated by densitometric analysis.
ELISA
Raw264.7, WT C57BL/6, or phox−/− BMDMs were seeded in a 96-well plate and infected with F. tularensis LVS, the sodBC mutant, or its transcomplemented strain for 10, 20, and 30 min. Cells were lysed, and protein concentrations were determined. Equal amounts of cell lysates were used in an ELISA. Antibodies against phosphorylated p38, JNK, and ERK were used to determine phosphorylation of these kinases using InstaOne ELISA (eBioscience) following the manufacturer's protocol.
Immunofluorescence Staining
Immunofluorescence staining was performed for detection of mitochondrial ROS (mROS), quantitation of macrophages exhibiting LC3 puncta, and subcellular localization of p65 subunit of NF-κB. For detection of mROS, Raw264.7 macrophages were infected with GFP expressing F. tularensis LVS and the sodBC mutant at an m.o.i. of 100 for 30 min in 8-well chamber slides and stained with Mitosox Red mitochondrial superoxide indicator (Molecular Probes) following the manufacturer's protocol.
For immunofluorescence staining for detection of LC3 puncta, Raw264.7 macrophages were infected with F. tularensis LVS, the sodBC mutant, or the transcomplemented strain at an m.o.i. of 100 for 12 and 24 h. The macrophages were then fixed with 4% paraformaldehyde in PBS at 4 °C for 15 min and permeabilized with 0.01% Triton X-100 in PBS for 12 min. Cultures were then stained for 2 h at room temperature with rabbit anti-mouse LC3 antibody (1:50; MBL International). After washing to remove excess primary antibodies, cultures were then incubated for 1 h at room temperature with an anti-rabbit IgG-Alexa488 secondary antibody (Jackson ImmunoResearch). The macrophages were also treated with rapamycin (500 nm), a classical inducer of autophagy, as a positive control. The cell were also either left untreated or treated with 10 mm 3-methyladenine (3MA), a classical inhibitor of autophagy, or 10 mm N-acetylcysteine (NAC) to inhibit ROS production in infected macrophages.
For nuclear localization of the p65 subunit of NF-κB, Raw264.7 macrophages were either left uninfected or infected with WT F. tularensis LVS, the sodBC mutant, or the transcomplemented strain at an m.o.i. of 100 for 30 min in 8-well chamber slides. These macrophages were either left untreated or were also treated with 10 mm NAC to inhibit ROS production. Immunofluorescence staining was performed for p65 localization as described earlier (46).
The cell nuclei were stained with SlowFade Gold antifade reagent with DAPI (Molecular Probes). The slides were dried and observed under Zeiss immunofluorescence microscope and images were captured at ×63 magnification. Formation of autophagosomes in macrophages infected with F. tularensis LVS or the sodBC mutant was determined by counting the cells showing LC3-positive autophagic vacuoles or the numbers of LC3 punctate dots using fluorescence microscopy. At least 200 cells were counted manually in randomly chosen fields for each condition. For quantification of localization of the p65 subunit of NF-κB, at least 100 cells were counted manually in randomly chosen fields.
Statistical Analysis
Results were expressed as means ± S.E. or S.D. Statistical significance between groups was determined by one-way ANOVA followed by Tukey-Kramer and Multiple Comparison test. Differences between the experimental groups were considered statistically significant at a p < 0.05 level.
Results
F. tularensis LVS Actively Suppresses Pro-inflammatory Cytokine Response
Several previous studies have demonstrated that F. tularensis SchuS4 induces a dampened pro-inflammatory cytokine response (41, 47, 48), and this is due to its ability to actively suppress an ongoing inflammatory response (48). However, it is not yet established whether F. tularensis LVS similarly exhibits such a phenomenon. Using an earlier reported F. novicida co-infection model (48), we investigated whether infection of macrophages with live or inactivated (UV-killed) F. tularensis LVS results in the suppression of pro-inflammatory cytokine response induced by F. novicida. Macrophages infected with F. novicida alone induced significantly higher levels of TNF-α (3279 ± 995 pg/ml) and IL-6 (7258 ± 1476 pg/ml) as compared with F. tularensis LVS-infected macrophages (1056 ± 525 and 586 ± 230 pg/ml, respectively). The macrophages that were infected with UV-killed F. novicida failed to induce any substantial amounts of TNF-α or IL-6, indicating that live F. novicida is required for induction of pro-inflammatory cytokine response. When macrophages were co-infected with F. novicida and F. tularensis LVS, the cytokine levels (TNF-α, 1602 ± 251 pg/ml, and IL-6, 751 ± 543 pg/ml) were reduced significantly (p < 0.001) to the levels similar to those observed for macrophages infected with F. tularensis LVS alone. These results indicated that F. tularensis LVS is capable of suppressing an ongoing pro-inflammatory cytokine response. Moreover, this suppression required the presence of live F. tularensis LVS, as UV-killed bacteria failed to suppress both TNF-α and IL-6 induced by live F. novicida (Fig. 1). Collectively, these results demonstrate that F. tularensis LVS possesses immunosuppressive properties similar to those reported for F. tularensis SchuS4 (48).
FIGURE 1.
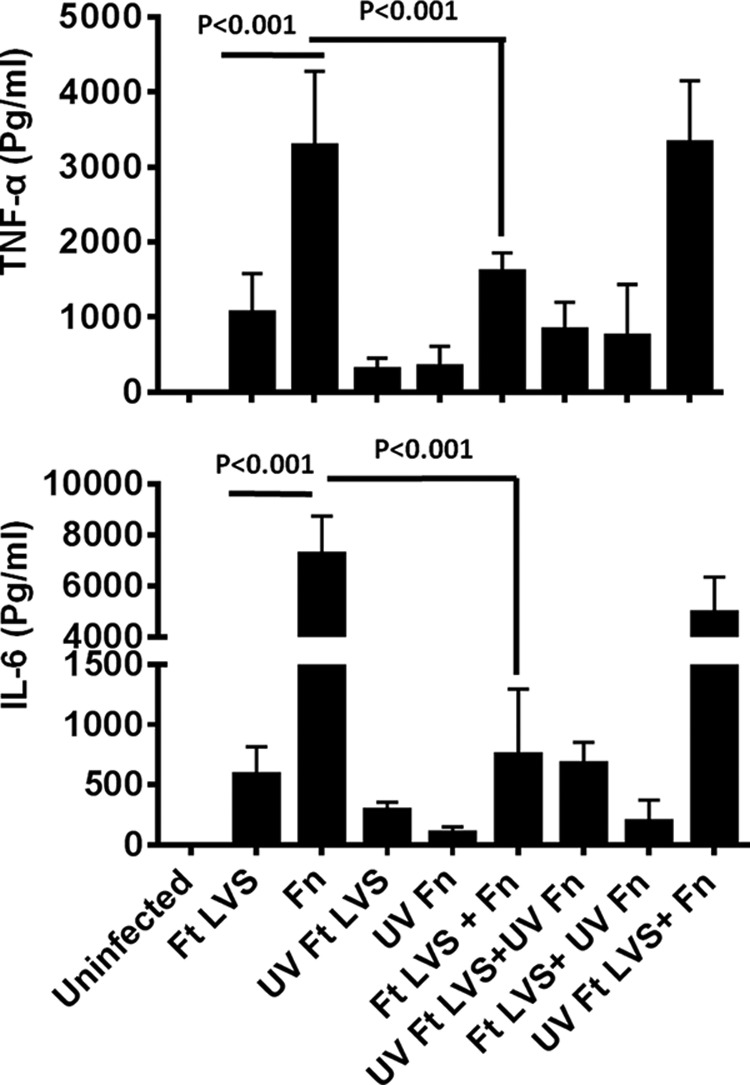
Live F. tularensis suppresses an ongoing inflammatory immune response induced by F. novicida. Primary BMDMs derived from C57BL/6 mice were infected with F. tularensis (Ft) LVS, F. novicida (Fn), UV-killed F. tularensis LVS (UV Ft LVS), UV-killed F. novicida (UV Fn), or a combination of these organisms at 100 m.o.i. (n = 3–5 biological replicates). The cell supernatants were collected at 24 h post-infection and analyzed for levels of TNF-α and IL-6. The data are representative of four independent experiments conducted with identical results. The data were analyzed by one-way ANOVA with Tukey-Kramer post-test, and a p value of 0.05 or less was considered significant.
sodBC Mutant of F. tularensis Fails to Suppress Pro-inflammatory Cytokine Response
The factors of F. tularensis that are required for the suppression of pro-inflammatory cytokine responses are not very well established. Our previous studies have shown that loss of catalase renders ΔkatG mutant hyper-inflammatory (29). We investigated whether antioxidant enzymes of F. tularensis play a role in suppression of pro-inflammatory cytokine response. We performed co-infection experiments using F. novicida and mutants of F. tularensis LVS in sodB, katG, sodC, and sodBC genes. It was observed that the sodB mutant alone neither induced higher levels of cytokines nor suppressed an ongoing pro-inflammatory cytokine response induced by F. novicida and exhibited a phenotype similar to that observed for wild type F. tularensis LVS. Unlike the sodB mutant, the ΔkatG and ΔsodC mutants induced higher levels of TNF-α as compared with those observed for F. tularensis LVS; however, similar to F. tularensis LVS, these mutants suppressed an ongoing pro-inflammatory cytokine response induced by F. novicida in a co-infection experiment (data not shown). Contrary to these antioxidant mutants of F. tularensis LVS, the sodBC mutant not only induced significantly (p < 0.01) higher levels of TNF-α as compared with the wild type F. tularensis LVS, but it failed to suppress higher levels of TNF-α induced in response to F. novicida in a co-infection experiment (Fig. 2). These results indicated that the antioxidant enzymes SodB and SodC together may have a role in suppression of pro-inflammatory cytokine response induced by F. tularensis. This notion was investigated further in this study.
FIGURE 2.
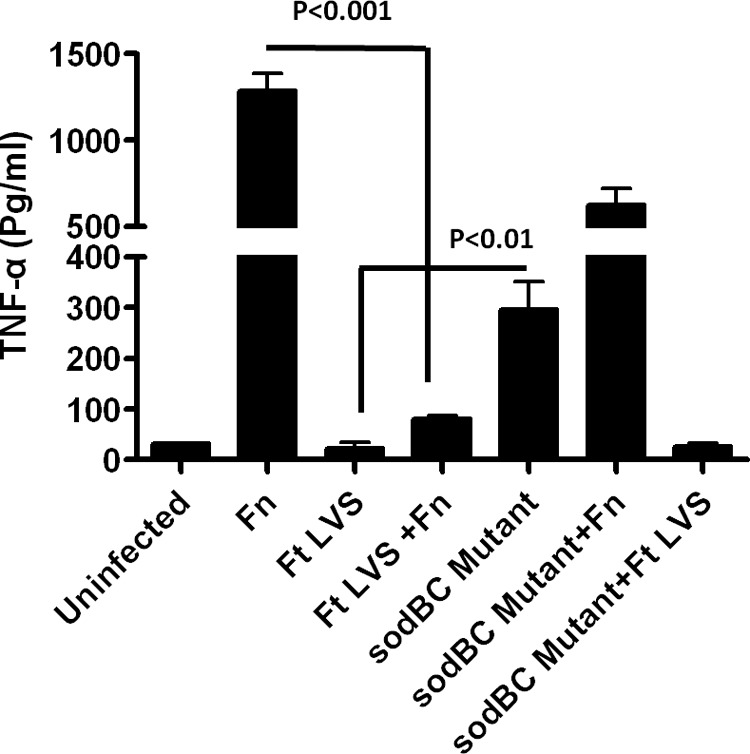
sodBC mutant of F. tularensis LVS has an impaired ability to suppress cytokine production. Raw264.7 macrophages were infected with F. novicida (Fn), wild type F. tularensis (Ft) LVS, or the sodBC mutants at 100 m.o.i. In co-infection experiments, 50 m.o.i. of each strain (total 100 m.o.i.) was used (n = 3 biological replicates). Cell supernatants were collected 24 h post-infection to measure TNF-α levels.
sodBC Mutant Induces Significantly Higher Levels of TNF-α and IL-6 in Vivo Which Are Dependent on NADPH Oxidase-generated ROS
Previously, we have reported that sodBC mutant of F. tularensis is attenuated for intramacrophage survival (30). To establish that higher levels of TNF-α observed in macrophages infected with the sodBC mutant were not due to its deficiency in intramacrophage survival, we corroborated these findings by performing in vivo experiments. Wild type and mice deficient in the gp91 component of NADPH-oxidase (phox−/−) on a C57BL/6 background were infected intranasally with sublethal (2 × 103 cfu) doses of F. tularensis LVS or the sodBC mutant. It was observed that in mice infected either with F. tularensis LVS or the sodBC mutant, the bacterial numbers increased from day 1 to 7 post-infection. However, no differences were observed in the numbers of F. tularensis LVS or sodBC mutant recovered from lungs of the wild type C57BL/6 mice on days 1, 5, and 7 post-infection (Fig. 3A). Additionally, except on day 7 post-infection, no differences in bacterial numbers were also observed in phox−/− mice. It was observed that both TNF-α and IL-6 levels (1030 ± 286 and 1633 ± 569 pg/ml, respectively) were significantly higher (p < 0.05) in lung homogenates from wild type C57BL/6 mice infected with the sodBC mutant than those observed for F. tularensis LVS-infected mice (576 ± 17 and 222 ± 42 pg/ml, respectively) on day 7 post-infection. On the contrary, the levels of both these pro-inflammatory cytokines in phox−/− mice infected with the sodBC mutant diminished to the levels observed for those infected with F. tularensis LVS (Fig. 3B). Collectively, these results indicated that antioxidant enzymes SodB and SodC of F. tularensis may have a role in redox-dependent suppression of pro-inflammatory cytokines.
FIGURE 3.
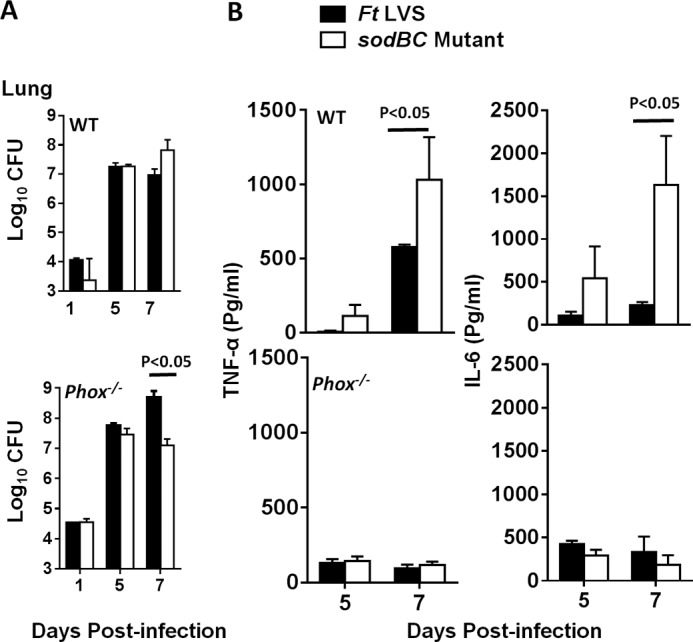
sodBC mutant induces NADPH oxidase-dependent production of TNF-α and IL-6 in vivo. A, wild type C57BL/6 and phox−/− mice were infected intranasally with 2 × 103 cfu of the sodBC mutant or F. tularensis (Ft) LVS. On days 1, 5, and 7 post-infection, mice (n = 4 per group) were euthanized, and their excised lungs were homogenized. Bacterial burdens were quantified in their lungs. Bacterial counts are expressed as log10 cfu. B, clear lung homogenates were used for quantification of TNF-α and IL-6 using cytometric bead array. The data are represented as mean ± S.D. The p values were determined using one-way ANOVA with Tukey-Kramer post-test, and a p value of 0.05 or less was considered significant. The data are representative of two independent experiments.
Antioxidant Enzymes of F. tularensis LVS Scavenge ROS Generated in Response to Infection
Our preceding results suggested that antioxidant enzymes SodB and SodC of F. tularensis restrict pro-inflammatory cytokine production in a redox-dependent fashion. We next quantitated the ROS generated in macrophages following infection with wild type F. tularensis LVS, the sodBC mutant, or the transcomplemented strain by staining the infected macrophages with redox-sensitive dyes DCFDA and DHE for ROS and O2˙̄, respectively. Significantly elevated levels of cellular ROS (p < 0.05) were observed in macrophages infected with the sodBC mutant as compared with those with F. tularensis LVS or the transcomplemented strain (Fig. 4A). Infection of macrophages with the sodBC mutant also revealed increased levels of O2˙̄, whereas the macrophages infected with F. tularensis LVS or the transcomplemented strain had O2˙̄ levels similar to those observed in uninfected controls (Fig. 4B). These results indicated that ROS rapidly accumulates in macrophages infected with the sodBC mutant, whereas on account of the capacity of the wild type F. tularensis LVS to scavenge ROS, the levels remain similar to those observed for uninfected controls.
FIGURE 4.
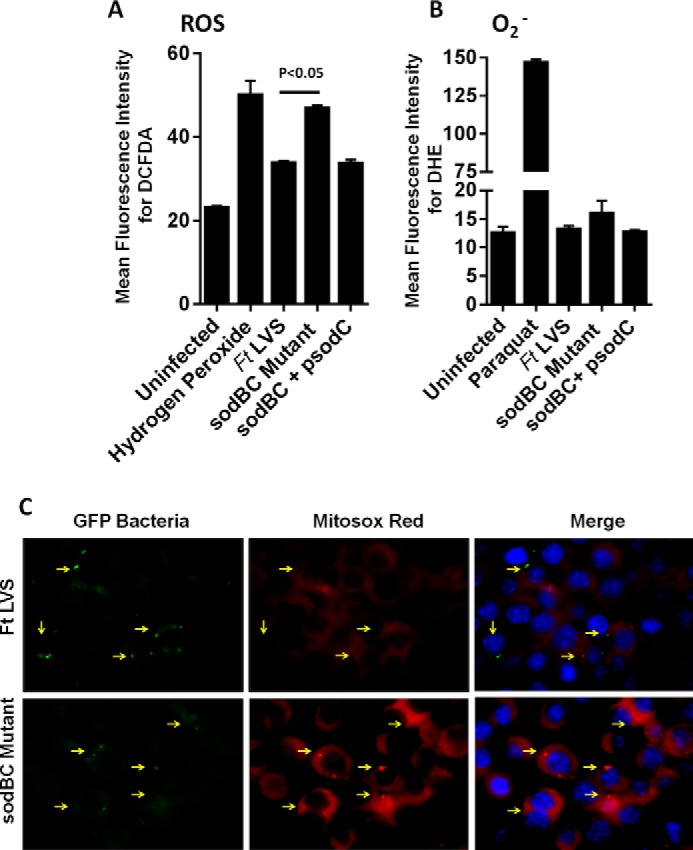
Antioxidant enzymes of F. tularensis prevent accumulation of ROS in infected macrophages. Raw264.7 macrophages were infected with F. tularensis (Ft) LVS, sodBC mutant, or its transcomplement (sodBC+psodC) for 30 min and treated with redox-sensitive fluorescent dyes DCFDA (for total ROS, A) and DHE (for superoxide radicals, O2˙̄, B) and analyzed by flow cytometry. Hydrogen peroxide (100 μm) and paraquat (100 μm) were used as positive controls in A and B, respectively. The results are expressed as mean fluorescence intensity. The data are representative of 3–4 independent experiments and were analyzed by one-way ANOVA, and p values were calculated. A p value of 0.05 or less was considered significant. C, macrophages were infected with GFP-expressing bacteria and stained with Mitosox-Red to detect mROS 30 min post-infection. Arrowheads indicate infected macrophages. Magnification, ×63.
We further investigated whether mROS is generated in response to infection of macrophages with F. tularensis LVS or the sodBC mutant. Macrophages infected with GFP expressing F. tularensis LVS or the sodBC mutants were stained with the mitochondrion-specific superoxide detector dye MitoSOX Red. It was observed that a higher level of mROS was produced in macrophages infected with the sodBC mutant as compared with those infected with F. tularensis LVS as indicated by enhanced immunofluorescence. Moreover, the MitoSOX Red staining co-localized with the sodBC mutant bacteria indicating that the mROS is generated in close bacterial proximity that was not observed in macrophages infected with F. tularensis LVS (Fig. 4C). Collectively, these results indicate that antioxidant enzymes of F. tularensis LVS scavenge the ROS generated in infected macrophages to maintain a redox environment similar to that observed for uninfected macrophages. However, loss of antioxidant enzymes SodB and SodC results in enhanced accumulation of ROS in macrophages infected with the sodBC mutant.
Activation of MAPK Signaling Is Observed in Macrophages Infected with the sodBC Mutant
We next investigated the downstream consequences of increased ROS production in macrophages infected with the sodBC mutant. We determined the activation state of p38, JNK, and ERK1/2 by ELISA and Western blot analysis. It was observed that infection of macrophages with F. tularensis LVS resulted in a transient activation of MAPK signaling. However, infection of macrophages with the sodBC mutant resulted in prolonged and significantly higher (p < 0.05) phosphorylated levels of p38, JNK, and ERK 30 min post-infection. Transcomplementation of sodBC mutant reversed these effects (Fig. 5, A and B). Furthermore, the temporal pattern of ROS production (O2˙̄ and H2O2) in the sodBC mutant-infected macrophages (Fig. 4) coincided with the phosphorylation of p38, JNK, and ERK with significantly higher levels being observed at 30 min post-infection when the ROS levels were also the highest. Collectively, these results indicate that loss of SodB and SodC antioxidant enzymes not only result in generation of an oxidizing environment due to accumulation of ROS, but also result in enhanced activation of MAPK signaling.
FIGURE 5.
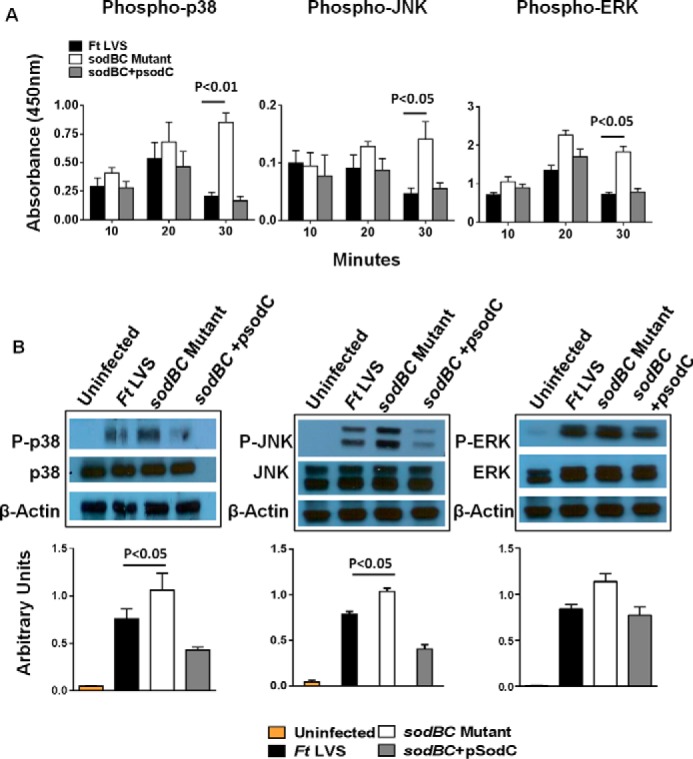
Activation of MAPK signaling is observed in macrophages infected with the sodBC mutant. A, quantitation of phosphorylated p38, JNK, and ERK at the indicated times. ELISA was performed using Raw264.7 macrophages infected with F. tularensis (Ft) LVS, sodBC mutant, or the transcomplemented (sodBC+psodC) strain. The data are representative of two independent experiments and were analyzed using one-way ANOVA, and p values were calculated. B, Raw264.7 macrophages were infected with F. tularensis LVS, sodBC mutant, or the sodBC+psodC strain for 30 min, lysed, separated by SDS-PAGE, and immunoblotted with phosphorylated (P) and total p38, JNK, and ERK. β-Actin was used as a loading control. Lower panels show quantitation of P-p38, P-JNK, and P-ERK bands. Uninfected macrophages were used as controls. The data were analyzed using one-way ANOVA, and p values were calculated (n = 2–3 blots).
Activation of MAPK Signaling Observed in Macrophages Infected with sodBC Mutant Is Dependent on ROS
A recent study has reported that BMDMs derived from phox−/− mice fail to produce any detectable levels of ROS in response to F. tularensis LVS infection (45). BMDMs from wild type C57BL/6 or phox−/− mice were used to determine whether ROS plays a critical role in the activation of MAPK signaling. Our results show that the levels of phosphorylated p38, JNK, and ERK were significantly higher in sodBC mutant-infected wild type BMDMs as compared with those infected with F. tularensis LVS or the transcomplemented strain. However, the levels of phospho-p38 were significantly reduced (p < 0.05), and JNKs were substantially reduced in phox−/− BMDMs. In contrast, the levels of p-ERK remained unchanged in phox−/− BMDMs (Fig. 6). Similar results were observed when macrophages were treated with ROS inhibitor NAC (data not shown). These results indicate that ROS activate MAPK signaling in sodBC-infected macrophages.
FIGURE 6.

Activation of MAPK signaling observed in macrophages infected with the sodBC mutant is dependent on NADPH oxidase-generated ROS. Quantitation of phosphorylated (Phospho) p38, JNK, and ERK at 30 min post-infection. ELISA was performed using wild type or phox−/− BMDMs infected with F. tularensis (Ft) LVS, sodBC mutant, or the transcomplemented (sodBC+psodC) strain. The data are representative of two independent experiments (n = 3 replicates) and were analyzed using one-way ANOVA, and p values were calculated.
ASK1 Serves as a Potential Trigger for Activation of MAPK Signaling in Macrophages Infected with the sodBC Mutant
Different ROS-dependent mechanisms exist for the activation of p38, JNK, and ERK MAPKs. It has been shown that a MAPK3 member, apoptosis signal regulating kinase-1 (ASK1), remains bound to reduced thioredoxin under homeostatic conditions. Under oxidizing conditions, ASK1 dissociates from thioredoxin and is activated by phosphorylation and oligomerization. This results in the phosphorylation and activation of downstream p38 and JNK (49). We determined the activation state of ASK1 by Western blotting analysis. A significantly higher level of phosphorylated ASK1 was observed in macrophages infected with the sodBC mutant as compared with the uninfected controls. The phosphorylated ASK1 levels were also higher in sodBC-infected macrophages than those observed in macrophages infected with F. tularensis LVS. However, these differences did not achieve significance (Fig. 7). Total ASK1 levels remained similar in macrophages infected either with wild type F. tularensis LVS or the sodBC mutant. Collectively, these results indicate that enhanced accumulation of ROS in macrophages infected with the sodBC mutant results in ASK1 activation that may serve as a potential trigger for the activation of downstream MAPK signaling.
FIGURE 7.
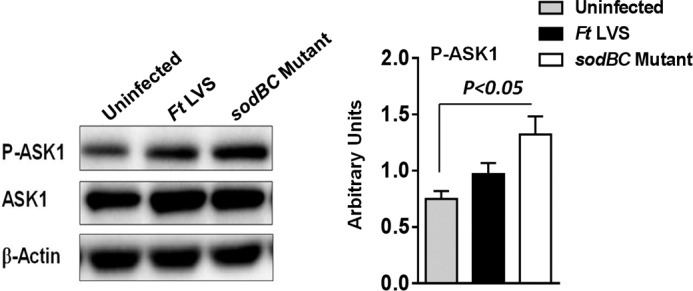
ASK1 serves as a potential trigger for activation of MAPK signaling in macrophages infected with the sodBC mutant. BMDMs were infected with F. tularensis (Ft) LVS or sodBC mutant for 15 min, lysed, and separated by SDS-PAGE, and immunoblotted with phosphorylated (P)-ASK1 (Thr845) and total ASK1 antibodies, and bands were quantitated using ImageJ (right panel, n = 3 blots). The data were analyzed using one-way ANOVA, and p values were calculated.
Infection of Macrophages with the sodBC Mutant Results in Enhanced Activation of NF-κB
Activation of MAPKs serves as important regulators of NF-κB activation and NF-κB-dependent gene expression. We determined the status of NF-κB activation by determining the translocation of the p65 subunit of NF-κB to the nucleus using immunofluorescence microscopy and by Western blot analysis of phosphorylated IKB-α or degradation of pIKB-α. Coincident with p38, JNK, and ERK activation, we observed a significantly higher (p < 0.05) percentage of cells (41 ± 7%) exhibiting nuclear translocation of the p65 subunit of NF-κB in sodBC mutant-infected macrophages as compared with those observed for F. tularensis LVS (10 ± 6%) or the transcomplemented strain (9 ± 2%) at 20 min post-infection. Treatment of macrophages with the ROS inhibitor NAC reduced the number of sodBC mutant-infected macrophages exhibiting nuclear localization of p65 subunit (11 ± 0%) (Fig. 8, A and B). These findings were also corroborated by Western blotting analysis. Increased phosphorylated levels of IKB-α were observed in macrophages infected with the sodBC mutant at 20 min post-infection. The levels of phosphorylated IKB-α were diminished and became similar to those observed for F. tularensis LVS when the macrophages were treated with the ROS inhibitor NAC indicating a ROS-dependent activation of NF-κB (Fig. 8C). Collectively, these results demonstrate that antioxidant enzymes SodB and SodC of F. tularensis not only impact activation of MAPK signaling but also interfere with activation of NF-κB signaling.
FIGURE 8.
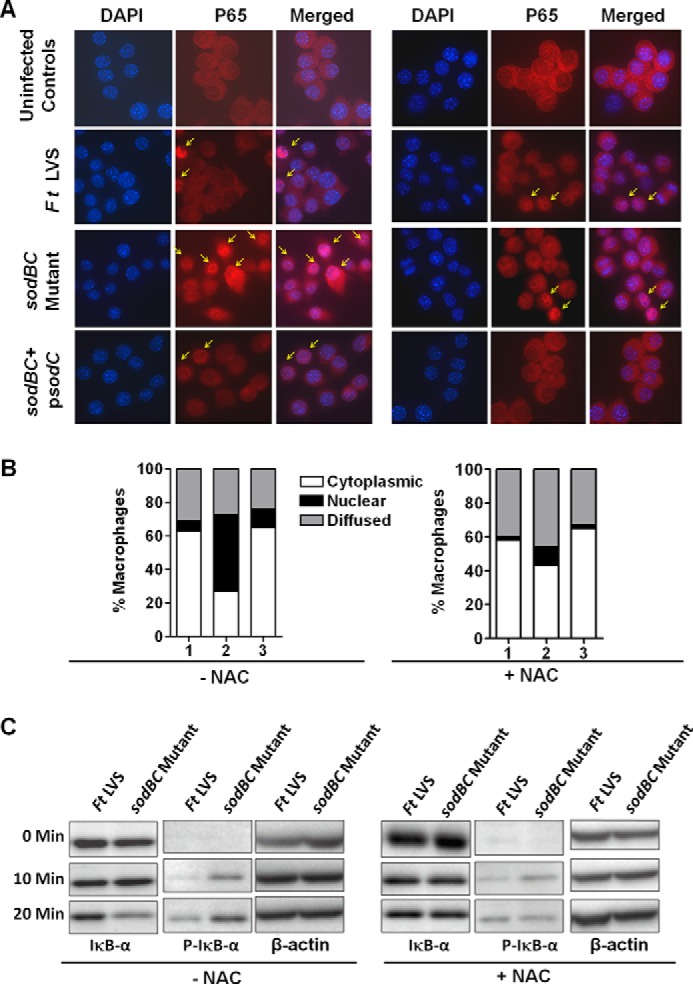
Infection of macrophages with the sodBC mutant results in an enhanced ROS-dependent activation of NF-κB. A, Raw264.7 macrophages were infected with F. tularensis LVS, sodBC mutant, or the sodBC+psodC strain for 20 min. The macrophages were either untreated or treated with 10 mm NAC. Immunofluorescence staining was performed to detect cellular localization of the p65 subunit of NF-κB (magnification ×63, red, p65; blue, nucleus). B, quantification of subcellular localization of p65 subunit is shown. At least 100 cells were counted manually in randomly chosen fields and expressed as percent macrophages with p65 staining. The data are representative of two independent experiments conducted. Lane 1, F. tularensis LVS; lane 2, sodBC mutant; lane 3, sodBC+psodC. C, BMDMs were infected with F. tularensis (Ft) LVS or the sodBC mutant in the absence or presence of ROS inhibitor NAC. The cell lysates were prepared at the indicated times and resolved by SDS-PAGE. Western blot analysis was performed using total IκB-α and phosphorylated (P)-IKB-α antibodies. β-Actin was used as a loading control. The data are representative of two independent experiments conducted. Arrows indicate cells with nuclear localization of p65 subunit of NF-κB.
Infection of Macrophages with sodBC Mutant Results in Enhanced ROS-dependent Autophagy
We next investigated the downstream consequences of an oxidizing environment, enhanced ROS-dependent MAPK and NF-κB signaling in macrophages infected with the sodBC mutant. We investigated the autophagy response in macrophages infected with F. tularensis LVS, the sodBC mutant, and its transcomplemented strain by staining the infected cells with anti-LC3 antibodies and counting the number of cells bearing LC3 punctate structures as a measure of autophagy. Higher numbers of punctate structures were observed in macrophages infected with the sodBC mutant than those observed in macrophages infected either with F. tularensis LVS or the transcomplemented strain (Fig. 9A). Quantification of macrophages positive for LC3 punctate structures revealed significantly higher percentage (79 ± 5%) of macrophages infected with the sodBC mutant to be positive for LC3 punctae as compared with those infected with F. tularensis LVS (47 ± 3%) or transcomplemented strain (63 ± 4%) at 24 h post-infection (Fig. 9B). When macrophages were treated with 3MA, an inhibitor of autophagy, the autophagic response was abrogated, and no differences were observed in numbers of macrophages bearing LC3 punctae or the numbers of LC3 punctae per cell irrespective of infection with F. tularensis LVS, sodBC mutant, or the transcomplemented strain (Fig. 9C). Similarly, the formation of LC3 punctae was abolished when macrophages were treated with the ROS inhibitor NAC (Fig. 9C). These results indicate that antioxidant enzymes SodB and SodC of F. tularensis inhibit autophagy in a ROS-dependent manner and demonstrate an antioxidant enzyme-mediated novel mechanism of autophagy inhibition by F. tularensis.
FIGURE 9.

Infection of macrophages with sodBC mutant results in enhanced ROS-dependent autophagy. A, detection of autophagy in Raw264.7 macrophages infected with F. tularensis LVS, sodBC mutant, or the transcomplemented strain sodBC+psodC by immunofluorescence staining 24 h post-infection (magnification ×63, green, LC3; blue nucleus). B, quantitation of autophagosomes by counting the number of cells bearing LC3 punctae at 12 and 24 h post-infection. C, macrophages were treated with 3MA, an inhibitor of autophagy, and NAC, a ROS inhibitor and infected with F. tularensis LVS, sodBC mutant, or the transcomplemented strain sodBC+psodC. Quantification of autophagosomes was done by counting the cells showing LC3-positive autophagic vacuoles or the numbers of LC3 punctate dots. At least 200 cells were counted manually in randomly chosen fields for each treatment group. The data are representative of 2–3 independent experiments and were analyzed by one-way ANOVA, and p values were calculated.
Targeting sodBC Mutant although FcRγ Results in Enhanced Production of Pro-inflammatory Cytokines in HMDMs
Our preceding results demonstrated that infection of macrophages with the sodBC mutant generates an oxidizing environment in macrophages resulting in an activation of MAPK and NF-κB signaling, enhanced pro-inflammatory cytokine response, autophagy, and bacterial clearance. We also observed that inhibition of ROS either by using phox−/− mice or macrophages or chemical inhibitors abrogated these responses. We next investigated whether potentiation of ROS production, instead of inhibition, would further enhance pro-inflammatory cytokine response in HMDMs. Because targeting opsonized F. tularensis LVS through FcγR has been shown to result in an enhanced ROS production (45), we utilized this approach for potentiation of ROS production. HMDMs were infected with F. tularensis LVS, the sodBC mutant, or its transcomplemented strain, unopsonized or opsonized with anti-F. tularensis LPS antibodies, and levels of TNF-α and IL-6 were determined 24 h later. Similar to that observed for murine macrophages, significantly higher levels of TNF-α and IL-6 were observed in HMDMs infected with unopsonized sodBC mutant. Opsonization of sodBC mutant with anti-F. tularensis LPS antibodies resulted in significantly higher levels of both of these pro-inflammatory cytokines as compared with those observed for opsonized F. tularensis LVS (Fig. 10A). Furthermore, treatment of HMDMs with ROS inhibitor apocynin significantly diminished the levels of both TNF-α and IL-6 in macrophages infected with the sodBC mutant (Fig. 10B). These results demonstrate that antioxidant enzymes of F. tularensis impact ROS-dependent production of pro-inflammatory cytokines in HMDMs in a fashion similar to murine macrophages. Additionally, potentiation of ROS production results in a further enhancement of pro-inflammatory cytokine response.
FIGURE 10.
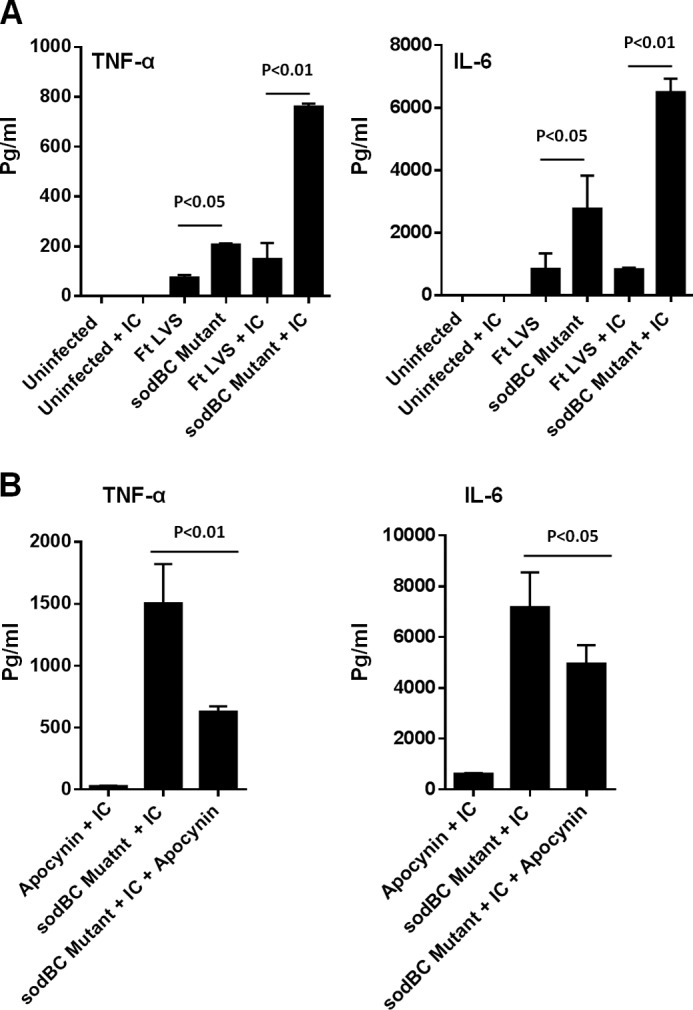
Targeting sodBC mutant though FcRγ results in an enhanced production of pro-inflammatory cytokines in HMDMs. HMDMs were infected with unopsonized F. tularensis (Ft) LVS or the sodBC mutant or opsonized with anti-F. tularensis LPS antibodies (5 μg/ml) (+IC) at an m.o.i. of 100 (A). HMDMs infected with opsonized F. tularensis LVS or the sodBC mutant (+IC) were also treated with ROS inhibitor apocynin (250 μm) (B). The culture supernatants collected at 24 h post-infection were analyzed for TNF-α and IL-6 levels. The data are representative of 2–3 independent experiments and were analyzed by one-way ANOVA, and p values were calculated.
Discussion
Historically, the production of ROS by phagocytes was mainly studied in the context of their microbicidal properties. Later, it was established that ROS also regulates normal physiologic “redox signaling” in many cell types. Macrophages are far less efficient in ROS production than neutrophils; therefore, the pro-inflammatory cytokine production via redox signaling is required for their microbicidal properties and innate immune functions. On the flip side, bacterial antioxidant enzymes neutralize macrophage-derived ROS to survive in macrophages, and these mechanisms have been well established. However, the impact of ROS neutralization by bacterial antioxidant enzymes on redox signaling pathways and innate immune responses remains understudied. Previously, we have demonstrated that antioxidant enzymes of F. tularensis are important virulence factors and play an essential role in the suppression of kinase signaling and pro-inflammatory cytokine production (29). In this study, we expose the link between efficient ROS scavenging capacity of F. tularensis and its ability to cause innate immune suppression. We demonstrate a novel role for antioxidant enzymes of F. tularensis in modulation of macrophage function.
Although F. tularensis genome encodes conventional antioxidant enzymes such as SodB, SodC, KatG, and AhpC homologs found in other bacterial pathogens, they differ in many ways with respect to their subcellular localizations. In a majority of bacterial pathogens, SodB localizes to the bacterial cytoplasm and protects bacteria from oxidative stress during aerobic growth; in F. tularensis, it is constitutively expressed and secreted (33, 26). Although KatG in Mycobacterium tuberculosis (50) and Burkholderia pseudomallei localizes to the bacterial cytoplasm, in F. tularensis it is located on the outer membrane (51). AhpC of Francisella and M. tuberculosis is located in the outer membrane, and it is located in the cytoplasm in Pseudomonas and Burkholderia. Additionally, unlike other bacterial pathogens, SoxR, an oxidative stress response regulator that regulates the expression of superoxide dismutases, and a manganese containing SodA are absent in F. tularensis. However, an oxidative stress regulator OxyR is present in F. tularensis that regulates the expression of KatG and AhpC.4 F. tularensis KatG, SodC, and AhpC are induced in response to oxidative stress (52) and are secreted in abundance in the extracellular milieu and into the cytosol of F. tularensis-infected macrophages (27). Expression of these primary antioxidant enzyme genes starts immediately upon phagocytosis of F. tularensis and remains significantly up-regulated during phagosomal and cytosolic phases (52), suggesting that F. tularensis experiences oxidative stress at both of these intracellular locations. We have also shown that both SodB and KatG are secreted by the type I secretion system of F. tularensis (33). Collectively, these observations point to the fact that antioxidant defense mechanisms of F. tularensis are unique and thus, in addition to their conventional role, may also contribute significantly to the immunopathogenesis of tularemia.
Results from this study demonstrate that F. tularensis LVS has immunosuppressive properties similar to the virulent SchuS4 strain. Furthermore, the ability of F. tularensis to repress an ongoing inflammatory response is associated with its antioxidant defense mechanisms. We observed that sodBC gene mutant not only elicited an enhanced pro-inflammatory cytokine response, it failed to restrict an ongoing inflammatory response induced by F. novicida. Our earlier studies have shown that the intramacrophage growth defect and virulence of the sodBC mutant is reversed either by chemical inhibition of NADPH oxidase or by infection in phox−/− mice (30). Concurrently, our in vivo findings from this study support the role of NADPH oxidase in the induction of enhanced levels of pro-inflammatory cytokines observed in mice infected with the sodBC mutant. Significantly higher levels of ROS accumulated in macrophages infected with the sodBC mutant that peaked at 30 min post-infection indicate that the redox environment of these macrophages is distinct from that of F. tularensis LVS-infected macrophages.
Mitochondrial electron transport chain and oxygen-metabolizing enzymatic reactions involving xanthine oxidase, NADPH oxidase, or the cytochrome p450 system are primarily involved in the production of ROS in macrophages (53). The results from our studies implicate NADPH oxidase as one of the sources of ROS in macrophages infected with the sodBC mutant. Additionally, in agreement with the recent findings that mROS co-localizes with intracellular bacterial pathogens (54), we also observed that mROS co-localized with sodBC mutant, but not with wild type F. tularensis LVS in infected macrophages. These results also support the notion that antioxidant enzymes of F. tularensis scavenge ROS emanating from different cellular sources to retain redox homeostasis similar to the one observed in uninfected macrophages.
ROS functions as a signaling messenger that activates redox-sensitive tyrosine kinases, phosphatases, MAPKs, or ion channels. The regulation of the redox-sensitive signaling components is influenced by the presence of cysteine residues or metal co-factors in kinases and phosphatases (55). MAPKs compose a family of ubiquitous protein-serine/threonine kinases that mediate intracellular signaling in response to extra- or intracellular stimuli. ERK1 and ERK2 isoforms, JNK and p38, are the members of MAPK family. Each of these MAPKs is activated through a cascade of sequential phosphorylations beginning at the levels of MAPK3s, MAPK2s, and MAPKs. The MAPKs are involved in the regulation of several cellular activities such as proliferation, differentiation, inflammatory responses, and apoptosis. ROS can activate MAPKs by the oxidative modification of signaling proteins or intracellular kinases such as MAPK3s (56).
ASK1 is one of several MAPK3s that is activated in response to pro-inflammatory stimuli such as ROS, leading to the activation of the MAPK2s-JNK/p38 cascades (49, 57,). Under cellular homeostatic conditions, ASK1 is kept inactive by association with thioredoxin 1 (Trx1). Under oxidative stress conditions, TXNIP competes with ASK1 to bind with Trx1, thereby releasing ASK1 from the Trx1-mediated inhibition (58). The activation of ASK1 is tightly regulated by phosphorylation of threonine residue (Thr838 in human and Thr845 in mouse) in its kinase domain and dephosphorylation at Ser967. Oxidation of ASK1 by H2O2 leads to its multimerization through the formation of interchain disulfide bonds that can be detected as slowly migrating species on non-reducing gels. It has also been shown that oxidation of ASK1 precedes its phosphorylation and is required for activation of downstream MAPK signaling. Phosphorylation of ASK1 activates cytochrome c release and transmission of oxidative stress-induced apoptotic signal. ROS-dependent activation of ASK1 has been demonstrated to be essential for the activation of the downstream MAPK pathway for the induction of pro-inflammatory cytokines during mycobacterial infection (59). ASK1 activation is also required for elimination of bacterial and viral pathogens (60, 61). Our results demonstrate a ROS-dependent activation of MAPK and NF-κB signaling in macrophages infected with the sodBC mutant, and ASK1, an upstream redox-sensitive MAPK3, serves as a potential trigger for the activation of downstream MAPKs. These findings suggest that antioxidant enzymes of F. tularensis restrict cytokine production in infected macrophages by limiting ROS. A similar association has been demonstrated for the attenuated BCG strain and other mutants of virulent M. tuberculosis; failure to control intramacrophage ROS production leads to an increased production of pro-inflammatory cytokines (62–66). Conversely, restriction of host ROS via overexpression of host superoxide dismutase results in the suppression of pro-inflammatory cytokines and impaired clearance of Listeria monocytogenes (67). These studies indicate a conserved theme of ROS-dependent cell signaling in the generation of innate immune responses and provide conceptual evidence that bacterial antioxidant enzymes prevent this response by inhibiting redox-sensitive signaling.
Avoiding autophagy is proposed as one of the immune evasion mechanisms adopted by F. tularensis (68). Studies conducted with M. tuberculosis have shown that ROS is required for autophagy-mediated elimination of intracellular BCG. M. tuberculosis encoded factor eis regulates autophagy, inflammation, and cell death in a redox-dependent fashion (69). Moreover, activation of JNK signaling facilitates the induction of non-canonical autophagy response (69). Additionally, increased oxidative stress results in endoplasmic reticulum stress, leading to autophagy and cell death via the activation of the JNK/p38 signaling pathway (70). Our results demonstrate that antioxidant enzymes of F. tularensis inhibit autophagy in a ROS-dependent manner, which may be mediated by keeping JNK/p38 MAPK in check. We believe that this is one of the novel mechanisms that F. tularensis adopts to inhibit autophagy and thus interfere with bacterial clearance. Further studies are warranted to understand the exact mechanism of ROS-mediated inhibition of autophagy by F. tularensis.
Finally, the results from this study also demonstrate that mechanisms of immunosuppression caused by antioxidant enzymes of F. tularensis are similar in both the murine and human macrophages. Potentiation of pro-inflammatory response by enhancing ROS in HMDMs further establish the role of redox signaling in modulation of innate immune function. Although we observed elevated TNF-α and IL-6 responses in HMDMs infected with opsonized sodBC mutant, the differences in the magnitude of TNF-α response varied between the experiments. We believe that these variations may have been due to variability in monocytes obtained from blood donors. Consistent with our findings, several previous studies have also reported that variability in susceptibility to infection and/or innate immune responses may arise due to variations in genetic and epigenetic background of each blood donor (71–73).
To conclude, this study demonstrates that antioxidant enzymes SodB and SodC of F. tularensis retain a homeostatic redox environment in infected macrophages to prevent activation of redox-sensitive MAPK signaling components, NF-κB, and production of pro-inflammatory cytokines. We also report that F. tularensis inhibits ROS-dependent autophagy to promote its intramacrophage survival. Collectively, this study reveals novel pathogenic mechanisms that F. tularensis adopts to modulate macrophage innate immune functions to create an environment permissive for its intracellular survival and growth. Overall, elucidating how bacterial antioxidant enzymes thwart macrophage antimicrobial mechanisms will lead to the design and implementation of effective therapeutics and vaccination strategies against Francisella and other intracellular bacterial pathogens.
Author Contributions
C. S. B., M. M., and J. A. M. conceived, designed, and coordinated the study. C. S. B. and M. M. wrote the paper. B. C. S. performed and analyzed the experiments shown in Fig. 2. S. M. R., Z. M., and S. V. C. performed and analyzed the experiments shown in Figs. 1, 4–7, 9, and 10. M. V. performed and analyzed the experiments shown in Figs. 3 and 8. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants P01AI056320 and R56AI101109 (to C. S. B.) and R15AI107698 (to M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
S. M. Rabadi and C. S. Bakshi, unpublished data.
- LVS
- live vaccine strain
- ROS
- reactive oxygen species
- RNS
- reactive nitrogen species
- ANOVA
- analysis of variance
- HMDM
- human monocyte-derived macrophage
- DHE
- dihydroethidium
- DCFDA
- 2′,7′-dichlorofluorescin diacetate
- m.o.i.
- multiplicity of infection
- BMDM
- bone marrow-derived macrophage
- mROS
- mitochondrial ROS
- HMDM
- human monocyte-derived macrophage
- 3MA
- 3-methyladenine
- NAC
- N-acetylcysteine.
References
- 1. Pechous R. D., McCarthy T. R., and Zahrt T. C. (2009) Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol. Mol. Biol. Rev. 73, 684–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barry E. M., Cole L. E., and Santiago A. E. (2009) Vaccines against tularemia. Hum. Vaccin. 5, 832–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oyston P. C., Sjostedt A., and Titball R. W. (2004) Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978 [DOI] [PubMed] [Google Scholar]
- 4. Dennis D. T., Inglesby T. V., Henderson D. A., Bartlett J. G., Ascher M. S., Eitzen E., Fine A. D., Friedlander A. M., Hauer J., Layton M., Lillibridge S. R., McDade J. E., Osterholm M. T., O'Toole T., Parker G., et al. (2001) Tularemia as a biological weapon: medical and public health management. JAMA 285, 2763–2773 [DOI] [PubMed] [Google Scholar]
- 5. Melillo A., Sledjeski D. D., Lipski S., Wooten R. M., Basrur V., and Lafontaine E. R. (2006) Identification of a Francisella tularensis LVS outer membrane protein that confers adherence to A549 human lung cells. FEMS Microbiol. Lett. 263, 102–108 [DOI] [PubMed] [Google Scholar]
- 6. Horzempa J., O'Dee D. M., Shanks R. M., and Nau G. J. (2010) Francisella tularensis ΔpyrF mutants show that replication in nonmacrophages is sufficient for pathogenesis in vivo. Infect. Immun. 78, 2607–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ben Nasr A., Haithcoat J., Masterson J. E., Gunn J. S., Eaves-Pyles T., and Klimpel G. R. (2006) Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J. Leukocyte Biol. 80, 774–786 [DOI] [PubMed] [Google Scholar]
- 8. Schwartz J. T., Barker J. H., Long M. E., Kaufman J., McCracken J., and Allen L. A. (2012) Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via complement receptors 1 and 3 in nonimmune serum. J. Immunol. 189, 3064–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sjöstedt A. (2006) Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 8, 561–567 [DOI] [PubMed] [Google Scholar]
- 10. Geier H., and Celli J. (2011) Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infect. Immun. 79, 2204–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desjardins M., Huber L. A., Parton R. G., and Griffiths G. (1994) Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124, 677–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patki V., Virbasius J., Lane W. S., Toh B. H., Shpetner H. S., and Corvera S. (1997) Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 7326–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simonsen A., Lippé R., Christoforidis S., Gaullier J. M., Brech A., Callaghan J., Toh B. H., Murphy C., Zerial M., and Stenmark H. (1998) EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394, 494–498 [DOI] [PubMed] [Google Scholar]
- 14. Clemens D. L., Lee B. Y., and Horwitz M. A. (2004) Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72, 3204–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Golovliov I., Baranov V., Krocova Z., Kovarova H., and Sjöstedt A. (2003) An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 71, 5940–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Checroun C., Wehrly T. D., Fischer E. R., Hayes S. F., and Celli J. (2006) Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deby C., and Goutier R. (1990) New perspectives on the biochemistry of superoxide anion and the efficiency of superoxide dismutases. Biochem. Pharmacol. 39, 399–405 [DOI] [PubMed] [Google Scholar]
- 18. Steinbeck M. J., Khan A. U., and Karnovsky M. J. (1993) Extracellular production of singlet oxygen by stimulated macrophages quantified using 9,10-diphenylanthracene and perylene in a polystyrene film. J. Biol. Chem. 268, 15649–15654 [PubMed] [Google Scholar]
- 19. Wink D. A., and Mitchell J. B. (1998) Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic. Biol. Med. 25, 434–456 [DOI] [PubMed] [Google Scholar]
- 20. Zahrt T. C., and Deretic V. (2002) Reactive nitrogen and oxygen intermediates and bacterial defenses: unusual adaptations in Mycobacterium tuberculosis. Antioxid. Redox Signal. 4, 141–159 [DOI] [PubMed] [Google Scholar]
- 21. Nathan C., and Shiloh M. U. (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halliwell B., and Gutteridge J. M. (1997) Lipid peroxidation in brain homogenates: the role of iron and hydroxyl radicals. J. Neurochem. 69, 1330–1331 [DOI] [PubMed] [Google Scholar]
- 23. Miller R. A., and Britigan B. E. (1997) Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rhee S. G., Chang T. S., Bae Y. S., Lee S. R., and Kang S. W. (2003) Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol. 14, S211–S215 [DOI] [PubMed] [Google Scholar]
- 25. Finkel T. (2001) Reactive oxygen species and signal transduction. IUBMB Life 52, 3–6 [DOI] [PubMed] [Google Scholar]
- 26. Bakshi C. S., Malik M., Regan K., Melendez J. A., Metzger D. W., Pavlov V. M., and Sellati T. J. (2006) Superoxide dismutase B gene (sodB)-deficient mutants of Francisella tularensis demonstrate hypersensitivity to oxidative stress and attenuated virulence. J. Bacteriol. 188, 6443–6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee B. Y., Horwitz M. A., and Clemens D. L. (2006) Identification, recombinant expression, immunolocalization in macrophages, and T-cell responsiveness of the major extracellular proteins of Francisella tularensis. Infect. Immun. 74, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindgren H., Shen H., Zingmark C., Golovliov I., Conlan W., and Sjöstedt A. (2007) Resistance of Francisella tularensis strains against reactive nitrogen and oxygen species with special reference to the role of KatG. Infect. Immun. 75, 1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melillo A. A., Bakshi C. S., and Melendez J. A. (2010) Francisella tularensis antioxidants harness reactive oxygen species to restrict macrophage signaling and cytokine production. J. Biol. Chem. 285, 27553–27560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melillo A. A., Mahawar M., Sellati T. J., Malik M., Metzger D. W., Melendez J. A., and Bakshi C. S. (2009) Identification of Francisella tularensis live vaccine strain CuZn superoxide dismutase as critical for resistance to extracellularly generated reactive oxygen species. J. Bacteriol. 191, 6447–6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dieppedale J., Sobral D., Dupuis M., Dubail I., Klimentova J., Stulik J., Postic G., Frapy E., Meibom K. L., Barel M., and Charbit A. (2011) Identification of a putative chaperone involved in stress resistance and virulence in Francisella tularensis. Infect. Immun. 79, 1428–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Llewellyn A. C., Jones C. L., Napier B. A., Bina J. E., and Weiss D. S. (2011) Macrophage replication screen identifies a novel Francisella hydroperoxide resistance protein involved in virulence. PLoS ONE 6, e24201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma Z., Banik S., Rane H., Mora V. T., Rabadi S. M., Doyle C. R., Thanassi D. G., Bakshi C. S., and Malik M. (2014) EmrA1 membrane fusion protein of Francisella tularensis LVS is required for resistance to oxidative stress, intramacrophage survival and virulence in mice. Mol. Microbiol. 91, 976–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allen L. A., and McCaffrey R. L. (2007) To activate or not to activate: distinct strategies used by Helicobacter pylori and Francisella tularensis to modulate the NADPH oxidase and survive in human neutrophils. Immunol. Rev. 219, 103–117 [DOI] [PubMed] [Google Scholar]
- 35. Mohapatra N. P., Balagopal A., Soni S., Schlesinger L. S., and Gunn J. S. (2007) AcpA is a Francisella acid phosphatase that affects intramacrophage survival and virulence. Infect. Immun. 75, 390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schulert G. S., McCaffrey R. L., Buchan B. W., Lindemann S. R., Hollenback C., Jones B. D., and Allen L. A. (2009) Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of LVS. Infect. Immun. 4, 1324–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bosio C. M. (2011) The subversion of the immune system by Francisella tularensis. Front. Microbiol. 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bosio C. M., Bielefeldt-Ohmann H., and Belisle J. T. (2007) Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178, 4538–4547 [DOI] [PubMed] [Google Scholar]
- 39. Chase J. C., Celli J., and Bosio C. M. (2009) Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect. Immun. 77, 180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rajaram M. V., Ganesan L. P., Parsa K. V., Butchar J. P., Gunn J. S., and Tridandapani S. (2006) Akt/protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J. Immunol. 177, 6317–6324 [DOI] [PubMed] [Google Scholar]
- 41. Butchar J. P., Cremer T. J., Clay C. D., Gavrilin M. A., Wewers M. D., Marsh C. B., Schlesinger L. S., and Tridandapani S. (2008) Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE 3, e2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fuller J. R., Craven R. R., Hall J. D., Kijek T. M., Taft-Benz S., and Kawula T. H. (2008) RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect. Immun. 76, 4934–4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bakshi C. S., Malik M., Mahawar M., Kirimanjeswara G. S., Hazlett K. R., Palmer L. E., Furie M. B., Singh R., Melendez J. A., Sellati T. J., and Metzger D. W. (2008) An improved vaccine for prevention of respiratory tularemia caused by Francisella tularensis SchuS4 strain. Vaccine 26, 5276–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mahawar M., Rabadi S. M., Banik S., Catlett S. V., Metzger D. W., Malik M., and Bakshi C. S. (2013) Identification of a live attenuated vaccine candidate for tularemia prophylaxis. PLoS ONE 8, e61539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Franchini A. M., Hunt D., Melendez J. A., and Drake J. R. (2013) FcγR-driven release of IL-6 by macrophages requires NOX2-dependent production of reactive oxygen species. J. Biol. Chem. 288, 25098–25108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahawar M., Atianand M. K., Dotson R. J., Mora V., Rabadi S. M., Metzger D. W., Huntley J. F., Harton J. A., Malik M., and Bakshi C. S. (2012) Identification of a novel Francisella tularensis factor required for intramacrophage survival and subversion of innate immune response. J. Biol. Chem. 287, 25216–25229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cremer T. J., Butchar J. P., and Tridandapani S. (2011) Francisella subverts innate immune signaling: focus on PI3K/Akt. Front. Microbiol. 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gillette D. D., Curry H. M., Cremer T., Ravneberg D., Fatehchand K., Shah P. A., Wewers M. D., Schlesinger L. S., Butchar J. P., Tridandapani S., and Gavrilin M. A. (2014) Virulent type A Francisella tularensis actively suppresses cytokine responses in human monocytes. Front. Cell. Infect. Microbiol. 4, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang C. S., Shin D. M., Lee H. M., Son J. W., Lee S. J., Akira S., Gougerot-Pocidalo M. A., El-Benna J., Ichijo H., and Jo E. K. (2008) ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell. Microbiol. 10, 741–754 [DOI] [PubMed] [Google Scholar]
- 50. Rezwan M., Lanéelle M. A., Sander P., and Daffé M. (2007) Breaking down the wall: fractionation of mycobacteria. J. Microbiol. Methods 68, 32–39 [DOI] [PubMed] [Google Scholar]
- 51. Huntley J. F., Conley P. G., Hagman K. E., and Norgard M. V. (2007) Characterization of Francisella tularensis outer membrane proteins. J. Bacteriol. 189, 561–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wehrly T. D., Chong A., Virtaneva K., Sturdevant D. E., Child R., Edwards J. A., Brouwer D., Nair V., Fischer E. R., Wicke L., Curda A. J., Kupko J. J. 3rd, Martens C., Crane D. D., Bosio C. M., et al. (2009) Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell. Microbiol. 11, 1128–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sarsour E. H., Kumar M. G., Chaudhuri L., Kalen A. L., and Goswami P. C. (2009) Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 11, 2985–3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. West A. P., Brodsky I. E., Rahner C., Woo D. K., Erdjument-Bromage H., Tempst P., Walsh M. C., Choi Y., Shadel G. S., and Ghosh S. (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ray P. D., Huang B. W., and Tsuji Y. (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Son Y., Kim S., Chung H. T., and Pae H. O. (2013) Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 528, 27–48 [DOI] [PubMed] [Google Scholar]
- 57. Ichijo H. (2007) ASK family proteins in stress and disease–“how cells sense stress”. Nihon Yakurigaku Zasshi 129, 89–93 [DOI] [PubMed] [Google Scholar]
- 58. Elgort M. G., O'Shea J. M., Jiang Y., and Ayer D. E. (2010) Transcriptional and translational downregulation of thioredoxin interacting protein is required for metabolic reprogramming during G(1). Genes Cancer 1, 893–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuk J. M., Shin D. M., Yang C. S., Kim K. H., An S. J., Rho J., Park J. K., and Jo E. K. (2009) Role of apoptosis-regulating signal kinase 1 in innate immune responses by Mycobacterium bovis bacillus Calmette-Guerin. Immunol. Cell Biol. 87, 100–107 [DOI] [PubMed] [Google Scholar]
- 60. Matsuzawa A., Saegusa K., Noguchi T., Sadamitsu C., Nishitoh H., Nagai S., Koyasu S., Matsumoto K., Takeda K., and Ichijo H. (2005) ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587–592 [DOI] [PubMed] [Google Scholar]
- 61. Hayakawa T., Kato K., Hayakawa R., Hisamoto N., Matsumoto K., Takeda K., and Ichijo H. (2011) Regulation of anoxic death in Caenorhabditis elegans by mammalian apoptosis signal-regulating kinase (ASK) family proteins. Genetics 187, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kurtz S., McKinnon K. P., Runge M. S., Ting J. P., and Braunstein M. (2006) The SecA2 secretion factor of Mycobacterium tuberculosis promotes growth in macrophages and inhibits the host immune response. Infect. Immun. 74, 6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Méndez-Samperio P., Pérez A., and Torres L. (2009) Role of reactive oxygen species (ROS) in Mycobacterium bovis bacillus Calmette Guerin-mediated up-regulation of the human cathelicidin LL-37 in A549 cells. Microb. Pathog. 47, 252–257 [DOI] [PubMed] [Google Scholar]
- 64. Miller J. L., Velmurugan K., Cowan M. J., and Briken V. (2010) The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-α-mediated host cell apoptosis. PLoS Pathog. 6, e1000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sadagopal S., Braunstein M., Hager C. C., Wei J., Daniel A. K., Bochan M. R., Crozier I., Smith N. E., Gates H. O., Barnett L., Van Kaer L., Price J. O., Blackwell T. S., Kalams S. A., and Kernodle D. S. (2009) Reducing the activity and secretion of microbial antioxidants enhances the immunogenicity of BCG. PLoS ONE 4, e5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Samuel L. P., Song C. H., Wei J., Roberts E. A., Dahl J. L., Barry C. E. 3rd., Jo E. K., and Friedman R. L. (2007) Expression, production and release of the Eis protein by Mycobacterium tuberculosis during infection of macrophages and its effect on cytokine secretion. Microbiology 153, 529–540 [DOI] [PubMed] [Google Scholar]
- 67. Break T. J., Jun S., Indramohan M., Carr K. D., Sieve A. N., Dory L., and Berg R. E. (2012) Extracellular superoxide dismutase inhibits innate immune responses and clearance of an intracellular bacterial infection. J. Immunol. 188, 3342–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Case E. D., Chong A., Wehrly T. D., Hansen B., Child R., Hwang S., Virgin H. W., and Celli J. (2014) The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell. Microbiol. 16, 862–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shin D. M., Jeon B. Y., Lee H. M., Jin H. S., Yuk J. M., Song C. H., Lee S. H., Lee Z. W., Cho S. N., Kim J. M., Friedman R. L., and Jo E. K. (2010) Mycobacterium tuberculosis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 6, e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Younce C. W., and Kolattukudy P. E. (2010) MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem. J. 426, 43–53 [DOI] [PubMed] [Google Scholar]
- 71. Travanty E., Zhou B., Zhang H., Di Y. P., Alcorn J. F., Wentworth D. E., Mason R., and Wang J. (2015) Differential susceptibilities of human lung primary cells to H1N1 influenza viruses. J. Virol. 89, 11935–11944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Orekhov A. N., Nikiforov N. G., Elizova N. V., Ivanova E. A., and Makeev V. J. (2015) Phenomenon of individual difference in human monocyte activation. Exp. Mol. Pathol. 99, 151–154 [DOI] [PubMed] [Google Scholar]
- 73. Yaqoob P., Newsholme E. A., and Calder P. C. (1999) Comparison of cytokine production in cultures of whole human blood and purified mononuclear cells. Cytokine 11, 600–605 [DOI] [PubMed] [Google Scholar]