

Abstract
Ligands of the tumor necrosis factor superfamily (TNFSF) interact with members of the TNF receptor superfamily (TNFRSF). TNFSF ligand-TNFRSF receptor interactions have been intensively evaluated by many groups. The affinities of TNFSF ligand-TNFRSF receptor interactions are highly dependent on the oligomerization state of the receptor, and cellular factors (e.g. actin cytoskeleton and lipid rafts) influence the assembly of ligand-receptor complexes, too. Binding studies on TNFSF ligand-TNFRSF receptor interactions were typically performed using cell-free assays with recombinant fusion proteins that contain varying numbers of TNFRSF ectodomains. It is therefore not surprising that affinities determined for an individual TNFSF ligand-TNFRSF interaction differ sometimes by several orders of magnitude and often do not reflect the ligand activity observed in cellular assays. To overcome the intrinsic limitations of cell-free binding studies and usage of recombinant receptor domains, we performed comprehensive binding studies with Gaussia princeps luciferase TNFSF ligand fusion proteins for cell-bound TNFRSF members on intact cells at 37 °C. The affinities of the TNFSF ligand G. princeps luciferase-fusion proteins ranged between 0.01 and 19 nm and offer the currently most comprehensive and best suited panel of affinities for in silico studies of ligand-receptor systems of the TNF family.
Keywords: cytokine, fusion protein, ligand-binding protein, NF-κB (NF-KB), protein complex, receptor regulation, TNF receptor-associated factor, tumor necrosis factor (TNF)
Introduction
The ligands and receptors of the tumor necrosis factor (TNF) family constitute two complementary families of proteins that regulate a huge variety of immune processes but are also involved in the control of tissue homeostasis and differentiation of certain cell types (1, 2). Assignment to the TNF ligand family is based on the presence of a conserved C-terminal TNF homology domain (THD),2 which mediates formation of homotrimeric molecules and binding to members of the TNF receptor family (1, 2). TNFSF ligands are typically type II or type III transmembrane proteins and thus contain in addition to the THD an N-terminal cytosolic domain and a single transmembrane domain that is separated from the THD by a stalk region of variable length and structure (1, 2). There are also soluble variants of TNFSF ligands that result from proteolytic processing in the stalk region or from alternative splicing. Because of the presence of the THD, these soluble molecules also form trimers that retain the receptor-binding ability of the membrane-bound ligand.
The TNFRSF receptors are characterized by the presence of one or more copies of a conserved cysteine-rich domain in the N-terminal part of these molecules (1, 2). According to structural and functional similarities, three subcategories of TNFRSF receptors can be defined. The first subgroup comprises the death receptors that are characterized by a conserved C-terminally located protein-protein interaction domain, called the death domain (3). The death domain enables most death receptors to induce apoptosis but is also required to transmit non-apoptotic signals. Second, there are the TNF receptor-associated factor (TRAF)-interacting receptors that possess one or more motifs in their cytoplasmic domain allowing direct interaction with members of the TRAF adapter protein family (4). The third subgroup consists of soluble and membrane-anchored TNFRSF receptors that act as decoy receptors of the death and TRAF-interacting receptors by competition for ligand binding and/or by forming signaling-incompetent heteromeric receptor complexes (5, 6).
The broad physiological and pathophysiological relevance of the ligands and receptors of the TNF family is, for example, evident from the fact that various inherited diseases, including autoimmune lymphoproliferative syndrome (ALPS), common variable immunodeficiency, TNFR1-associated periodic fever syndrome, X-linked hyper-IgM syndrome, and X-linked hypohidrotic ectodermal dysplasia, can be caused by mutations in genes encoding ligand and receptors of the TNF family (CD40L (X-linked hyper-IgM syndrome), CD95L (ALPS), ectodysplasin-A1 (EDA-A1; XEDA), CD95 (ALPS), TACI (common variable immunodeficiency), TNFR1 (TNFR1-associated periodic fever syndrome)) (7). Moreover, chronic production of TNF and the deregulated activity of receptor activator of NF-κB ligand have been identified as crucial factors in the development of autoimmune diseases and the progression of bone destructive processes (7). The specificity of TNFSF ligand-TNFRSF receptor interactions and the extracellular accessibility of these molecules make them highly attractive and straightforward targets for therapeutic interventions. Indeed, recombinant TNF is approved for the treatment of soft tissue sarcoma by isolated limb perfusion, and TNF-specific antibodies and a TNFR2 ectodomain Fc-fusion protein are an established and broadly used option in the therapy of various immune diseases, including rheumatoid arthritis and Crohn disease (7). Furthermore, neutralizing antibodies specific for receptor activator of NF-κB ligand and BAFF are approved for the treatment of osteoporosis and systemic lupus erythematosus, and several more molecules targeting ligands and receptors of the TNF family are under consideration in clinical trials.
The development and analysis of drugs modulating the activity of a certain TNFSF ligand-TNFRSF receptor pair as well as the understanding of the principles of TNF receptor activation highly benefit from knowledge of the affinities of these interactions. With few exceptions, however, experimental data on the receptor affinity of TNF ligands derived from analysis with intact cells are rare. Indeed, the majority of available binding data on TNFSF ligand-TNFRSF receptor interactions are gained with cell-free methods that are based on the usage of distinct experimental methods and recombinant receptor variants containing varying numbers of receptor molecules (Table 1). Therefore, it is not really unexpected that affinities published for a certain ligand-receptor interaction can vary considerably (Table 1). In particular, values derived from cell free assays are often quite different from binding parameters obtained by cellular binding studies (Table 1). Presumably, this dissatisfying situation is mainly caused by the fact that the formation of TNFSF ligand-TNFRSF receptor complexes on the cell surface is controlled by several cell intrinsic factors, such as dynamic self-assembly of TNFRSF receptors in the absence of ligand and interactions with the cytoskeleton that cannot be adequately gathered by cell-free techniques (8).
TABLE 1.
Literature survey of TNFSF ligand-TNFRSF receptor affinities
The abbreviations used are as follows: CBS, cellular binding study; SPR, surface plasmon resonance; ITC, isothermal titration calorimetry; HRTF, homogenous time-resolved fluorescence; n.i., not indicated.


a 41BBed-AP is a fusion protein of the extracellular domain of 41BB with alkaline phosphatase, and the latter forms dimers.
b Endogenous/transfected 41BBL of high and low affinity binding sites.
c Solution phase BIAcore binding assay (for details see Ref.)
Here, we generated highly bioluminescent and functionally not compromised ligand variants of all TNFSF members by N-terminal fusion with the luciferase from Gaussia princeps (GpL). By help of these fusion proteins, we performed a systematic and comprehensive study on the interactions between TNFSF ligands and their cell surface-expressed receptors.
Experimental Procedures
Cloning, Production, and Purification of Recombinant Proteins
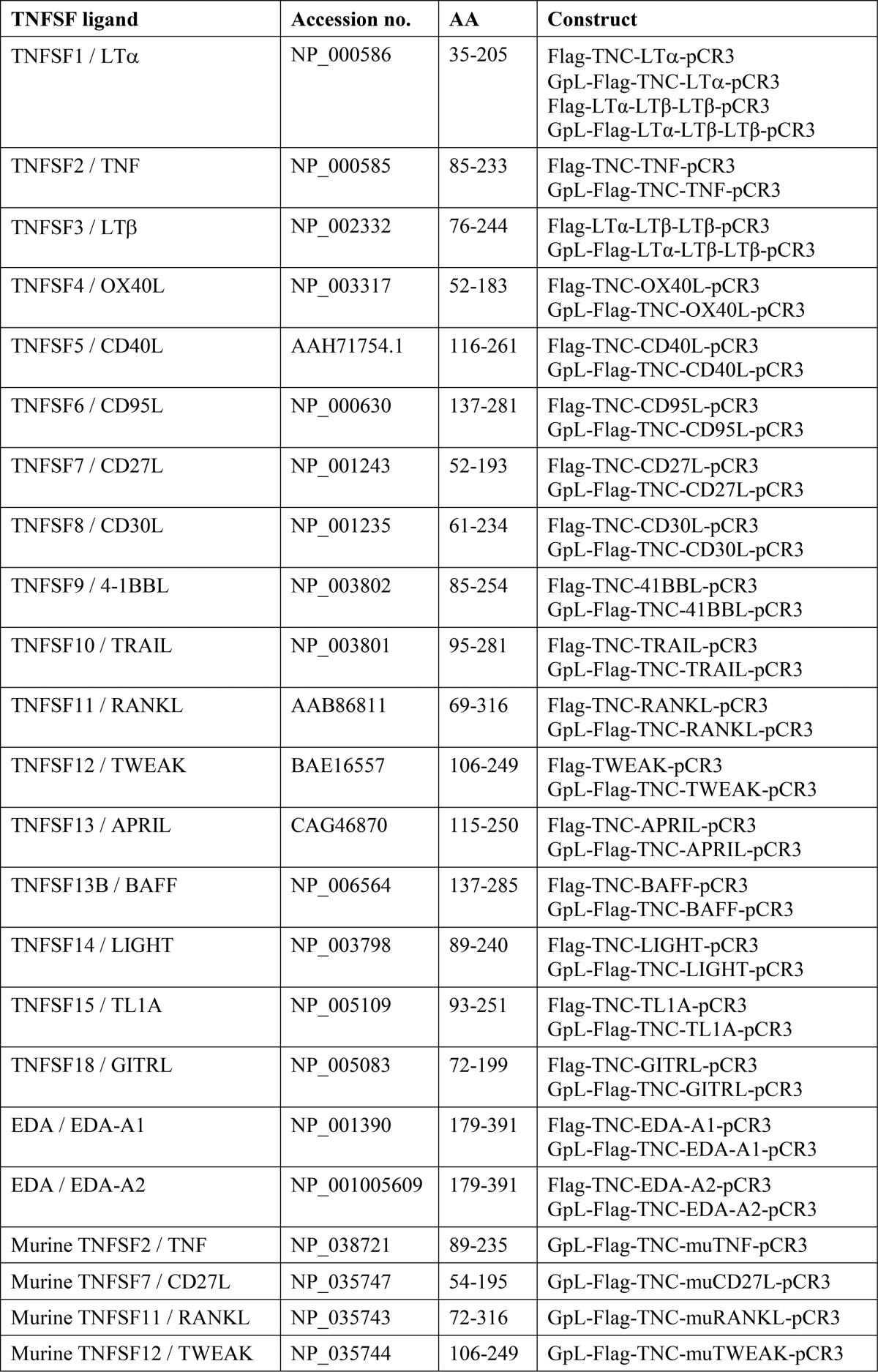
Cloning of GpL-FLAG-TNC-TNF, GpL-FLAG-TNC-TWEAK, and GpL-fLAG-TNC-CD95L has been described elsewhere (9, 10). The expression plasmids encoding the GpL-FLAG-TNC variants of the other TNF ligands used in this study have been obtained by exchange of the TNF-encoding fragment of the pCR3-based GpL-FLAG-TNC-TNF expression plasmid with PCR amplicons encoding THD-encompassing soluble versions of the other TNFSF ligand types (Table 2) by help of flanking EcoRI (5′ end) and XbaI (3′ end) restriction sites. A single synthetic DNA fragment encoding a QLGGGS linker followed by a FLAG epitope and an LTα protomer (amino acids 35–205) connected with an LTβ protomer (amino acids 76–244) between two (GGGS)4 linkers was used to replace the FLAG-TNC part of GpL-FLAG-TNC-LTβ encoding amino acids 76–244 of LTβ to obtain the expression plasmid encoding GpL-FLAG-scLTαβ2. The single chain-encoded LTαβ2 (scLTαβ2) part has been designed in analogy to single chain-encoded constructs for TNF (11) and is largely similar to a C-terminally His-tagged scLTαβ2 variant encoding LTα amino acids 62–205 and LTβ amino acids 87–243 separated by short GGSG linkers that have been published elsewhere (12). The conventional FLAG-TNC-TNFSF ligand expression constructs have been obtained by replacement of the TNF domain in the FLAG-TNC-TNF-encoding expression plasmid with THD-encompassing DNA fragments/amplicons. FLAG-scLTαβ2 was obtained by replacing the FLAG-TNC-TNF cassette with the FLAG-scLTαβ2 cassette of GpL-FLAG-scLTαβ2. HEK293 cells transiently or stably transfected with expression plasmids encoding the recombinant protein of interest were grown near confluency and were then cultivated for 5–7 days in low serum medium (0.5–2%). Supernatants containing the recombinant proteins were collected and cleared by centrifugation and then either used directly for experiments or subjected to anti-FLAG affinity purification.
TABLE 2.
Amino acid residues of full-length TNFSF ligands contained in the soluble TNFSF ligand variants
SDS-PAGE, Silver Staining, and Western Blot Analysis
For evaluation of the purity and protomer size of recombinant soluble TNFSF ligands, the protein samples were separated on 12.5% (w/v) polyacrylamide gels according to Laemmli. To visualize the proteins, the gels were then subjected to silver staining by help of a commercially available kit (Pierce® silver stain kit, Thermo Scientific, Braunschweig, Germany). For Western blot analysis of p100 to p52 processing, cells were washed once with PBS and scraped into PBS using a rubber policeman. Cells were pelleted and resuspended in 4× Laemmli sample buffer (∼1 × 106 cells/100 μl of buffer) supplemented with complete protease inhibitor (Roche Applied Science) and phosphatase inhibitor mixtures II (Sigma). To improve cell disintegration, samples were sonicated for 15 s with maximum amplitude (UP100H Ultrasonic Processor, Hielscher, Germany), heated at 95 °C for 5 min, and centrifuged for 5 min to remove residual insoluble material. Lysates were further processed by SDS-PAGE and standard Western blotting procedures. Finally, p100 processing was determined using anti-p100/p52 from Millipore (05-361), horseradish peroxidase-conjugated anti-mouse IgG (Dako), and the ECL Western blotting detection reagents and analysis system (Amersham Biosciences).
Binding Studies with GpL-TNFSF Ligand Fusion Proteins
Cells (typically 2 × 105 per well in 24-well plates) were cultured overnight to ensure adherence. For equilibrium binding experiments, the cells in half of the wells were dedicated for determination of nonspecific binding and were preincubated for 1 h at 37 °C with either an excess of the “GpL-less” variant of the soluble TNFSF ligand investigated or with an antibody blocking access to the membrane-bound TNFRSF receptor. Untreated cells (= total binding) and “blocked” cells were then pairwise incubated with the GpL-TNFSF ligand fusion protein of interest for an additional hour at 37 °C. Unbound GpL-TNFSF ligands were removed by 10 rapid washes in ice-cold PBS, and cells were scratched with a rubber policeman in 50 μl of culture medium (0.5% FCS, penicillin/streptomycin). Cell-associated GpL activity was finally measured in black 96-well plates using a commercial Gaussia luciferase assay kit (New England Biolabs GmbH, Frankfurt a.M., Germany). To keep errors caused by the comparatively rapid decay of bioluminescent activity of GpL (∼T½ = 4 min) below 3%, substrate/buffer solution was applied to only one column of a 96-well plate at one time, and light emission was immediately measured (1 s per sample, Lucy 2 Luminometer, Anthos Labtec Instruments, Krefeld, Germany). In the cases where TNFRSF receptor transfectants have been used for the determination of total binding, the corresponding parental and receptor-negative transfectants have been used for the evaluation of nonspecific binding without further “blocking” treatment.
TNFSF Ligand-induced Apoptosis and IL8 Production
To evaluate the activity of soluble TNFSF ligands and GpL-TNFSF ligand fusion proteins, HT1080 cells, or HT1080 transfectants of the TNFRSF of interest, were seeded in 96-well tissue culture plates (2 × 104/well) and cultured overnight. The following day, cells were stimulated in triplicate with the recombinant ligands. In experiments where oligomerized ligand molecules have been used, ligands were preincubated on a separate 96-well plate with 1 μg/ml FLAG-specific mAb M2 irrespective of the ligand concentration and were transferred to the cells after 15–30 min. In cases where IL8 induction has been investigated, medium was exchanged prior to ligand stimulation to minimize the background of constitutively produced IL8. Next day, IL8 production and cellular viability were measured. In cell death experiments, cells were sensitized for apoptosis induction by co-treatment with 2.5 μg/ml cycloheximide. IL8 in cell culture supernatants was quantified using a commercially available ELISA kit (BD Biosciences, Heidelberg, Germany) according to the manufacturer's instructions, and cellular viability was quantified by crystal violet staining.
Results
Generation, Production, and Purification of a Panel of Soluble GpL Fusion Proteins Covering the Human Members of the TNFSF Ligand Family
A variety of studies with soluble fusion proteins containing the THD of TNFSF ligands suggest that the functionality of the THD with respect to receptor binding and receptor activation is largely insensitive to N-terminal linkage of additional protein domains (3). We recently took advantage of this fact and generated GpL fusion proteins of the TNFSF ligands CD95L, TNF, and TWEAK for cellular binding studies (9, 10). These GpL-TNFSF ligand fusion proteins were proved to be highly traceable and functionally not distinguishable from their conventional counterparts. We extended this work and generated a panel of GpL fusion protein covering all human members of the TNFSF ligand family for the systematic and comprehensive analysis of TNFSF ligand-TNFRSF receptor interactions on intact cells (Fig. 1, A and B). The GpL-TNFSF ligand fusion proteins consist of an N-terminal GpL domain and C-terminally of the THD of the various human TNFSF ligands. To facilitate purification of the GpL-TNFSF ligand fusion proteins, we furthermore introduced an internal FLAG epitope between the GpL and TNFSF ligand domain. Soluble TNFSF ligands typically assemble with high efficiency into trimeric molecules and interact with members of the TNFRSF receptors. However, in some cases soluble TNF ligand variants are unstable and tend to form not only trimers but to a varying extent inactive misfolded aggregates (13, 14). We and others have previously found that the formation of the latter can be reduced by introduction of a heterologous trimerization domain, such as a modified leucine zipper or the tenascin-C trimerization domain (13–15). Therefore, we also introduced the trimerization domain of TNC preceding the THD to ensure proper production and trimer assembly of all TNFSF ligands used in this study (Fig. 1A, left panel). To obtain LTα-LTβ heterotrimers of defined 1:2 stoichiometry, we encoded peptide linker-connected LTα and LTβ protomers (single-chain LTαβ2, scLTαβ2) by a single DNA expression cassette (Fig. 1B, right panel). Supernatants collected from HEK293 cells transiently transfected with expression plasmids encoding the various GpL-FLAG-TNC-TNFSF ligand fusion proteins typically yielded 30–120 μg of recombinant protein per 15-cm cell culture Petri dish. GpL-FLAG-TNC-TRAIL showed the poorest productivity and only yielded ∼10 μg per 15-cm cell culture Petri dish despite supplementing the culture medium with zinc ions, which are required by this TNF ligand for proper folding (16). SDS-PAGE and silver staining of anti-FLAG affinity chromatography-purified GpL-FLAG-TNC-TNFSF ligand fusion proteins revealed one to three bands that correspond in size to the deduced molecular weight of the non-modified ligand molecules or to moderately glycosylated forms (Fig. 1B). Luciferase activity of all GpL-FLAG-TNC-TNFSF ligand fusion proteins was between 940 and 1360 relative light units per s and fmol. Thus, there was no evidence for a TNFSF ligand-specific impact of the THD in the various GpL-FLAG-TNC-TNFSF ligand fusion proteins on the activity of the GpL domain. As found before for the GpL fusion proteins of CD95L, luciferase activity increased linearly over several orders of magnitude with the concentration of the GpL-FLAG-TNC-TNFSF ligand fusion proteins (data not shown).
FIGURE 1.

Structure and purification of GpL-TNFSF ligand fusion proteins. A, general domain architecture of the soluble TNFSF ligand variants used in this study. Left panel, TNC domain-stabilized soluble TNFSF ligands with and without the GpL domain. Right panel, single-chain encoded LTαβ2 heterotrimeric molecules. F, FLAG epitope; TNC, trimerization domain of tenascin-C. B, 100 ng of the indicated anti-FLAG M2-agarose affinity-purified TNFSF ligands were separated by SDS-PAGE and visualized by silver staining. Lane M, molecular weight marker.
GpL Fusion Proteins of Soluble TNFSF Ligands Display Unaltered Receptor Stimulating Activities
Next, we investigated for a subset of the novel GpL-FLAG-TNC-TNFSF ligands whether the GpL domain interferes with the receptor stimulating activity of the THD contained in these molecules. For this purpose, we determined side-by-side the dose dependence of induction of apoptosis, IL8 production, or p100 processing by the GpL fusion proteins of 4–1BBL, CD27L, OX40L, CD40L, GITRL, TRAIL, TNF, CD95L, LTα, LIGHT, and scLTαβ2 and the corresponding conventional ligands without GpL domain. We observed in no case major differences in the corresponding ED50 values between the GpL domain-containing and the GpL domain-less variants (Figs. 2 and 3). For example, GpL-FLAG-TNC-TRAIL induced apoptosis in HT1080 cells with an ED50 value of 250 pm (= 20 ng/ml), whereas conventional FLAG-TNC-TRAIL showed an ED50 value of 43 pm (= 6 ng/ml) (Fig. 2A). Likewise, GpL-FLAG-TNC-LTα, GpL-FLAG-TNC-GITRL, GpL-FLAG-TNC-CD40L, GpL-FLAG-TNC-41BBL, GpL-FLAG-TNC-CD95L, GpL-FLAG-TNC-OX40L, GpL-FLAG-TNC-CD27L, and GpL-FLAG-TNC-LIGHT triggered IL8 production in HT1080 cells with comparable efficacy as their GpL domain-less counterparts (Fig. 2B). Induction of p100 processing by GpL-FLAG-TNC-CD27L, GpL-FLAG-TNC-LIGHT, GpL-FLAG-TNC-OX40L, GpL-FLAG-TNC-CD40L, and GpL-FLAG-scLTαβ2, on the one hand, and FLAG-TNC-CD27L, FLAG-TNC-LIGHT, FLAG-TNC-OX40L, FLAG-TNC-CD40L, and FLAG-scLTαβ2, on the other hand, also occurred with similar efficacy (Fig. 3). It is well established that some members of the TNF receptor superfamily require oligomerization of their corresponding soluble trimeric ligands to become properly activated. Importantly, the presence of a GpL domain had no influence or only a minor influence on this requirement (Figs. 2 and 3). Together, the dose-response analysis experiments indicate that genetic fusion of the GpL domain to the N-terminal end of the THD of soluble TNFSF ligands has no major impact on the TNFSF ligand-TNFRSF receptor interaction. Thus, N-terminal fusion of the GpL domain is a generally applicable strategy for labeling soluble TNFSF ligands for studies on TNFSF ligand-TNFRSF receptor interactions in living cells.
FIGURE 2.

Induction of apoptosis and IL8 production by GpL-TNFSF ligand fusion proteins and conventional soluble TNFSF ligands. A, HT1080 cells were grown in 96-well plates (20,000 cells per well) overnight, sensitized for apoptosis by treatment with 2.5 μg/ml cycloheximide and then challenged for an additional day with the indicated concentrations of GpL-FLAG-TNC-TRAIL and GpL-FLAG-TNC-TNF in the presence and absence of 1 μg/ml anti-FLAG mAb M2. Cell viability was finally quantified by crystal violet staining. B, HT1080 cells (for analysis of CD95L, TRAIL, LIGHT, LTα, and LTαβ2) and HT1080 transfectants expressing 4-1BB, CD27, GITR, CD40, or OX40 were seeded in 96-well plates (10,000 cells per well). The next day, medium was changed to reduce the background of constitutively produced IL8, and cells were then challenged overnight in triplicate with increasing concentrations of the indicated TNFSF ligand variants in the absence and presence of 1 μg/ml of the FLAG-specific mAb M2. Finally, the IL8 content of supernatants was determined by ELISA analysis.
FIGURE 3.

Induction of p100 processing by GpL-TNFSF ligand fusion proteins and conventional soluble TNFSF ligands. Cells (HT1080 for LIGHT and LTαβ2, HT1080 TNFRSF receptor transfectants for CD40L, CD27L, and OX40L) were challenged with increasing concentrations of the indicated TNFSF ligands in the presence and absence of the anti-FLAG mAb M2 (1 μg/ml). After overnight incubation, total cell lysates were prepared and subjected to Western blot analysis to evaluate p100 to p52 expression.
Binding Studies with GpL-FLAG-TNC-tagged TNFSF Ligand Fusion Proteins
To determine binding affinities of the various TNFSF ligands at 37 °C to their cell-expressed TNFRSF receptors, saturation binding studies were performed using the various GpL-TNFSF ligand fusion proteins. Depending on the particular TNFRSF receptor-TNFSF ligand pair that has been investigated, nonspecific binding was determined in two different ways. For a few TNF receptors (CD40, TNFR2, CD27, OX40, 41BB, GITR, and CD30) that are not endogenously expressed in HT1080 or HeLa cells, we had available stable transfectants from other work. In these cases, specific binding was calculated as the difference of total binding of a GpL-TNFSF ligand fusion protein to the corresponding receptor-expressing transfectant and its nonspecific binding to the receptor expression-negative parental cell line. Similarly, binding studies with GpL-FLAG-TNC-BAFF, GpL-FLAG-TNC-APRIL, GpL-FLAG-TNC-EDA-A2, GpL-FLAG-TNC-TL1A, and GpL-FLAG-TNC-TRAIL were performed using HEK293 cells transiently transfected with expression vectors encoding BCMA, BAFFR, TACI, XEDAR, DR3, and the TRAIL receptors (TRAILR1, TRAILR2, TRAILR3, and TRAILR4) and mock-transfected control cells. HEK293 cells express moderate amounts of TRAIL receptors, but this barely affected the binding studies due to the much higher number of ectopically expressed TRAIL receptors. In the remaining cases, nonspecific binding was determined by preincubating cells either with an excess of the GpL-less ligand variant or with a blocking TNFRSF receptor-specific antibody. Specific binding was then calculated again by subtracting the nonspecific binding from total binding. The affinities obtained ranged over 3 orders of magnitude reaching from low concentrations of 0.010 and 0.017 nm for LTαβ2 and LIGHT for binding to the LTβR to concentrations of 7.1 and 19.2 nm for the CD40L-CD40 interaction and the CD30L-CD30 interaction (Figs. 4 and 5 and Table 3).
FIGURE 4.

Saturation binding studies with GpL-TNFSF ligand fusion proteins and endogenously expressed TNFRSF receptors. Cells of the indicated cell lines (2 × 105 cells/well) were cultivated overnight in 24-well plates. The next day, half of the wells were preincubated with an excess (2 μg/ml) of the “GpL-less” soluble TNFSF ligand or in the case of the interaction of TNF and LTα with TNFR1 with the mAb H398 (10 μg/ml) to block access to the membrane-bound TNFRSF receptor. Untreated cells (= total binding) and blocked cells (= nonspecific binding) were then pairwise incubated with the GpL-TNFSF ligand fusion proteins for an additional hour at 37 °C. After removal of unbound molecules, the nonspecific binding values were subtracted from the corresponding total binding values to obtain specific binding values that were fitted by non-linear regression to a single binding site interaction plot using the GraphPad Prism5 software. One representative experiment is shown for each of the TNFSF ligand-TNFRSF receptor interactions. For the number of experiments and statistics of each interaction, see Table 3.
FIGURE 5.

Saturation binding studies with GpL-TNFSF ligand fusion proteins and TNFRSF receptor transfectants. HT1080 and HeLa transfectants stably expressing the indicated TNFRSF receptors and HEK293 transiently transfected with expression plasmids encoding the indicated TNFRSF receptor along with corresponding control cells (HT1080, HeLa, and mock-transfected HEK293 cells and HeLa-TNFR2 transfectants in experiments with HeLa-TNFR2-CD95 double transfectants) were cultivated overnight in 24-well plates (half-plate control cells and half-plate receptor transfectants). The next day, cells were pairwise-incubated with increasing concentrations of the GpL-TNFSF ligand fusion proteins for an additional hour at 37 °C. After removal of unbound molecules, the nonspecific binding values (= HT1080, HeLa, and HEK293 cells and HeLa-TNFR2 transfectants in experiments with HeLa-TNFR2-CD95 double transfectants) were subtracted from the total binding values (= HT1080, HeLa, and HEK293 receptor transfectants, HeLa-TNFR2-CD95 double transfectants) to obtain specific binding values. The latter were fitted by non-linear regression to a single binding site interaction plot with the GraphPad Prism5 software. One representative experiment is shown for each of the TNFSF ligand-TNFRSF receptor interactions. For the number of experiments and statistics of each interaction, see Table 3.
TABLE 3.
TNFSF ligand-TNFRSF receptor affinities derived from cellular binding studies at 37 ° C and their comparison with literature values
For references of affinity constants, see Table 1. All binding studies considered were performed at 37 °C and showed R2 values for non-linear regression of specific binding data of >0.95.


a In this study, binding to full-length DR3 has been determined, and in the cited study binding to a death domain deletion mutant of DR3 has been investigated.
b Our GpL-FLAG-TNC-TRAIL preparation contained significant impurities (see Fig. 1). Folding, integrity, and thus specific activity of recombinant TRAIL preparations differ notoriously, depending on the process of production and purification. The affinities of soluble GpL-TNC-FLAG-TRAIL for the various cell-expressed TRAIL receptorsindicated here could therefore be even higher.
c Functional data have been acquired with transfectants expressing an artificial BCMA-CD95 chimeric receptor.
d The enhancing effect observed in this study depends on the TWEAK-induced pathway considered. Oligomerization lowered the EC50 value for classical NF-κB signaling for 2 orders of magnitude and more but had no effect on triggering p100 processing.
e Activity data were obtained with a soluble trimeric EDA-A1 variant without the oligomerizing collagen domain of this molecule and transfectants expressing an artificial EDAR-CD95 chimeric receptor.
In view of the great relevance of mouse models for basic and preclinical research in the TNFSF/TNFRSF field, we also spot-checked a couple of murine TNFSF ligand-TNFRSF receptor interactions. In all five interactions investigated, there was no difference or only a very minor difference in the affinity of the human and murine ligand-receptor pair (Fig. 6 and Table 4). This suggests that not only the specificities of the TNFSF ligand-TNFRSF receptor interactions are largely conserved between mouse and men but also the strength of these interactions.
FIGURE 6.

Saturation binding studies of murine TNFSF ligand-TNFRSF receptor interactions. A, 100 ng of the indicated anti-FLAG M2-agarose affinity-purified murine GpL-TNFSF ligands were separated by SDS-PAGE and visualized by silver staining. B, HEK293 transiently transfected with expression plasmids encoding the indicated murine TNFRSF receptors along with mock-transfected HEK293 cells were cultivated overnight in 24-well plates (half-plate control cells and half-plate receptor transfectants). The next day, cells were pairwise-incubated with increasing concentrations of the indicated GpL-TNFSF ligand fusion proteins for an additional hour at 37 °C. After removal of unbound molecules, the nonspecific binding values (= mock transfectants) were subtracted from the total binding values (receptor transfectants) to obtain specific binding values. To analyze the murine TWEAK-Fn14 interaction, Colon-26 cells were cultivated in 24-well plates. Half of the wells were preincubated with an excess (2 μg/ml) of FLAG-TWEAK to block Fn14 binding. Cells were then pairwise incubated with GpL-FLAG-TNC-muTWEAK for an additional hour at 37 °C, and after removal of unbound molecules, specific binding was again calculated as the difference of total binding (untreated cells) and nonspecific binding (blocked cells). Specific binding values were finally fitted by non-linear regression to a single binding site interaction plot with the GraphPad Prism5 software. One representative experiment is shown for each of the murine TNFSF ligand-TNFRSF receptor interactions. For the number of experiments and statistics of each interaction, see Table 4.
TABLE 4.
Murine TNFSF ligand-TNFRSF receptor affinities derived from cellular binding studies at 37 °C
All binding studies considered were performed at 37 °C and showed R2 values for non-linear regression of specific binding data >0.95.
| Interaction | Sequence identity in ectodomain (%) |
KDa (nm) |
No. of experimentsa |
|||
|---|---|---|---|---|---|---|
| Ligand | Receptor | Mouse | Human | Mouse | Human | |
| TNF-TNFR1 (TNFSF2-TNFRSF1A) | 76 | 71 | 0.086 ± 0.008 | 0.040 ± 0.010 | 4 | 5 |
| TNF-TNFR2 (TNFSF2-TNFRSF1B) | 76 | 57 | 0.089 ± 0.012 | 0.082 ± 0.028 | 4 | 4 |
| CD27L-CD27 (TNFSF7-TNFRSF7) | 63 | 61 | 1.186 ± 0.111 | 0.801 ± 0.14 | 3 | 4 |
| RANKL-RANK (TNFSF11-TNFRSF11A) | 85 | 78 | 0.444 ± 0.118 | 0.337 ± 0.048 | 4 | 8 |
| TWEAK-Fn14 (TNFSF12-TNFRSF12A) | 89 | 78 | 0.051 ± 0.015 | 0.152 ± 0.05 | 3 | 3 |
a Data of the human interactions are from Table 3.
Discussion
Although soluble TNFSF ligand variants comprising the THD typically retain the capability to interact with TNFSF receptors, in several cases this is not sufficient to ensure robust receptor activation. Noteworthy, this seems not to reflect an intrinsic insufficiency of the soluble ligand molecules but rather defines different requirements of TNFRSF receptors for the way the ligands are presented. This is evident from the interactions of TNF with TNFR1 and TNFR2 as well as from studies with APRIL and the receptors TACI and BAFFR. Soluble TNF interacts efficiently with TNFR1 and TNFR2 (Tables 1 and 3) (17), but only TNFR1 becomes strongly activated this way, although proper TNFR2 activation requires stimulation by membrane TNF (18, 19). Likewise, activation of TACI occurs in response to proteoglycan-attached APRIL but not upon binding of soluble APRIL lacking the proteoglycan-binding site of the molecule (20, 21). There are several other TNFRSF receptors that despite ligand binding are not or are only poorly activated by soluble ligand trimers, e.g. CD95, TRAILR2, OX40, CD27, 41BB, and GITR (14, 22–24). A latent capability to stimulate strong receptor signaling, however, is also present in “poorly active” TNFSF ligand-TNFRSF receptor complexes. First, strong receptor activation by poorly active soluble ligand trimers can be restored by multimerization, e.g. by oligomerization with antibodies recognizing an epitope/tag not interfering with receptor binding or by genetically enforced formation of hexameric, nonameric, or dodecameric molecules (14, 19, 22, 23, 25–30). Second, inefficient receptor activation by soluble ligand trimers can be overcome by binding to the extracellular matrix or by artificial cell surface immobilization using genetically engineered trimeric variants of soluble TNF ligands (31). A quite simple and straightforward explanation for the enhanced activity of oligomerized soluble TNFSF ligands would be that the increase in avidity that is associated with the oligomerization process results in a higher apparent affinity that compensates for low affinity of certain TNFSF-TNFRSF interactions. Similarly, membrane anchoring might compensate for low affinities by reducing ligand mobility and increasing local ligand concentrations. However, this view is not supported by the affinities we found for the various TNFSF ligand-TNFRSF receptor interactions. Our set of data shows no correlation between the affinities of the TNFSF ligand-TNFRSF receptor interactions and the activity-enhancing effect of ligand oligomerization. For example, TNF binds with high affinities of 0.04 and 0.082 nm to TNFR1 and TNFR2, but although oligomerization of soluble TNF does not enhance TNFR1 signaling, TNFR2 activation has been shown to be highly dependent on TNF oligomerization (23, 32). Furthermore, soluble CD40L, which benefits only moderately from ligand oligomerization, has a relatively poor affinity of 7.1 nm, whereas EDA-A1, which interacts with EDAR with an affinity of 0.05 nm, still gains activity upon oligomerization (33). Indeed, we recently addressed the relevance of ligand oligomerization for affinity by help of GpL fusion proteins for soluble TWEAK, which poorly stimulate Fn14-mediated induction of the classical NFκB target IL8, and for soluble CD95L, which fails to trigger robust apoptosis induction (9, 10). In these two cases, we noticed no major effect of ligand oligomerization on receptor occupancy and apparent affinity. Noteworthy, phylogenetic subgroups of the TNFRSF such as TNFR1 and TNFR2, the various TRAIL receptors, or TACI, BCMA, and BAFFR have quite similar affinities but nevertheless can differ in their ligand oligomerization requirement for activation. This is evident from the already mentioned TNFR1-TNFR2 system but also from the TACI-BCMA-BAFFR group. Here, as discussed above, TACI and BCMA activation highly benefits from oligomerization of its ligands APRIL and BAFF, although this plays no role in BAFFR stimulation by BAFF (21, 34).
There is increasing evidence in the literature that the superior response of the TNFRSF receptors discussed above to oligomerized or membrane-anchored soluble ligand trimers is due to the secondary clustering of the initially formed TNFSF ligand3-TNFRSF receptor3 complexes. In the supramolecular TNFSF ligand-TNFRSF receptor clusters, certain pathways may then be more robustly activated by trans-activation of signaling proteins associated with the primary TNFSF ligand3-TNFRSF receptor3 complexes (8). The molecular mechanisms that drive/assist this secondary interaction are poorly understood but may involve receptor-receptor interaction via the PLAD and/or topological factors (spatial pre-orientation of membrane-bound molecules, association with the cytoskeleton, limited diffusibility). Thus, the KD values of soluble TNFSF ligands for cell-expressed TNFRSF receptors, as determined in this study, alone are not in any case sufficient to allow modeling of receptor activity. Nevertheless, these values are also an essential prerequisite for in silico analysis of activation of TNFRSF receptors superiorly stimulated by membrane-bound ligands. First, the KD values of the soluble ligand molecules could be used to initially calculate binding of membrane TNFSF ligand to cell-expressed TNFRSF receptors by considering the membrane-localized molecules as soluble molecules in a spherical shell of the thickness of 1–2 receptor molecules. Second, also TNFSF ligand molecules that primarily act in their membrane-bound form are often shed. Now, the resulting soluble trimers may activate specific signaling pathways that do not need secondary clustering of TNFSF ligand3-TNFRSF receptor3 complexes (there is evidence for this possibility for TWEAK and CD95L (8)) and for such cases the KD values determined in our study would be the relevant parameter to describe receptor activation. Moreover, as has been demonstrated for CD95L (35), soluble TNFSF ligands may act as inhibitors of their corresponding membrane-bound form. In these cases the affinity of the soluble molecule would again be a functional relevant parameter. It is worth mentioning that binding studies with GpL-TNFSF ligand fusion proteins also easily allow the determination of association and dissociation rate constants and the mean lifetime of ligand-receptor complexes (9, 10). The systematic evaluation of these parameters may give new insights into the question of how the dynamics/stability of the ligand-receptor complex contributes to the quality and quantity by which a certain TNFRSF receptor type activates intracellular signaling pathways. The simplicity of cellular binding studies with GpL-TNFSF ligand fusion proteins obviously has considerable potential for the evaluation and screening of substances interfering with the TNFSF ligand-TNFRSF receptor interactions.
Affinities of the huge majority of the known TNFSF ligand-TNFRSF interactions can be found in the scientific literature. However, the affinities available for a distinct TNFSF ligand-TNFRSF interaction often differ by several orders of magnitude. This inconsistency may not only limit in silico analysis of TNFSF ligand-TNFRSF receptor systems comprising several ligands and receptors under non-saturating conditions but might also affect conclusions that are based on pharmacological data. The major issue that could explain this inconsistency is the fact that in the various binding studies receptor molecules with a different number of protomers have been considered. In most studies using cell-free methods recombinant soluble TNFRSF receptor fusion proteins containing 1 or 2 receptor protomers have been used. Crystallographic studies, however, show that a TNFSF ligand trimer typically interacts with three receptor molecules (8).
In the absence of ligand, receptor dimers/trimers have been observed for several members of the TNFRSF, including TNFR1, TNFR2, CD40, CD95, and the TRAIL death receptors that are formed due to the autoaffinity of the PLAD. The autoaffinity of the PLAD of TNFRSF receptors is however rather low. This is evident from the fact that recombinant ectodomains of TNFRSF receptors typically form monomers. Indeed, even the PLAD of TNFR1, a TNFRSF receptor with high autoaggregating activity, is below 1 μm (36). We therefore assume that the majority of cell-expressed TNFRSF receptors occur in the form of monomers with low ligand affinity, which are in equilibrium with a minor species of dimeric/trimeric receptors with higher avidity and thus higher apparent affinity. As the dimeric/trimeric receptor species preferentially interacts with the trimeric TNFSF ligand molecules under formation of much more stable ligand-receptor complexes, the ligand-free dimeric/trimeric receptor species are continuously removed from the equilibrium with their monomers until the equilibrium between trimeric ligand and dimeric/trimeric receptor species has been reached. As a consequence, although initially only a few receptor molecules might bind their ligand with avidity and high affinity, over time a significant fraction of TNFRSF receptors becomes liganded despite low ligand concentrations, and this is what we have measured in our equilibrium binding studies. Thus, the low affinities determined in cell-free assays with monomeric recombinant receptor variants presumably correspond to the ligand affinity of cell-expressed monomeric TNFRSF receptors that typically may barely contribute to total ligand binding. In the case of TNFRSF receptor types with a rather small difference in ligand affinity between a single receptor protomer and receptor dimer/trimers, the low affinity binding of trimeric TNFSF ligands to monomeric receptors might significantly contribute to ligand binding especially when the receptor type has a poor autoaffinity.
A second issue that might contribute to the higher affinities that are typically found in cellular binding studies is the fact that the formation and stability of TNFSF ligand-TNFRSF complexes in intact cells can be supported by auxiliary processes, such as e.g. interaction with the cytoskeleton or lipid rafts.
We overcame the limitations of cell free-binding studies by performing cellular binding assays at 37 °C and ascertained a comprehensive set of apparent affinities for most TNFSF ligand-TNFRSF receptor interactions. We have used the same method of TNFSF ligand labeling, namely genetic fusion with the luciferase of G. princeps that not only improved the comparability of the data obtained for different interactions but also ensured high reproducibility from batch to batch. It is also worth mentioning that the genetic fusion of the GpL domain results in only one defined molecular species, whereas the majority of biochemical labeling methods, e.g. with iodine-131 or fluorochromes, results in a mixture of molecular species with a different degree of label load and unknown activity. In contrast to the use of such biochemically labeled ligands, the use of the GpL domain in our studies ensures that the activities measured in functional assays are indeed a property of the molecular species that is responsible for receptor occupation in corresponding binding studies. In sum, we think that our systematic study offers the scientific community the currently most comprehensive and best suited panel of affinities of TNFSF ligand-TNFRSF receptor interactions for in silico studies of ligand-receptor systems of the TNF family.
Author Contributions
I. L. performed binding studies and functional studies, analyzed data, and wrote the manuscript. S. F., A. W., A. F., J. T., and J. A. C. A. performed binding studies and functional studies and analyzed data. V. S. and D. W. cloned and produced the various recombinant proteins used in the study. H. W. designed the project, analyzed data, and wrote the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft Grants Wa 1025/19-2 and Wa 1025/24-1. The authors declare that they have no conflicts of interest with the contents of this article.
- THD
- TNF homology domain
- TNFSF
- tumor necrosis factor superfamily
- TNFRSF
- TNF receptor superfamily
- GpL
- G. princeps luciferase
- TRAF
- TNF receptor-associated factor
- ALPS
- autoimmune lymphoproliferative syndrome
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- PLAD
- pre-ligand assembly domain.
References
- 1. Bodmer J. L., Schneider P., and Tschopp J. (2002) The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 27, 19–26 [DOI] [PubMed] [Google Scholar]
- 2. Locksley R. M., Killeen N., and Lenardo M. J. (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 [DOI] [PubMed] [Google Scholar]
- 3. Wajant H. (2003) Death receptors. Essays Biochem. 39, 53–71 [DOI] [PubMed] [Google Scholar]
- 4. Xie P. (2013) TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lemke J., von Karstedt S., Zinngrebe J., and Walczak H. (2014) Getting TRAIL back on track for cancer therapy. Cell Death Differ. 21, 1350–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin W. W., and Hsieh S. L. (2011) Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem. Pharmacol. 81, 838–847 [DOI] [PubMed] [Google Scholar]
- 7. Aggarwal B. B., Gupta S. C., and Kim J. H. (2012) Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119, 651–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wajant H. (2015) Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 22, 1727–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fick A., Lang I., Schäfer V., Seher A., Trebing J., Weisenberger D., and Wajant H. (2012) Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14). J. Biol. Chem. 287, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lang I., Fick A., Schäfer V., Giner T., Siegmund D., and Wajant H. (2012) Signaling active CD95 receptor molecules trigger co-translocation of inactive CD95 molecules into lipid rafts. J. Biol. Chem. 287, 24026–24042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krippner-Heidenreich A., Grunwald I., Zimmermann G., Kühnle M., Gerspach J., Sterns T., Shnyder S. D., Gill J. H., Männel D. N., Pfizenmaier K., and Scheurich P. (2008) Single-chain TNF, a TNF derivative with enhanced stability and antitumoral activity. J. Immunol. 180, 8176–8183 [DOI] [PubMed] [Google Scholar]
- 12. Sudhamsu J., Yin J., Chiang E. Y., Starovasnik M. A., Grogan J. L., and Hymowitz S. G. (2013) Dimerization of LTβR by LTα1β2 is necessary and sufficient for signal transduction. Proc. Natl. Acad. Sci. U.S.A. 110, 19896–19901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berg D., Lehne M., Müller N., Siegmund D., Münkel S., Sebald W., Pfizenmaier K., and Wajant H. (2007) Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ. 14, 2021–2034 [DOI] [PubMed] [Google Scholar]
- 14. Wyzgol A., Müller N., Fick A., Munkel S., Grigoleit G. U., Pfizenmaier K., and Wajant H. (2009) Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J. Immunol. 183, 1851–1861 [DOI] [PubMed] [Google Scholar]
- 15. Rozanov D. V., Savinov A. Y., Golubkov V. S., Rozanova O. L., Postnova T. I., Sergienko E. A., Vasile S., Aleshin A. E., Rega M. F., Pellecchia M., and Strongin A. Y. (2009) Engineering a leucine zipper-TRAIL homotrimer with improved cytotoxicity in tumor cells. Mol. Cancer Ther. 8, 1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hymowitz S. G., O'Connell M. P., Ultsch M. H., Hurst A., Totpal K., Ashkenazi A., de Vos A. M., and Kelley R. F. (2000) A unique zinc-binding site revealed by a high-resolution x-ray structure of homotrimeric Apo2L/TRAIL. Biochemistry 39, 633–640 [DOI] [PubMed] [Google Scholar]
- 17. Grell M., Wajant H., Zimmermann G., and Scheurich P. (1998) The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. U.S.A. 95, 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grell M., Douni E., Wajant H., Löhden M., Clauss M., Maxeiner B., Georgopoulos S., Lesslauer W., Kollias G., Pfizenmaier K., and Scheurich P. (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80-kDa tumor necrosis factor receptor. Cell 83, 793–802 [DOI] [PubMed] [Google Scholar]
- 19. Rauert H., Wicovsky A., Müller N., Siegmund D., Spindler V., Waschke J., Kneitz C., and Wajant H. (2010) Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 285, 7394–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hendriks J., Planelles L., de Jong-Odding J., Hardenberg G., Pals S. T., Hahne M., Spaargaren M., and Medema J. P. (2005) Heparan sulfate proteoglycan binding promotes APRIL-induced tumor cell proliferation. Cell Death Differ. 12, 637–648 [DOI] [PubMed] [Google Scholar]
- 21. Ingold K., Zumsteg A., Tardivel A., Huard B., Steiner Q. G., Cachero T. G., Qiang F., Gorelik L., Kalled S. L., Acha-Orbea H., Rennert P. D., Tschopp J., and Schneider P. (2005) Identification of proteoglycans as the APRIL-specific binding partners. J. Exp. Med. 201, 1375–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Müller N., Wyzgol A., Münkel S., Pfizenmaier K., and Wajant H. (2008) Activity of soluble OX40 ligand is enhanced by oligomerization and cell surface immobilization. FEBS J. 275, 2296–2304 [DOI] [PubMed] [Google Scholar]
- 23. Schneider P., Holler N., Bodmer J. L., Hahne M., Frei K., Fontana A., and Tschopp J. (1998) Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 187, 1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wajant H., Moosmayer D., Wüest T., Bartke T., Gerlach E., Schönherr U., Peters N., Scheurich P., and Pfizenmaier K. (2001) Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 20, 4101–4106 [DOI] [PubMed] [Google Scholar]
- 25. Gupta S., Clark E. S., Termini J. M., Boucher J., Kanagavelu S., LeBranche C. C., Abraham S., Montefiori D. C., Khan W. N., and Stone G. W. (2015) DNA vaccine molecular adjuvants SP-D-BAFF and SP-D-APRIL enhance anti-gp120 immune response and increase HIV-1 neutralizing antibody titers. J. Virol. 89, 4158–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haswell L. E., Glennie M. J., and Al-Shamkhani A. (2001) Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur. J. Immunol. 31, 3094–3100 [DOI] [PubMed] [Google Scholar]
- 27. Holler N., Tardivel A., Kovacsovics-Bankowski M., Hertig S., Gaide O., Martinon F., Tinel A., Deperthes D., Calderara S., Schulthess T., Engel J., Schneider P., and Tschopp J. (2003) Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 23, 1428–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanagavelu S., Termini J. M., Gupta S., Raffa F. N., Fuller K. A., Rivas Y., Philip S., Kornbluth R. S., and Stone G. W. (2014) HIV-1 adenoviral vector vaccines expressing multi-trimeric BAFF and 4–1BBL enhance T cell mediated anti-viral immunity. PLoS ONE 9, e90100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanagavelu S. K., Snarsky V., Termini J. M., Gupta S., Barzee S., Wright J. A., Khan W. N., Kornbluth R. S., and Stone G. W. (2012) Soluble multi-trimeric TNF superfamily ligand adjuvants enhance immune responses to a HIV-1 Gag DNA vaccine. Vaccine 30, 691–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stone G. W., Barzee S., Snarsky V., Kee K., Spina C. A., Yu X. F., and Kornbluth R. S. (2006) Multimeric soluble CD40 ligand and GITR ligand as adjuvants for human immunodeficiency virus DNA vaccines. J. Virol. 80, 1762–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wajant H., Gerspach J., and Pfizenmaier K. (2013) Engineering death receptor ligands for cancer therapy. Cancer Lett. 332, 163–174 [DOI] [PubMed] [Google Scholar]
- 32. Rauert H., Stühmer T., Bargou R., Wajant H., and Siegmund D. (2011) TNFR1 and TNFR2 regulate the extrinsic apoptotic pathway in myeloma cells by multiple mechanisms. Cell Death Dis. 2, e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swee L. K., Ingold-Salamin K., Tardivel A., Willen L., Gaide O., Favre M., Demotz S., Mikkola M., and Schneider P. (2009) Biological activity of ectodysplasin A is conditioned by its collagen and heparan sulfate proteoglycan-binding domains. J. Biol. Chem. 284, 27567–27576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bossen C., Cachero T. G., Tardivel A., Ingold K., Willen L., Dobles M., Scott M. L., Maquelin A., Belnoue E., Siegrist C. A., Chevrier S., Acha-Orbea H., Leung H., Mackay F., Tschopp J., and Schneider P. (2008) TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 111, 1004–1012 [DOI] [PubMed] [Google Scholar]
- 35. Suda T., Hashimoto H., Tanaka M., Ochi T., and Nagata S. (1997) Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J. Exp. Med. 186, 2045–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cao J., Meng F., Gao X., Dong H., and Yao W. (2011) Expression and purification of a natural N-terminal pre-ligand assembly domain of tumor necrosis factor receptor 1 (TNFR1 PLAD) and preliminary activity determination. Protein J. 30, 281–289 [DOI] [PubMed] [Google Scholar]
- 37. Corcoran A. E., Barrett K., Turner M., Brown A., Kissonerghis A. M., Gadnell M., Gray P. W., Chernajovsky Y., and Feldmann M. (1994) Characterization of ligand binding by the human p55 tumour-necrosis-factor receptor. Involvement of individual cysteine-rich repeats. Eur. J. Biochem. 223, 831–840 [DOI] [PubMed] [Google Scholar]
- 38. Gray P. W., Barrett K., Chantry D., Turner M., and Feldmann M. (1990) Cloning of human tumor necrosis factor (TNF) receptor cDNA and expression of recombinant soluble TNF-binding protein. Proc. Natl. Acad. Sci. U.S.A. 87, 7380–7384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marsters S. A., Frutkin A. D., Simpson N. J., Fendly B. M., and Ashkenazi A. (1992) Identification of cysteine-rich domains of the type 1 tumor necrosis factor receptor involved in ligand binding. J. Biol. Chem. 267, 5747–5750 [PubMed] [Google Scholar]
- 40. Pitti R. M., Marsters S. A., Lawrence D. A., Roy M., Kischkel F. C., Dowd P., Huang A., Donahue C. J., Sherwood S. W., Baldwin D. T., Godowski P. J., Wood W. I., Gurney A. L., Hillan K. J., Cohen R. L., et al. (1998) Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 396, 699–703 [DOI] [PubMed] [Google Scholar]
- 41. Zhan C., Patskovsky Y., Yan Q., Li Z., Ramagopal U., Cheng H., Brenowitz M., Hui X., Nathenson S. G., and Almo S. C. (2011) Decoy strategies: the structure of TL1A:DcR3 complex. Structure 19, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Armitage R. J., Sato T. A., Macduff B. M., Clifford K. N., Alpert A. R., Smith C. A., and Fanslow W. C. (1992) Identification of a source of biologically active CD40 ligand. Eur. J. Immunol. 22, 2071–2076 [DOI] [PubMed] [Google Scholar]
- 43. Zhou Z., Kim S., Hurtado J., Lee Z. H., Kim K. K., Pollok K. E., and Kwon B. S. (1995) Characterization of human homologue of 4–1BB and its ligand. Immunol. Lett. 45, 67–73 [DOI] [PubMed] [Google Scholar]
- 44. Rabu C., Quéméner A., Jacques Y., Echasserieau K., Vusio P., and Lang F. (2005) Production of recombinant human trimeric CD137L (4–1BBL). Cross-linking is essential to its T cell co-stimulation activity. J. Biol. Chem. 280, 41472–41481 [DOI] [PubMed] [Google Scholar]
- 45. Alderson M. R., Smith C. A., Tough T. W., Davis-Smith T., Armitage R. J., Falk B., Roux E., Baker E., Sutherland G. R., and Din W. S. (1994) Molecular and biological characterization of human 4–1BB and its ligand. Eur. J. Immunol. 24, 2219–2227 [DOI] [PubMed] [Google Scholar]
- 46. Al-Shamkhani A., Mallett S., Brown M. H., James W., and Barclay A. N. (1997) Affinity and kinetics of the interaction between soluble trimeric OX40 ligand, a member of the tumor necrosis factor superfamily, and its receptor OX40 on activated T cells. J. Biol. Chem. 272, 5275–5282 [DOI] [PubMed] [Google Scholar]
- 47. Newton P., Harrison P., and Clulow S. (2008) A novel method for determination of the affinity of protein: protein interactions in homogeneous assays. J. Biomol. Screen. 13, 674–682 [DOI] [PubMed] [Google Scholar]
- 48. Hargreaves P. G., and Al-Shamkhani A. (2002) Soluble CD30 binds to CD153 with high affinity and blocks transmembrane signaling by CD30. Eur. J. Immunol. 32, 163–173 [DOI] [PubMed] [Google Scholar]
- 49. Smith C. A., Gruss H. J., Davis T., Anderson D., Farrah T., Baker E., Sutherland G. R., Brannan C. I., Copeland N. G., and Jenkins N. A. (1993) CD30 antigen, a marker for Hodgkin's lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell 73, 1349–1360 [DOI] [PubMed] [Google Scholar]
- 50. Harrop J. A., McDonnell P. C., Brigham-Burke M., Lyn S. D., Minton J., Tan K. B., Dede K., Spampanato J., Silverman C., Hensley P., DiPrinzio R., Emery J. G., Deen K., Eichman C., Chabot-Fletcher M., et al. (1998) Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J. Biol. Chem. 273, 27548–27556 [DOI] [PubMed] [Google Scholar]
- 51. Morishige T., Yoshioka Y., Inakura H., Tanabe A., Yao X., Tsunoda S., Tsutsumi Y., Mukai Y., Okada N., and Nakagawa S. (2010) Creation of a LIGHT mutant with the capacity to evade the decoy receptor for cancer therapy. Biomaterials 31, 3357–3363 [DOI] [PubMed] [Google Scholar]
- 52. Eldredge J., Berkowitz S., Corin A. F., Day E. S., Hayes D., Meier W., Strauch K., Zafari M., Tadi M., and Farrington G. K. (2006) Stoichiometry of LTβR binding to LIGHT. Biochemistry 45, 10117–10128 [DOI] [PubMed] [Google Scholar]
- 53. Nelson C. A., Warren J. T., Wang M. W., Teitelbaum S. L., and Fremont D. H. (2012) RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 20, 1971–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Willard D., Chen W. J., Barrett G., Blackburn K., Bynum J., Consler T., Hoffman C., Horne E., Iannone M. A., Kadwell S., Parham J., and Ellis B. (2000) Expression, purification, and characterization of the human receptor activator of NF-κB ligand (RANKL) extracellular domain. Protein Expr. Purif. 20, 48–57 [DOI] [PubMed] [Google Scholar]
- 55. Schneeweis L. A., Willard D., and Milla M. E. (2005) Functional dissection of osteoprotegerin and its interaction with receptor activator of NF-κB ligand. J. Biol. Chem. 280, 41155–41164 [DOI] [PubMed] [Google Scholar]
- 56. Vitovski S., Phillips J. S., Sayers J., and Croucher P. I. (2007) Investigating the interaction between osteoprotegerin and receptor activator of NF-κB or tumor necrosis factor-related apoptosis-inducing ligand: evidence for a pivotal role for osteoprotegerin in regulating two distinct pathways. J. Biol. Chem. 282, 31601–31609 [DOI] [PubMed] [Google Scholar]
- 57. Emery J. G., McDonnell P., Burke M. B., Deen K. C., Lyn S., Silverman C., Dul E., Appelbaum E. R., Eichman C., DiPrinzio R., Dodds R. A., James I. E., Rosenberg M., Lee J. C., and Young P. R. (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 273, 14363–14367 [DOI] [PubMed] [Google Scholar]
- 58. Truneh A., Sharma S., Silverman C., Khandekar S., Reddy M. P., Deen K. C., McLaughlin M. M., Srinivasula S. M., Livi G. P., Marshall L. A., Alnemri E. S., Williams W. V., and Doyle M. L. (2000) Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J. Biol. Chem. 275, 23319–23325 [DOI] [PubMed] [Google Scholar]
- 59. Bittner S., Knoll G., Fullsack S., Kurz M., Wajant H., and Ehrenschwender M. (2015) Soluble TL1A is sufficient for activation of Death Receptor 3. FEBS J. 10.1111/febs.13576, in press [DOI] [PubMed] [Google Scholar]
- 60. Kanakaraj P., Migone T. S., Nardelli B., Ullrich S., Li Y., Olsen H. S., Salcedo T. W., Kaufman T., Cochrane E., Gan Y., Hilbert D. M., and Giri J. (2001) BLyS binds to b cells with high affinity and induces activation of the transcription factors NF-κB and elf-1. Cytokine 13, 25–31 [DOI] [PubMed] [Google Scholar]
- 61. Day E. S., Cachero T. G., Qian F., Sun Y., Wen D., Pelletier M., Hsu Y. M., and Whitty A. (2005) Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry 44, 1919–1931 [DOI] [PubMed] [Google Scholar]
- 62. Patel D. R., Wallweber H. J., Yin J., Shriver S. K., Marsters S. A., Gordon N. C., Starovasnik M. A., and Kelley R. F. (2004) Engineering an APRIL-specific B cell maturation antigen. J. Biol. Chem. 279, 16727–16735 [DOI] [PubMed] [Google Scholar]
- 63. Wu Y., Bressette D., Carrell J. A., Kaufman T., Feng P., Taylor K., Gan Y., Cho Y. H., Garcia A. D., Gollatz E., Dimke D., LaFleur D., Migone T. S., Nardelli B., Wei P., et al. (2000) Tumor necrosis factor (TNF) receptor superfamily member TACI is a high affinity receptor for TNF family members APRIL and BLyS. J. Biol. Chem. 275, 35478–35485 [DOI] [PubMed] [Google Scholar]
- 64. Wiley S. R., Cassiano L., Lofton T., Davis-Smith T., Winkles J. A., Lindner V., Liu H., Daniel T. O., Smith C. A., and Fanslow W. C. (2001) A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 15, 837–846 [DOI] [PubMed] [Google Scholar]
- 65. Brown S. A., Hanscom H. N., Vu H., Brew S. A., and Winkles J. A. (2006) TWEAK binding to the Fn14 cysteine-rich domain depends on charged residues located in both the A1 and D2 modules. Biochem. J. 397, 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou Z., Song X., Berezov A., Zhang G., Li Y., Zhang H., Murali R., Li B., and Greene M. I. (2008) Human glucocorticoid-induced TNF receptor ligand regulates its signaling activity through multiple oligomerization states. Proc. Natl. Acad. Sci. U.S.A. 105, 5465–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chattopadhyay K., Ramagopal U. A., Mukhopadhaya A., Malashkevich V. N., Dilorenzo T. P., Brenowitz M., Nathenson S. G., and Almo S. C. (2007) Assembly and structural properties of glucocorticoid-induced TNF receptor ligand: Implications for function. Proc. Natl. Acad. Sci. U.S.A. 104, 19452–19457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chattopadhyay K., Ramagopal U. A., Brenowitz M., Nathenson S. G., and Almo S. C. (2008) Evolution of GITRL immune function: murine GITRL exhibits unique structural and biochemical properties within the TNF superfamily. Proc. Natl. Acad. Sci. U.S.A. 105, 635–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Trebing J., El-Mesery M., Schäfer V., Weisenberger D., Siegmund D., Silence K., and Wajant H. (2014) CD70-restricted specific activation of TRAILR1 or TRAILR2 using scFv-targeted TRAIL mutants. Cell Death Dis. 5, e1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roos C., Wicovsky A., Müller N., Salzmann S., Rosenthal T., Kalthoff H., Trauzold A., Seher A., Henkler F., Kneitz C., and Wajant H. (2010) Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-κB pathway. J. Immunol. 185, 1593–1605 [DOI] [PubMed] [Google Scholar]