

Abstract
The low density lipoprotein receptor-related protein 1 (LRP1) is a ubiquitously expressed cell surface receptor that protects from intracellular cholesterol accumulation. However, the underlying mechanisms are unknown. Here we show that the extracellular (α) chain of LRP1 mediates TGFβ-induced enhancement of Wnt5a, which limits intracellular cholesterol accumulation by inhibiting cholesterol biosynthesis and by promoting cholesterol export. Moreover, we demonstrate that the cytoplasmic (β) chain of LRP1 suffices to limit cholesterol accumulation in LRP1−/− cells. Through binding of Erk2 to the second of its carboxyl-terminal NPXY motifs, LRP1 β-chain positively regulates the expression of ATP binding cassette transporter A1 (ABCA1) and of neutral cholesterol ester hydrolase (NCEH1). These results highlight the unexpected functions of LRP1 and the canonical Wnt5a pathway and new therapeutic potential in cholesterol-associated disorders including cardiovascular diseases.
Keywords: ABC transporter, atherosclerosis, cholesterol regulation, transgenic mice, Wnt pathway, lipoprotein receptor-related protein 1 (LRP1)
Introduction
Cholesterol is a major component of mammalian cell membranes that accumulates in the vascular wall during atherosclerosis, the leading cause of death in industrialized societies (1, 2). The low density lipoprotein receptor-related protein 1 (LRP1),3 a cell surface receptor that belongs to the LDL receptor family, endocytoses multiple ligands (3). It consists of an 85-kDa membrane-bound carboxyl fragment (β chain) and a non-covalently attached 515-kDa (α chain) amino-terminal fragment (4). We previously demonstrated that LRP1 limits cholesterol accumulation in the arterial wall. Mice deficient for LRP1 in vascular smooth muscle cells (vSMCs) (smLRP1 mice) develop vSMCs proliferation, cholesterol accumulation (5), and massive foam cell formation when fed a cholesterol-rich diet (6–10). Whereas LRP1 integrates the platelet-derived growth factor (PDGF-BB) (8, 9) and transforming growth factor-β (TGF-β) at the plasma membrane, two pathways known to regulate vSMCs proliferation (7), the physiological importance and function of LRP1 in regulating intracellular cholesterol homeostasis is still poorly understood. Several mechanisms have been proposed. LRP1 has been shown to promote cholesterol export in vSMCs through induction of ATP binding cassette transporter A1 (ABCA1) levels (5) and to induce a Wnt5a/β-catenin pathway to limit cholesterol overload in mouse embryonic fibroblasts (11). Moreover, smLRP1 mice express very low levels of Wnt5a in vSMCs (12). TGF-β also stimulates a non-canonical Wnt5a pathway in airway smooth muscle cells (13). These data strongly suggest that a TGF-β/LRP1/Wnt5a pathway limits intracellular cholesterol accumulation.
How Wnt5a interferes with cholesterol homeostasis is unknown. It might increase cholesterol export and/or block cholesterol synthesis. ABCA1 and the ATP binding cassette transporter G1 (ABCG1) are two proteins that promote cholesterol efflux. Cholesterol synthesis is tightly regulated by a feedback system that senses the level of cholesterol and modulates the transcription of genes encoding enzymes of cholesterol biosynthesis and uptake (14, 15). For instance, when cholesterol levels rise in cells, the membrane-embedded protein of the endoplasmic reticulum (ER), Scap, senses the increase and binds to Insigs, proteins located to the ER. Insigs then limit cleavage and nuclear translocation of sterol regulatory element-binding proteins (SREBPs), in particular SREBP-2, an activator of cholesterol synthesis in liver and adipose tissue of mice (16). This reduces 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase transcript levels and decreases cholesterol synthesis.
In the current study we sought to determine how LRP1 regulates Wnt5a and how Wnt5a prevents intracellular cholesterol accumulation. Our results show that a TGFβ/LRP1 signaling pathway positively regulates Wnt5a mRNA and protein levels. We further demonstrate that Wnt5a protects against intracellular cholesterol accumulation by interfering with its biosynthesis through down-regulation of HMG-CoA reductase and by interfering with cholesterol export through up-regulation of ABCG1. Finally, we found that the proximal NPXY motif (N-Ter) within the LRP1 β chain is critical to activate the expression of ABCA1 and the neutral cholesterol ester hydrolase (NCEH), an enzyme that hydrolyzes cholesterol esters, the initial step toward elimination of cholesterol (17).
Experimental Procedures
Animals and Diets
All animal experimentations and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Strasbourg, France, and conformed to the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The generation of Wnt5a transgenic animals was achieved by expressing Wnt5a from the fatty acid binding protein-4 (FABP4/aP2) promoter. The FABP4/aP2 protein is predominantly expressed in adipose tissue. The transgenic vector contains the FABP4/aP2 promoter, the mouse Wnt5a cDNA, a β-globin intron fragment for splicing, and a polyadenylation sequence. Genotyping of the wild type and Wnt5a mutant mice by polymerase chain reaction (PCR) was performed as described (6) using primers specific for Wnt5a (primers are available upon request). Animals were maintained on a 12-h light/12-h dark cycle. For feeding studies, mutants and control mice were fed a caloric-rich diet for 24 weeks as described previously (6). For the isolation of tissue for further analysis, the agents used for euthanasia were ketamine (750 mg/kg) and xylazine (50 mg/kg) intraperitoneally.
Cell Culture
Mouse embryonic fibroblasts (MEFs) and human embryonic kidney (HEK) cells were grown in monolayer cultures at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% newborn calf serum and with 10% fetal bovine serum (FBS), respectively. Adipocyte differentiation was induced using a mixture of insulin, dexamethasone, isobutylmethylxanthine (Sigma), and rosiglitazone (Applied Biochemical Technology) (11). Medium was changed every 2 days, and after 10 days cells were fixed in 10% formaldehyde, and neutrals lipids were stained with Oil Red O (Sigma). For experiments with conditioned medium from cells over expressing Wnt5a (L-MTK, CRL-2814; American Type Culture Collection, Manassas, VA), the medium was maintained for 3 days in contact with the confluents cells and then removed and cleared by centrifugation. The medium was supplemented with 5% FBS before use. For TGFβ treatment, MEFs were set up at 60,000 cells per well in DMEM, 10% newborn calf serum. When they reached contact inhibition they were starved in DMEM supplemented with 0.5% FBS for 18 h. 5 ng/ml TGFβ were added in the medium during the indicated times. The cells were scrapped in radioimmune precipitation lysis buffer for protein extraction or in TRIzol reagent for RNA isolation. For cell fractionation, monolayers of HEK 293 cells were transfected with plasmids as described (18). The cells were incubated for 20 h in medium containing 5% sterol-depleting serum with 50 μm compactin and 50 μm sodium mevalonate in the absence or the presence of sterols as indicated in the legends. Sterol mixtures contained 0.1–1 μg/ml of 25 hydroxycholesterol plus 1–10 μg/ml cholesterol as described previously (19). Thereafter, the cells received N-acetyl-leucinal-leucinal-norleucinal at a final concentration of 25 μg/ml, and the cells were harvested 3 h later as described (18). Cells were then fractionated as described (20). PLA2α inhibitor N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide, HCl, was from Calbiochem (catalog no. 525143).
Protein and mRNA Expression Analysis
RNA was isolated using TRIzol reagent (Sigma) according to the manufacturer's instructions. 50 μg of RNA were converted to cDNA using the high capacity cDNA Archive kit (Applied Biosystems, Foster City, CA). PCR amplification was performed using SYBR Green PCR master mix (Applied Biosystems) according to the manufacturer's instructions. Primers sequences are available upon request. SDS-polyacrylamide gel electrophoresis, and immunoblot analysis was performed according to standard procedures. Proteins were transferred onto nitrocellulose membranes, and immunoblot analyses were carried out using antibodies directed against Wnt5a (R&D Systems), HMG-CoA reductase (a kind gift from Russell de Bose-Boyd, UT Southwestern Medical Center, Dallas, TX), ABCA1 (Santa Cruz, CA), ABCG1 (Santa Cruz, CA), p44/42 Erk (Cell Signaling Technology), c-myc (Sigma), p-AKT (Cell Signaling Technology), or GAPDH (Sigma). For mass spectrometry analysis, peptides were analyzed by nanoflow liquid chromatography coupled to tandem mass spectrometry (nanoLC-MS/MS). All MS/MS data were interpreted with two different search engines (Mascot and OMSSA) using several protein sequence databases (NCBI, SwissProt). For immunostaining and histology experiments, adipose tissue was fixed with 4% paraformaldehyde in phosphate-buffered saline, embedded in paraffin, and cut in 5-μm slices as described (6). Sections were stained with hematoxylin and eosin. The ABCG1 promoter A, which is located upstream exon 1 (−670 to −10), or the ABCG1 promoter B, which is located upstream exon 5 (−610 to −10) (21), or an empty vector linked to a luciferase reporter were transiently transfected in LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (LRP1−/−, Wnt5a MEFs). At 48 h after the transfection, cells were collected, and reporter gene assays were carried out using the Promega dual-luciferase reporter assay system (Promega).
Plasmids and Probes
HEK cells were transfected with a plasmid coding SREBP-2 under the control of a thymidine kinase promoter and carrying the gene for Geneticin resistance (kindly provided by Russel de Bose-Boyd). Transfections were performed with FuGENE 6 Reagent (Promega) according the manufacturer's protocol. Briefly, 60% confluent cell medium was switched with DMEM + 0.5% FBS, and the FuGENE 6 mixed with the plasmid was added for 5 h. Medium was replaced by DMEM with 10% FBS for 24–48 h. For stable cell lines medium was then switched to DMEM + 10% FBS containing the selective antibiotic G418 at a concentration of 1.8 mg/ml for selection then 0.6 mg/ml.
Cholesterol Measurement
Total cholesterol from white adipose tissue of Wnt5a transgenic mice was quantified using a cholesterol/cholesteryl ester quantitation kit (Calbiochem-EMD Biosciences, San Diego, CA) according to the manufacturer's protocol. Briefly, 20 mg of tissue was dissolved in chloroform 1% Triton X-100 and centrifuge at 14,000 rpm for 10 min at 20 °C. The organic phase was transfer in a new Eppendorf tube and was air dried at 55 °C to remove chloroform. The dried lipids were dissolved in 200 μl of cholesterol reaction buffer by vortexing. Cellular cholesterol was measured by incubating MEFs in medium containing serum (10% FBS) or with [1,2-3H]cholesterol (specific activity 50 mCi/ml) (PerkinElmer Life Sciences, final radioactivity cholesterol, [1,2-3H]cholesterol, 1 mCi (37 MBq)) for 36 h in a CO2 incubator. For equilibration of cellular free cholesterol pools, cells were starved for 18 h in DMEM supplemented with 0.3% BSA. AcylCoA:cholesterol acyltransferase (ACAT) inhibitor (TMP153 Sigma) was added to both the prelabeling and equilibration media to prevent cholesterol esterification at a final concentration of 220 nm. For cholesterol efflux, cells were washed and incubated for 3 h at 37 °C in serum-free medium containing 6 μg/ml lipid free apoA-I. The medium was collected and centrifuged for 15 min at 4 °C at 10,000 × g, and aliquots of supernatant were counted in a β-counter. Cells were washed with cold PBS, harvested, lysed in 0.3 m NaOH for 3 h in ice, and cell-associated radioactivity was counted. Cholesterol efflux was expressed as the proportion of [3H]cholesterol transferred from cells to medium.
Statistical Analysis
Values are reported as the mean ± S.E. of at least triplicate determinations. Statistical significance (p < 0.05) was determined using an unpaired Student's t test or analysis of variance (GraphPad Prism, Abacus Concepts, Berkeley, CA).
Results
The Extracellular Domain of LRP1 Mediates a TGF-β-induced Wnt5a Pathway
To test whether TGF-β induces Wnt5a, we treated MEFs with TGF-β and found an induction of Wnt5a protein much more pronounced in LRP1+/+ MEFs than in LRP1−/− MEFs (Fig. 1A). The induction occurs after 30 min of treatment (Fig. 1A). Similarly, Wnt5a transcript levels were increased 3-fold upon 30 min of TGF-β stimulation (Fig. 1B) and remained significantly higher than controls even after 24 h of treatment. No induction was observed in LRP1−/− MEFs (Fig. 1B), demonstrating that LRP1 is required for TGF-β-induced Wnt5a transcript levels. Interestingly, TGF-β did not increase mRNA levels of Wnt5a in cells that express only the cytoplasmic domain (β-chain) of LRP1 (Fig. 1B). These data indicate that the α-chain of LRP1 is required for TGF-β-mediated induction of Wnt5a.
FIGURE 1.
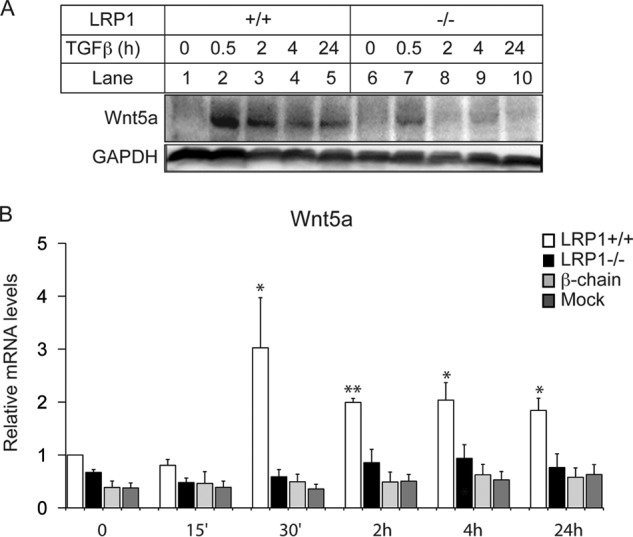
TGFβ stimulates Wnt5a expression via the LRP1 extracellular domain. A, Western blot analysis of Wnt5a upon TGFβ treatments (5 ng/ml) in LRP1+/+ and LRP1−/− MEFs (n = 4). B, quantitative RT-PCR analysis of Wnt5a upon TGFβ treatments in LRP1+/+ and LRP1−/− MEFs and in LRP1−/− MEFs stably transfected with an expression vector coding for the cytoplasmic tail of LRP1 (β-chain), or the mock control (Mock) (n = 4). Values are means ± S.E. with *, p < 0.05.
Wnt5a Enhances ABCG1 Expression and Promotes Cholesterol Efflux
To test whether Wnt5a stimulates cholesterol export, we treated LRP1−/− MEFs stably re-transfected with a Wnt5a expression vector and LRP1−/− MEFs control that do not express Wnt5a with an adipogenic mixture for 10 days. Wnt5a-re-transfected LRP1−/− MEFs exported about two times more cholesterol than LRP1−/− MEFs transfected with an empty vector (Mock) (Fig. 2A). Overexpression of Wnt5a in LRP1−/− MEFs enhanced mRNA (Fig. 2B) and protein levels of ABCG1 (Fig. 2C-D) but not those of ABCA1 (Fig. 2C and data not shown), two proteins that promote cholesterol efflux (22–24). Similarly, murine L-MTK cells stably transfected with Wnt5a contained much higher amounts of ABCG1 transcripts (Fig. 2E) and proteins (Fig. 2F) than wild type cells. Conversely, ABCG1 protein expression is decreased in 3T3-L1 preadipocytes and in human vSMCs silenced for Wnt5a (Fig. 2G). Thus, Wnt5a enhances ABCG1 expression.
FIGURE 2.

Wnt5a stimulates cholesterol efflux and ABCG1 expression. Cells were untreated (T0) or treated with an adipogenic mixture for 10 days (T10) to stimulate cholesterol accumulation. A, cholesterol efflux in LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (LRP1−/−, Wnt5a) or the mock control (LRP1−/−, Mock) (n = 4). B, quantitative RT-PCR analysis of ABCG1 in LRP1+/+ and LRP1−/− MEFs and LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (LRP1−/−, Wnt5a) or the mock control (LRP1−/−, Mock) (n = 7). C, Western blot analysis of the indicated genes in LRP1+/+, LRP1−/− MEFs and LRP1−/− MEFs stably transfected with Wnt5a (LRP1−/−, Wnt5a) or mock (LRP1−/−, Mock) (n = 3). D, immunostaining of ABCG1 (green) in LRP1−/− MEFs stably transfected with Wnt5a (LRP1−/−, Wnt5a) or mock (LRP1−/−, Mock). The nucleus is stained with DAPI in blue (n = 6). Quantitative RT-PCR (E) and representative Western blot (F) analysis of ABCG1 in wild type (WT) or Wnt5a stably transfected (Wnt5a+) LMTK cells (n = 3). G, representative Western blot analysis of ABCG1 in 3T3-L1 preadipocytes and cultured human vSMCs treated with siWnt5a versus control (n = 3). H, quantitative RT-PCR analysis of the indicated genes in LMTK stably transfected with a Wnt5a expression vector (Wnt5a+) and in wild type cells (Wt) (n = 4). I, effects of 6 and 8 h of actinomycin D treatments on ABCG1 transcripts in LRP1+/+ and LRP1−/− MEFs and in LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (LRP1−/−, Wnt5a) or with a mock control (LRP1−/−, Mock) (n = 4). J, LRP1−/−, Wnt5a MEFs transiently transfected with luciferase reporter containing the ABCG1 promoter A, B, or an empty vector in untreated cells (n = 3). Values are the means ± S.E. with p < 0.05 (*).
Liver X receptors (LXRs) play a key role in cholesterol efflux. They form obligate heterodimers with retinoid X receptors (RXR). LXR/RXR heterodimers bind to lipogenic target gene promoters such as apoE, SREBP1c, ABCA1, and ABCG1. In LMTK cells stably transfected with Wnt5a, mRNA levels of LXRβ remained unchanged (Fig. 2H) and mRNA levels of LXRα are decreased (Fig. 2H). Transcript levels of LXRα also remained unchanged in LRP1−/− MEFs and in LRP1−/− MEFs stably transfected with Wnt5a (data not shown). Moreover, mRNA levels of SREBP1c (Fig. 2H) and apoE (Fig. 2H) were not modified by Wnt5a overexpression in LMTK cells. These data indicate that Wnt5a induces ABCG1 expression independently of LXR levels. In cells, mRNA levels are determined by the relative rates of RNA production and degradation. To determine whether Wnt5a modifies ABCG1 mRNA degradation, MEFs were stimulated with an adipogenic mixture to induce cholesterol accumulation and treated with actinomycin D for various time periods to block transcription. Whereas ABCG1 mRNA levels in Wnt5a re-transfected MEFs were ∼3× higher than controls (Mock), Fig. 2I shows that upon 6–8 h of actinomycin D, ABCG1 mRNA levels in these cells returned to levels of controls. The ABCG1 gene has been shown to have two promoters that are located upstream of exon 1 and of exon 5 and have been designated as promoter A and promoter B, respectively (21). Fig. 2J shows that LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a stimulated luciferase expression driven by the promoter B of ABCG1 (21) but not by its promoter A. These data indicate that Wnt5a promotes cholesterol efflux through induction of transcription of ABCG1.
Wnt5a Decreases SREBP-2 Nuclear Translocation and Down-regulates HMG-CoA Reductase Expression
Next, we determined whether Wnt5a interferes with cholesterol biosynthesis. Upon treatment with an adipogenic mixture for 10 days, we observed in LRP1−/− MEFs stably transfected with Wnt5a a 70% decrease of the mRNA levels of the HMG-CoA reductase encoding the rate-limiting enzyme of the mevalonate pathway (Fig. 3A). SREBPs, in particular SREBP-2, activate the transcription of several genes that encode enzymes required for cholesterol synthesis, including HMG-CoA reductase (16). We thus postulated that Wnt5a might decrease HMG-CoA reductase expression through decreased SREBP-2 nuclear translocation. To test this, we transfected HEK 293 cells stably expressing tagged SREBP-2 cDNA under the control of a TK promoter (20) with a vector coding for Wnt5a. We found that overexpression of Wnt5a in HEK decreased SREBP-2 nuclear translocation (Fig. 3B, lanes 6 and 8). In addition to cholesterol, the mevalonate pathway is also regulated by oxysterols (25). For instance, SREBP-2 nuclear translocation is inhibited when oxysterols are added to cultured cells (16). We thus postulated that Wnt5a might potentiate the effects of 25-hydroxycholesterol on SREBP-2 nuclear location. Fig. 3, C and D, shows in HEK 293 cells overexpressing SREBP-2 the effect of various concentrations of 25 hydroxycholesterol on the amount of the mature nuclear form of SREBP-2. Whereas in cells non-transfected with Wnt5a treatments, 0.3 μg/ml sterols modestly decreased SREBP-2 levels in the nucleus, in cells that overexpressed Wnt5a treatments, 0.3 μg/ml of sterols markedly decreased SREBP-2 in the nucleus (Fig. 3, C, lane 8, and D). When treated with 0.3 μg/ml of sterols, the decrease was similar to that seen at 1 μg/ml of sterols in Wnt5a non-transfected cells (Fig. 3C, lane 2 and 8). In a complementary approach, we treated the HEK 293 overexpressing SREBP-2 with a Wnt5a-enriched conditioned medium (11). Cell fractionation experiments revealed that SREBP-2 nuclear translocation was decreased after 6 h (Fig. 3E, lanes 4 and 5) and 24 h of treatment (Fig. 3E, lanes 6 and 7).
FIGURE 3.

Wnt5a-mediated inhibition of SREBP-2 processing and cholesterol biosynthesis. A, quantitative RT-PCR analysis of the HMG-CoA reductase in LRP1+/+ and LRP1−/− MEFs and in LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (−/−, Wnt5a) or the mock control (Mock) upon adipogenesis (n = 7). B, HEK 293 cells were stably transfected with an expression vector coding for SREBP-2 and transiently transfected with an expression vector coding for Wnt5a or with an empty vector (mock). Cell lysate, membrane (Mb), and nuclear (N) extracts were subjected to SDS-PAGE followed by immunoblot analysis. Precursors and Nuclear forms denote the uncleaved membrane precursor and cleaved nuclear forms of SREBP-2, respectively (n = 5). C, HEK 293 cells stably transfected with an expression vector coding for SREBP-2 were incubated at 37 °C in standard medium. After transient transfection with an expression vector coding for Wnt5a or mock, cells were incubated in medium containing 5% sterol-depleting serum and treated for 20 h with a mixture of sterols containing the indicated final concentration of 25-hydroxycholesterol as described under “Experimental Procedures”. Nuclear extracts and membrane fractions were prepared, and an aliquot of each fraction (60 μg of protein) was subjected to SDS-PAGE and immunoblot analysis (n = 3). D, densitometric scanning of the nuclear extracts (n = 3). E, HEK 293 cells stably transfected with an expression vector coding for SREBP-2 were incubated at 37 °C in standard medium. After incubation for the indicated times with Wnt5a or mock-conditioned medium membrane (Mb) and nuclear (N) extracts were subjected to 10% SDS-PAGE followed by immunoblot analysis (n = 5). F, LRP1+/+ (+/+) and LRP1−/− MEFs stably transfected with an expression vector coding for Wnt5a (−/−, Wnt5a) or a mock control (−/−, Mock) were transfected with pTK-Insig1-Myc. On day 0, cells were set up at 5 × 105 cells per 100-mm dish in medium containing 5% sterol-depleting serum. Cells were then switched to fresh medium containing 50 mm cycloheximide (CHX) in the presence of 1 μg/ml 25-hydroxycholesterol (25-HC). At the indicated time after cycloheximide addition, cells were harvested and fractionated, and aliquots of whole-cell lysates were subjected to 10% SDS-PAGE and immunoblot analysis with monoclonal anti-Myc IgG against Insig-1 (n = 4). Values are means ± S.E. with *, p < 0.05.
Insig-1 is an ER protein known to limit cleavage and nuclear translocation of membrane-bound SREBP-2 in mice (16). Whereas Insig-1 retains Scap/SREBP in the ER, a Golgi-localized membrane protein progestin and adipoQ receptors 3 (PAQR3) was recently identified as interacting with Scap/SREBP to tether them to the Golgi (26). To determine whether Wnt5a modifies Insig-1 levels, we used SDS-PAGE and immunoblotting to follow the disappearance of total Insig-1 after protein synthesis was blocked by cycloheximide. We also treated the cells with oxysterols because degradation of Insig-1 is inhibited when oxysterols are added to cultured cells (16). Fig. 3F shows that treatment with sterols stabilized Insig-1 expression in LRP1−/− MEFs transfected with Wnt5a (−/−, Wnt5a) but not in mock controls that do not express Wnt5a (−/−, Mock). This indicates that sterols and Wnt5a are both required to stabilize Insig-1 levels. It retains SREBP-2 in the ER and decreases HMG-CoA reductase levels.
Reduced Cholesterol Accumulation in Adipocytes of Mice Overexpressing Wnt5a in Adipose Tissue
To show that Wnt5a limits intracellular cholesterol accumulation in vivo, we generated mice that overexpress Wnt5a selectively in adipocytes (aTgWnt5a). In addition to its role in storage of excess energy in form of triglycerides, the adipose tissue contains the largest pool of cholesterol (27, 28). Consistent with our previous observation in MEFs, ABCG1 mRNA (Fig. 4A) and protein levels (Fig. 4B) were increased in white adipose tissue from aTgWnt5a mice. ABCG1 mRNA levels were also significantly increased in purified white adipocytes (Fig. 4C). No difference in macrophage infiltration of adipose tissue was observed between aTgWnt5a mice and controls (Fig. 4D), excluding that increased ABCG1 mRNA levels resulted from an increased number of macrophages. As observed in MEFs, ABCA1 mRNA levels were only moderately increased by Wnt5a overexpression (Fig. 4E). SREBP-2, LXRα, and RXRα as well as LXR target genes such as SREBP-1c were unchanged in aTgWnt5a mice (data not shown). We also tested whether HMG-CoA reductase expression was altered in aTgWnt5a mice. In agreement with our in vitro data, mRNA (Fig. 4E) and protein levels (Fig. 4B) of HMG-CoA reductase were 5–6-fold lower in white adipose tissue from aTgWnt5a mice compared with controls. Moreover, mRNA levels HMG-CoA synthase, the enzyme upstream of the mevalonate pathway, were 80–90% decreased compared with controls (Fig. 4E). This was accompanied by a 6–8-fold increase of Insig-1 mRNA levels (Fig. 4F) in white adipose tissue of aTgWnt5a mice, whereas no difference in mRNA levels of PAQR3 was seen (Fig. 4F). Similarly, a 2-fold decrease of total cholesterol (Fig. 4G) in white adipose tissue of aTgWnt5a mice was observed without difference in the total amount of triglycerides (data not shown). Histological analysis did not reveal any difference in white adipocyte size (Fig. 4H), and no difference in body weight (data not shown) was observed between transgenic and control mice even after 24 weeks of a high fat diet. LDL receptor protein expressions were similar in white adipose tissue from aTgWnt5a and controls (Fig. 4I), suggesting that cholesterol uptake was not affected. Thus, overexpression of Wnt5a in adipose tissue of mice decreases the expression of the rate-limiting enzymes for cholesterol biosynthesis and increases the expression of proteins that promote cholesterol efflux resulting in decreased intracellular cholesterol levels.
FIGURE 4.

Adipose tissue in aTgWnt5a mice display inhibition of the mevalonate pathway and increased ABCG1 expression. A, quantitative RT-PCR analysis of ABCG1 in white adipose tissue (WAT) from mice transgenic for Wnt5a (aTgWnt5a) and controls (Ctrl). B, Western blot analysis of the indicated proteins in white adipose tissue from aTgWnt5a and control mice. C, quantitative RT-PCR analysis of ABCG1 in purified white adipocytes. D, densitometry of Western blot analysis of macrophage markers in white adipose tissue. E, quantitative RT-PCR analysis of the indicated genes in adipose tissue from aTgWnt5a and controls. F, quantitative RT-PCR analysis of Insig-1 and PAQR3 in adipose tissue from aTgWnt5a and control mice. G, cellular cholesterol in white adipose tissue from aTgWnt5a and controls. H, white adipocyte size in aTgWnt5a and controls. I, Western blot analysis of the LDL receptor (LDLr) in white adipose tissue from aTgWnt5a and controls. Data represent the mean ± S.E. from five-seven mice in each group. *, p < 0.05.
The Cytoplasmic Tail of LRP1 Prevents Cholesterol Intracellular Accumulation Independently of Wnt5a Signaling
LRP1 is known to induce ABCA1 expression (5) and thus could also limit intracellular cholesterol accumulation through a Wnt5a-independent pathway. To test this, we transfected an expression vector coding for the LRP1 β-chain (Fig. 5A) in LRP1−/− MEFs and subjected these cells to an adipogenic mixture for 10 days. Under these conditions TGF-β cannot bind to LRP1 and did not induce Wnt5a (Fig. 1, A and B). We found that the LRP1 β-chain sufficed to reduce intracellular cholesterol accumulation (Fig. 5B). Interestingly, transfection of the LRP1 β-chain in LRP1−/− MEFs was sufficient to increase mRNA levels of ABCA1 by 50% (Fig. 5C). Because tyrosine phosphorylation of the LRP1 cytoplasmic domain increases its affinity for some adaptor proteins that are involved in signaling pathways, we replaced the Y of the NPXY motifs located within the cytoplasmic tail of LRP1 with an F residue (Fig. 6A). Expression vectors encoding these mutant forms of LRP1 were stably transfected in LRP1−/− MEFs, and cells analyzed for their ability to accumulate cholesterol upon adipogenesis. Cholesterol accumulation was abolished when the second NPXY (C-Ter) motif was mutated, whereas when the first NPXY (N-Ter) or both motifs (N-Ter/C-Ter) were mutated cells accumulated large amounts of cholesterol (Fig. 6B). MEFs bearing a mutation on the C-Ter motif behaved like LRP1 wild type cells and expressed large amounts of ABCA1 mRNA (Fig. 6D) and of the phospholipoprotein transfer protein (PLTP) known to potentiate ABCA1 activity (29) (Fig. 6E). They also exported about two times more cholesterol than when the first NPXY (N-Ter) or both motifs (N-Ter/C-Ter) were mutated (Fig. 6F). On the other hand, because these cells do not express the external domain of LRP1, they cannot mediate TGFβ signaling. As a consequence, ABCG1 mRNA (Fig. 6H) and protein levels (data not shown) and Wnt5a proteins levels (Fig. 6C) were not affected.
FIGURE 5.
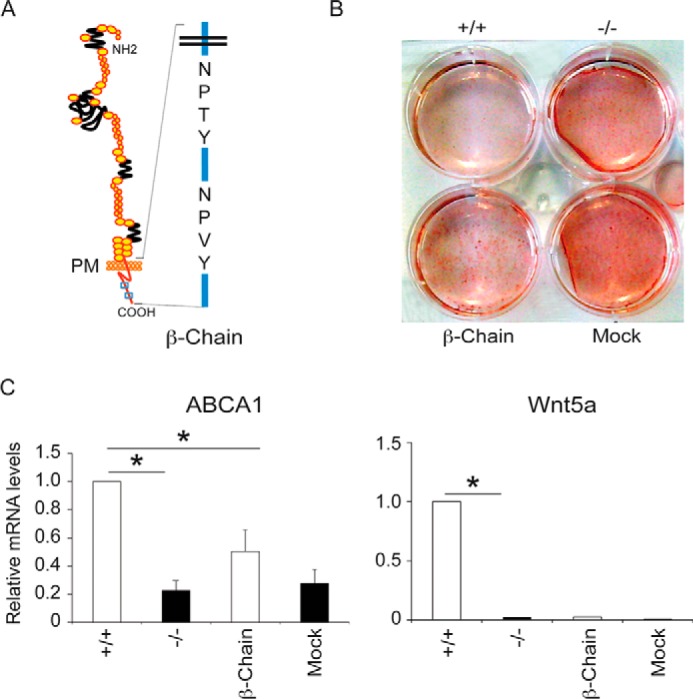
The cytoplasmic tail of LRP1 blocks cholesterol accumulation. Cells were treated with the adipogenic mixture for 10 days to stimulate cholesterol accumulation. A, schematic representation of the LRP1 cytoplasmic tail (β-Chain) with NPXY motifs. B, plates of LRP1 +/+, LRP1 −/− MEFs, and MEFs expressing the LRP1 β-chain or mock. The extent of cellular lipid accumulation was determined by Oil Red O staining (n = 5). C, quantitative RT-PCR analysis of the indicated genes in LRP1 +/+ and LRP1 −/− MEFs and in MEFs expressing the β-chain of LRP1 or mock (n = 5). Values are the means ± S.E. with p < 0.05 (*).
FIGURE 6.

The second NPXY motif within the cytoplasmic tail of LRP1 blocks cholesterol accumulation. Cells were treated with the adipogenic mixture for 10 days to stimulate cholesterol accumulation. A, schematic representation of the LRP1 cytoplasmic tail with the NPXY motif mutations. PM, plasma membrane. B, plates of LRP1−/− MEFs stably transfected with an expression vector coding for the β-chain of LRP1 and bearing the proximal NPXY (N-Ter), the distal NPXY (C-Ter), or both NPXY motifs (N-Ter + C-Ter) mutated (n = 8). The extent of cellular lipid accumulation was determined by Oil Red O staining. Shown is Western blot analysis of the indicated proteins (representative of n = 3) (C) and quantitative RT-PCR analysis of ABCA1 (D) and phospholipoprotein transfer protein (PLTP) (E) in the N-Ter, the C-Ter, the N-Ter + C-Ter mutants, and the mock control (n = 4). F, cholesterol efflux in the N-Ter, the C-Ter, the N-Ter + C-Ter mutants, and the mock control (n = 3). G, treatments of the N-Ter and the C-Ter mutants with a p-cPLA2α inhibitor (400 μm). The extent of cellular lipid accumulation was determined by Oil Red O staining. A representative experiment is shown (n = 3). Quantitative RT-PCR analysis of ABCG1 (n = 7) (H) and NCEH1 (I) in the N-Ter, the C-Ter, the N-Ter + C-Ter mutants, and the mock control (n = 4). Values are the means ± S.E. with p < 0.05 (*). NS = not significant.
To identify the protein complex that binds to LRP1 β-chain and inhibits intracellular cholesterol accumulation, we stably transfected LRP1−/− MEFs with a vector expressing a tagged LRP1 β-chain bearing the mutations in the C-Ter and N-Ter motifs described above. Proteins that bind to the tail of LRP1 were immunoprecipitated from lysates with antibodies against the tag and identified using a comparative proteomic analysis. Upon adipogenesis, mass spectrometry analysis showed that the second NPXY (C-Ter), but not the first NPXY motif, is required for binding of Erk2. In agreement with this, unlike cells bearing the mutated C-Ter motif, MEFs bearing the mutated N-Ter motif expressed large amounts of p-Erk1/2 and phosphorylated cytosolic phospholipase A2 (p-cPLA2; Fig. 6C), which are known to release arachidonic acid and antagonize ABCA1 expressions (30). In addition, inhibition of p-cPLA2 activity in these cells impaired neutral lipid accumulation, as evaluated by Oil Red O staining (Fig. 6G). Interestingly, MEFs bearing both the N-Ter and C-Ter mutations accumulated large amounts of cholesterol (Fig. 6B) and had low ABCA1 (Fig. 6D) and phospholipoprotein transfer protein (PLTP; Fig. 6E) transcript levels.
Because hydrolysis of cholesterol esters is the initial step of cholesterol export and only free cholesterol is available for its cellular efflux (31, 32), we tested whether LRP1 modifies the expression of the neutral cholesterol ester hydrolase (NCEH) (17). Fig. 6I shows that LRP1−/− MEFs contained lower NCEH1 transcript levels than LRP1+/+ MEFs. Moreover, MEFs bearing the mutated C-Ter motif expressed higher NCEH1 mRNA levels than MEFs bearing the mutated N-Ter mutation (Fig. 6I). Thus, the first (N-Ter) and the second NPXY motif (C-Ter) within the LRP1 β-chain have opposed effects on ABCA1 and NCEH1 mRNA levels. The first motif activates, whereas the second motif, through the binding of Erk2, decreases transcript levels of these two regulators of free cholesterol export.
Discussion
We investigated in this study how the ubiquitously expressed transmembrane receptor LRP1 prevents intracellular cholesterol accumulation. We show that two pathways are involved. TGFβ stimulates Wnt5a expression that down-regulates cholesterol biosynthesis and increases cholesterol efflux. This pathway requires the extracellular domain of LRP1. A second mechanism involves the C-terminal NPXY motif within the cytoplasmic domain of LRP1, which recruits Erk2. This is accompanied by an increase in the expression of NCEH1 and ABCA1 that allows cholesteryl esters to be hydrolyzed and free cholesterol to be exported from cells. This dual role of LRP1 concurs to efficiently maintain cholesterol homeostasis in cells (Fig. 7).
FIGURE 7.
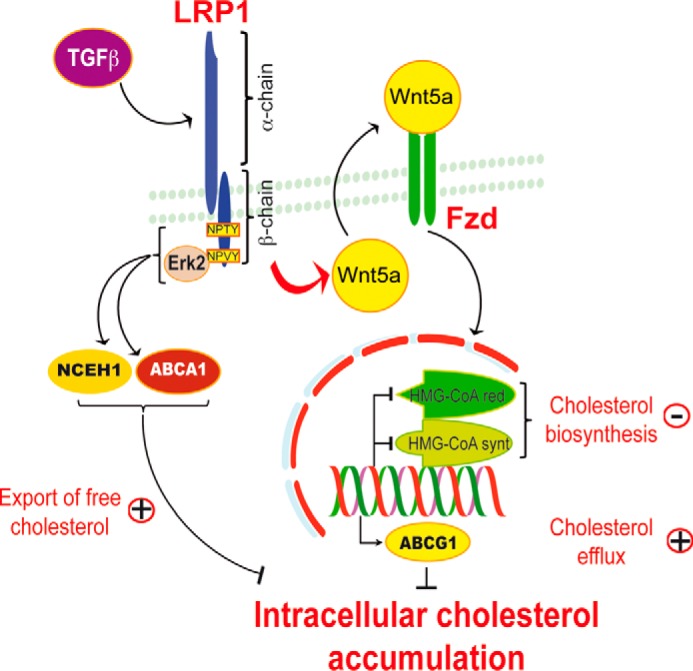
A schematic diagram of LRP1/Wnt5a-dependent inhibition of intracellular cholesterol accumulation. TGFβ through the LRP1 extracellular domain positively regulates a canonical Wnt5a signaling pathway. Wnt5a stimulates ABCG1-mediated cholesterol export. Wnt5a also inhibits SREBP-2 processing and cholesterol biosynthesis. Furthermore, the tail of LRP1 independently of Wnt5a blocks cholesterol accumulation through the binding of Erk2 to the second NPXY motif and the concomitant stimulation of ABCA1 and NCEH1 expressions. The synergy of the mechanisms that signal through both the extracellular domain and the cytoplasmic tail of LRP1 limits cholesterol intracellular accumulation. Conversely, in the absence of LRP1, Wnt5a is down-regulated, and cells accumulate cholesterol.
Our data show that Wnt5a restricts the cholesterol biosynthetic pathway by preventing proteolytic activation of SREBP-2 and decreasing HMG-CoA synthase and reductase expressions. Furthermore, Wnt5a stabilizes Insig-1 expression and triggers the action of oxysterols on the regulatory pathway. However, how Wnt5a interferes with sterols is unknown. Because there is no sterol-sensing domain on Wnt ligands, it is unlikely that Wnt5a binds directly oxysterols. Similarly, in the Insig-1·Scap·sterol complex, oxysterols do not bind to Scap, and a separate (not yet identified) sterol-binding protein might mediate the effects of sterols on Scap. Wnt5a might activate a sterol-binding protein that binds to Scap and stabilizes the Insig-1·Scap· cholesterol complex.
Wnt5a, which mediates a non-canonical Wnt signaling, is expressed at low levels in atherosclerotic plaques (33), and its expression increases in advanced lesions (34) and in lesions that undergo vascular calcification (12). Wnt5a is also associated with macrophage inflammatory responses (35) and plaque instability (36). Thus, the increase in Wnt5a levels in advanced lesions could be an attempt to block intracellular cholesterol accumulation, and this might ultimately lead to the deleterious effect of calcifying the lesions.
ABCG1 is widely expressed in many tissues and cell types, including macrophages (37–40), adipose tissue (41), neurons (42), and vascular smooth muscle cells (5). As reported by Zhou et al. (5), LRP1−/− primary cultured murine vSMCs with low levels of endogenous Wnt5a expressed very low levels of ABCG1. Here we show that deletion of Wnt5a in cultured human vSMCs or in 3T3-L1 preadipocytes using siRNA technology also decreases ABCG1 expression (Fig. 2G). In agreement with these results, ABCG1 mRNA and protein levels are increased in L-MTK cells and in MEFs stably overexpressing Wnt5a (Fig. 2, B–F). On the other hand, in LRP1+/+ MEFs we have not seen major differences in ABCG1 expressions compared with LRP1−/− MEFs. This indicates that Wnt5a induces ABCG1 expression in a cell-specific manner. ABCG1 is induced and translocated from the ER to the plasma membrane by LXR/RXR heterodimers (43–45), and this promotes the efflux of cellular cholesterol to circulating HDL (42–44, 46). Nonetheless, an earlier study reported that ABCG1 mRNA expression can be induced by LXR/RXR-independent mechanisms (47). Indeed, ABCG1 can show both intracellular and plasma membrane localizations even in the absence of LXR/RXR activators (47). Our results are consistent with this observation and show that Wnt5a stimulates ABCG1 mRNA and protein levels independently of LXRs. This stimulation, by increasing the level of protein in the cell might contribute to the redistribution of ABCG1 from the endoplasmic reticulum or another cellular compartment (48) into the plasma membrane. TGFβ is known to increase the expression of ABCG1, ABCA1, and cholesterol efflux in macrophage derived foam cells, with a particularly strong effect on ABCG1 (49). Because TGFβ participates in the inhibition of macrophage foam cell formation induced by very low density lipoprotein remnants (50), these results suggest an antiatherogenic role for the TGFβ/LRP1/Wnt5a pathway.
In the present study we also show that independently of the Wnt5a pathway the cytoplasmic tail of LRP1 is sufficient to limit cholesterol accumulation. We identified the second NPXY motif within the cytoplasmic tail of LRP1 as being critically involved in the regulation of ABCA1 expression, another determinant protein for cholesterol export. We show that mutation of this domain blocks cholesterol accumulation in MEFs upon induction of adipogenesis. We also show that the second NPXY motif within the cytoplasmic tail of LRP1 is required for the binding of Erk2 and that Erk1/2 is phosphorylated when cholesterol does not accumulate. Prior investigations have shown that p-Erk1/2 induces phosphorylation of the cPLA2. Activated cPLA2 then releases arachidonic acid from the phospholipid pool and suppresses ABCA1 expression (5). Here we show that the cytoplasmic tail of LRP1 not only recruits Erk2 but also mediates cPLA2 phosphorylation in MEFs. Interestingly, MEFs bearing the N-Ter mutation or both the N-Ter and C-Ter mutations accumulate large amounts of cholesterol and behave like LRP1−/− MEFs. They also exhibit low transcript levels of ABCA1, phospholipoprotein transfer protein, and NCEH1. This suggests that upon adipogenesis, a yet unidentified activator of ABCA1 binds to the N-Ter NPXY motif of the LRP1 β-chain. In conclusion, these regulatory events, which we have shown here in MEFs, could be modulated further in a cell type-specific manner in vivo depending on the presence and activity of interacting proteins and ancillary factors.
How LRP1 induces Wnt5a expression remains unclear. It might involve cleavage and nuclear translocation of the cytoplasmic domain of LRP1, a mechanism known to stimulate transcription. Likewise, upon LPS treatment intramembranous processing of LRP1 releases its cytoplasmic domain from the plasma membrane and promotes its translocation to the nucleus where it binds to and represses IFN-γ transcription (51).
Collectively, the data presented in our study demonstrate that Wnt5a and LRP1 have a remarkable synergistic effect on the regulation of intracellular cholesterol homeostasis. When LRP1 is deleted, cells accumulate large amounts of cholesterol due to the loss of ABCA1, NCEH1, and Wnt5. TGF-β-stimulated LRP1-mediated induction of Wnt5a is an event that requires the extracellular domain of LRP1 (Fig. 7). Wnt5a inhibits endogenous cholesterol biosynthesis and increases expression of ABCG1, one of the main proteins for cholesterol export (Fig. 7). In contrast, the expression of ABCA1 and NCEH1 is under the control of the first and the second NPXY motifs within the LRP1 β-chain (Fig. 7). Characterization of new factors interfering with the TGFβ/LRP1/Wnt5a pathway is a prerequisite to develop suitable new treatment strategies that will complement lipid-lowering therapies and help to fight against cholesterol accumulation diseases such as atherosclerosis.
Author Contributions
Z. E. A. and J. T. contributed equally to this work. Z. E. A., J. T., M. J., L. H., H. J., R. L. M., M. M., E. S., A. Z., and V. B. performed immunofluorescence, cell fractionation, cholesterol measurements, and immunoblot experiments. M. M., V. B., J. T., and J.-M. G. generated and analyzed the LRP1 mutant cells. Z. E. A., J. T., R. L. M., V. B., L. H., and H. J. characterized the aTgWnt5a mice. Z. E. A., J. T., V. B., E. S., and M. J. performed the quantitative real-time-PCR experiments. J.-M. G. and C. B. generated recombinant proteins. J. T. performed cholesterol efflux and luciferase experiments. V. B., J. T., R. L. M., M. J., and Z. E. A. performed all the statistical analyses. D. B., D. T., C. S., and A. V. D. performed the mass spectrometry analysis. All authors analyzed and discussed the data. P. B. coordinated the project. J. H., V. B., and P. B. supervised the project and wrote the manuscript.
Acknowledgments
We are grateful to Jean Marc Bornert (IGBMC, University of Strasbourg) for technical advice and expertise, Christian Bronner (IGBMC, University of Strasbourg) for human vascular smooth muscle cells, Daniel Metzger (IGBMC, University of Strasbourg) and Russell DeBose-Boyd and Jin Ye (Department of Molecular Genetics, UT Southwestern Medical Center, Dallas, TX) for critical reading of the manuscript.
This work was supported by grants from Fondation de France, Fondation pour la Recherche Médicale (FRM), and the Agence Nationale de la Recherche (ANR-06-Physio-032-01 and ANR-09-BLAN-0121-01). This work was also supported by National Institutes of Health Grant HL63762 (to J. H.). The authors declare that they have no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LRP1
- lipoprotein receptor-related protein 1
- vSMC
- vascular smooth muscle cell
- ABC
- ATP binding cassette transporter
- ER
- endoplasmic reticulum
- SREBP
- sterol regulatory element-binding protein
- HMG-CoA
- 3-hydroxy-3-methylglutaryl-coenzyme A
- NCEH
- neutral cholesterol ester hydrolase
- MEF
- mouse embryonic fibroblast
- LXR
- liver X receptor
- RXR
- retinoid X receptor
- PAQR3
- progestin and adipoQ receptors 3
- C-Ter
- second NPXY
- N-Ter
- proximal NPXY motif
- cPLA2
- cytosolic phospholipase A2.
References
- 1. WRITING GROUP MEMBERS, Lloyd-Jones D., Adams R. J., Brown T. M., Carnethon M., Dai S., De Simone G., Ferguson T. B., Ford E., Furie K., Gillespie C., Go A., Greenlund K., Haase N., Hailpern S., Ho P. M., Howard V., Kissela B., Kittner S., Lackland D., Lisabeth L., Marelli A., McDermott M. M., Meigs J., Mozaffarian D., Mussolino M., Nichol G., Roger V. L., Rosamond W., Sacco R., Sorlie P., Stafford R., Thom T., Wasserthiel-Smoller S., Wong N. D., Wylie-Rosett J., and American Heart Association Statistics Committee and Stroke Statistics Subcommittee (2010) Heart disease and stroke statistics-2010 update: a report from the American Heart Association. Circulation 121, e46–e215 [DOI] [PubMed] [Google Scholar]
- 2. Williams K. J., and Tabas I. (1995) The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 15, 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herz J., Hamann U., Rogne S., Myklebost O., Gausepohl H., and Stanley K. K. (1988) Surface location and high affinity for calcium of a 500-kDa liver membrane protein closely related to the LDL-receptor suggest a physiological role as lipoprotein receptor. The EMBO J. 7, 4119–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. May P., Woldt E., Matz R. L., and Boucher P. (2007) The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann. Med. 39, 219–228 [DOI] [PubMed] [Google Scholar]
- 5. Zhou L., Choi H. Y., Li W. P., Xu F., and Herz J. (2009) LRP1 controls cPLA2 phosphorylation, ABCA1 expression and cellular cholesterol export. Plos ONE 4, e6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boucher P., Gotthardt M., Li W. P., Anderson R. G., and Herz J. (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332 [DOI] [PubMed] [Google Scholar]
- 7. Boucher P., Li W. P., Matz R. L., Takayama Y., Auwerx J., Anderson R. G., and Herz J. (2007) LRP1 functions as an atheroprotective integrator of TGFβ and PDFG signals in the vascular wall: implications for Marfan syndrome. Plos ONE 2, e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boucher P., Liu P., Gotthardt M., Hiesberger T., Anderson R. G., and Herz J. (2002) Platelet-derived growth factor mediates tyrosine phosphorylation of the cytoplasmic domain of the low Density lipoprotein receptor-related protein in caveolae. J. Biol. Chem. 277, 15507–15513 [DOI] [PubMed] [Google Scholar]
- 9. Loukinova E., Ranganathan S., Kuznetsov S., Gorlatova N., Migliorini M. M., Loukinov D., Ulery P. G., Mikhailenko I., Lawrence D. A., and Strickland D. K. (2002) Platelet-derived growth factor (PDGF)-induced tyrosine phosphorylation of the low density lipoprotein receptor-related protein (LRP): evidence for integrated co-receptor function between LRP and the PDGF. J. Biol. Chem. 277, 15499–15506 [DOI] [PubMed] [Google Scholar]
- 10. Zhou L., Takayama Y., Boucher P., Tallquist M. D., and Herz J. (2009) LRP1 regulates architecture of the vascular wall by controlling PDGFRβ-dependent phosphatidylinositol 3-kinase activation. PLoS ONE 4, e6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Terrand J., Bruban V., Zhou L., Gong W., El Asmar Z., May P., Zurhove K., Haffner P., Philippe C., Woldt E., Matz R. L., Gracia C., Metzger D., Auwerx J., Herz J., and Boucher P. (2009) LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J. Biol. Chem. 284, 381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Woldt E., Terrand J., Mlih M., Matz R. L., Bruban V., Coudane F., Foppolo S., El Asmar Z., Chollet M. E., Ninio E., Bednarczyk A., Thiersé D., Schaeffer C., Van Dorsselaer A., Boudier C., Wahli W., Chambon P., Metzger D., Herz J., and Boucher P. (2012) The nuclear hormone receptor PPARγ counteracts vascular calcification by inhibiting Wnt5a signalling in vascular smooth muscle cells. Nat. Commun. 3, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumawat K., Menzen M. H., Bos I. S., Baarsma H. A., Borger P., Roth M., Tamm M., Halayko A. J., Simoons M., Prins A., Postma D. S., Schmidt M., and Gosens R. (2013) Noncanonical WNT-5A signaling regulates TGF-β-induced extracellular matrix production by airway smooth muscle cells. FASEB J. 27, 1631–1643 [DOI] [PubMed] [Google Scholar]
- 14. Goldstein J. L., DeBose-Boyd R. A., and Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 15. Horton J. D., Goldstein J. L., and Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horton J. D., Shimomura I., Brown M. S., Hammer R. E., Goldstein J. L., and Shimano H. (1998) Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J. Clin. Invest. 101, 2331–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sekiya M., Osuga J., Igarashi M., Okazaki H., and Ishibashi S. (2011) The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J. Atheroscler. Thromb. 18, 359–364 [DOI] [PubMed] [Google Scholar]
- 18. Hua X., Sakai J., Ho Y. K., Goldstein J. L., and Brown M. S. (1995) Hairpin orientation of sterol regulatory element-binding protein-2 in cell membranes as determined by protease protection. J. Biol. Chem. 270, 29422–29427 [DOI] [PubMed] [Google Scholar]
- 19. Sakai J., Duncan E. A., Rawson R. B., Hua X., Brown M. S., and Goldstein J. L. (1996) Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046 [DOI] [PubMed] [Google Scholar]
- 20. Hua X., Sakai J., Brown M. S., and Goldstein J. L. (1996) Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 271, 10379–10384 [DOI] [PubMed] [Google Scholar]
- 21. Lorkowski S., Kratz M., Wenner C., Schmidt R., Weitkamp B., Fobker M., Reinhardt J., Rauterberg J., Galinski E. A., and Cullen P. (2001) Expression of the ATP-binding cassette transporter gene ABCG1 (ABC8) in Tangier disease. Biochem. Biophys. Res. Commun. 283, 821–830 [DOI] [PubMed] [Google Scholar]
- 22. Wang N., Lan D., Chen W., Matsuura F., and Tall A. R. (2004) ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 9774–9779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vedhachalam C., Duong P. T., Nickel M., Nguyen D., Dhanasekaran P., Saito H., Rothblat G. H., Lund-Katz S., and Phillips M. C. (2007) Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J. Biol. Chem. 282, 25123–25130 [DOI] [PubMed] [Google Scholar]
- 24. Vedhachalam C., Ghering A. B., Davidson W. S., Lund-Katz S., Rothblat G. H., and Phillips M. C. (2007) ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler. Thromb. Vasc. Biol. 27, 1603–1609 [DOI] [PubMed] [Google Scholar]
- 25. Russell D. W. (2000) Oxysterol biosynthetic enzymes. Biochim. Biophys. Acta 1529, 126–135 [DOI] [PubMed] [Google Scholar]
- 26. Xu D., Wang Z., Zhang Y., Jiang W., Pan Y., Song B. L., and Chen Y. (2015) PAQR3 modulates cholesterol homeostasis by anchoring Scap/SREBP complex to the Golgi apparatus. Nat. Commun. 6, 8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krause B. R., and Hartman A. D. (1984) Adipose tissue and cholesterol metabolism. J. Lipid Res. 25, 97–110 [PubMed] [Google Scholar]
- 28. Kovanen P. T., Nikkilä E. A., and Miettinen T. A. (1975) Regulation of cholesterol synthesis and storage in fat cells. J. Lipid Res. 16, 211–223 [PubMed] [Google Scholar]
- 29. Oram J. F., Wolfbauer G., Vaughan A. M., Tang C., and Albers J. J. (2003) Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J. Biol. Chem. 278, 52379–52385 [DOI] [PubMed] [Google Scholar]
- 30. Ou J., Tu H., Shan B., Luk A., DeBose-Boyd R. A., Bashmakov Y., Goldstein J. L., and Brown M. S. (2001) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. U.S.A. 98, 6027–6032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghosh S., St Clair R. W., and Rudel L. L. (2003) Mobilization of cytoplasmic CE droplets by overexpression of human macrophage cholesteryl ester hydrolase. J. Lipid Res. 44, 1833–1840 [DOI] [PubMed] [Google Scholar]
- 32. Soccio R. E., and Breslow J. L. (2004) Intracellular cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 24, 1150–1160 [DOI] [PubMed] [Google Scholar]
- 33. Christman M. A. 2nd, Goetz D. J., Dickerson E., McCall K. D., Lewis C. J., Benencia F., Silver M. J., Kohn L. D., and Malgor R. (2008) Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 294, H2864–H2870 [DOI] [PubMed] [Google Scholar]
- 34. Malgor R., Bhatt P. M., Connolly B. A., Jacoby D. L., Feldmann K. J., Silver M. J., Nakazawa M., McCall K. D., and Goetz D. J. (2014) Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflamm. Res. 63, 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pereira C. P., Bachli E. B., and Schoedon G. (2009) The wnt pathway: a macrophage effector molecule that triggers inflammation. Curr. Atheroscler. Rep. 11, 236–242 [DOI] [PubMed] [Google Scholar]
- 36. Tsaousi A., Mill C., and George S. J. (2011) The Wnt pathways in vascular disease: lessons from vascular development. Curr. Opin. Lipidol. 22, 350–357 [DOI] [PubMed] [Google Scholar]
- 37. Moore K. J., Sheedy F. J., and Fisher E. A. (2013) Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13, 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosenson R. S., Brewer H. B. Jr., Davidson W. S., Fayad Z. A., Fuster V., Goldstein J., Hellerstein M., Jiang X. C., Phillips M. C., Rader D. J., Remaley A. T., Rothblat G. H., Tall A. R., and Yvan-Charvet L. (2012) Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125, 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adorni M. P., Zimetti F., Billheimer J. T., Wang N., Rader D. J., Phillips M. C., and Rothblat G. H. (2007) The roles of different pathways in the release of cholesterol from macrophages. J. Lipid Res. 48, 2453–2462 [DOI] [PubMed] [Google Scholar]
- 40. Phillips M. C. (2014) Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 289, 24020–24029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchmann J., Meyer C., Neschen S., Augustin R., Schmolz K., Kluge R., Al-Hasani H., Jürgens H., Eulenberg K., Wehr R., Dohrmann C., Joost H. G., and Schürmann A. (2007) Ablation of the cholesterol transporter adenosine triphosphate-binding cassette transporter G1 reduces adipose cell size and protects against diet-induced obesity. Endocrinology 148, 1561–1573 [DOI] [PubMed] [Google Scholar]
- 42. Tarr P. T., and Edwards P. A. (2008) ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J. Lipid Res. 49, 169–182 [DOI] [PubMed] [Google Scholar]
- 43. Wang N., Ranalletta M., Matsuura F., Peng F., and Tall A. R. (2006) LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler. Thromb. Vasc. Biol. 26, 1310–1316 [DOI] [PubMed] [Google Scholar]
- 44. Tarling E. J., and Edwards P. A. (2011) ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. U.S.A. 108, 19719–19724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tarling E. J., and Edwards P. A. (2012) Dancing with the sterols: critical roles for ABCG1, ABCA1, miRNAs, and nuclear and cell surface receptors in controlling cellular sterol homeostasis. Biochim. Biophys. Acta 1821, 386–395 [DOI] [PubMed] [Google Scholar]
- 46. Ranalletta M., Wang N., Han S., Yvan-Charvet L., Welch C., and Tall A. R. (2006) Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arterioscler. Thromb. Vasc. Biol. 26, 2308–2315 [DOI] [PubMed] [Google Scholar]
- 47. Xie Q., Engel T., Schnoor M., Niehaus J., Hofnagel O., Buers I., Cullen P., Seedorf U., Assmann G., and Lorkowski S. (2006) Cell surface localization of ABCG1 does not require LXR activation. Arterioscler. Thromb. Vasc. Biol. 26, e143–144 [DOI] [PubMed] [Google Scholar]
- 48. Maxfield F. R., and Tabas I. (2005) Role of cholesterol and lipid organization in disease. Nature 438, 612–621 [DOI] [PubMed] [Google Scholar]
- 49. Hu Y. W., Wang Q., Ma X., Li X. X., Liu X. H., Xiao J., Liao D. F., Xiang J., and Tang C. K. (2010) TGF-β1 up-regulates expression of ABCA1, ABCG1 and SR-BI through liver X receptor α signaling pathway in THP-1 macrophage-derived foam cells. J. Atheroscler. Thromb. 17, 493–502 [DOI] [PubMed] [Google Scholar]
- 50. Argmann C. A., Van Den Diepstraten C. H., Sawyez C. G., Edwards J. Y., Hegele R. A., Wolfe B. M., and Huff M. W. (2001) Transforming growth factor-β1 inhibits macrophage cholesteryl ester accumulation induced by native and oxidized VLDL remnants. Arterioscler. Thromb. Vasc. Biol. 21, 2011–2018 [DOI] [PubMed] [Google Scholar]
- 51. Zurhove K., Nakajima C., Herz J., Bock H. H., and May P. (2008) γ-Secretase limits the inflammatory response through the processing of LRP1. Sci. Signal. 1, ra15. [DOI] [PMC free article] [PubMed] [Google Scholar]